

Abstract
The main NFκB transcription factor family members RelA-p50 heterodimer and RelA homodimer have different biological functions and show different transcriptional activation profiles. To investigate whether the two family members adopt a similar conformation in their free states, we performed hydrogen-deuterium exchange mass spectrometry (HDXMS), all-atom molecular dynamics (MD) simulations and stopped-flow binding kinetics experiments. Surprisingly, the N-terminal DNA binding domains adopt an open conformation in RelA-p50 but a closed conformation in RelA homodimer. Both HDXMS and MD simulations indicate the formation of an interface between the N-terminal DNA binding domains only in the RelA homodimer. Such an interface would be expected to impede DNA binding and stopped-flow binding kinetics show that association of DNA is slower for the homodimer as compared to the heterodimer. Our results show that the DNA-binding cavity in the RelA-p50 heterodimer is open for DNA binding whereas in the RelA homodimer, it is occluded.
Graphical abstract

Introduction
The nuclear factor kappa B (NFκB) family of transcription factors responds to a large number of extracellular stress stimuli, including factors controlling inflammation and the immune response1; 2; 3. Dysregulation of NFκB results in numerous disease states, particularly cancer4. NFκB is a member of the Rel homology domain-containing (RHD) family of proteins which are composed of two immunoglobulin-like (Ig-like) β-barrel subdomains connected by a linker. The family of NFκB proteins includes RelA, p50, p52, cRel and RelB, which form homo- and heterodimers. Utilizing mostly irregular loops, both Ig-like subdomains of both monomers engage the DNA major groove. The N-terminal Ig-like domains are typically referred to as the DNA-binding domains (DBDs) due to their large surface engaged in DNA-binding. The C-terminal Ig-like domains are referred to as the dimerization domains, but they also contribute substantial DNA binding affinity through phosphate-ribose contacts by amino acids from five ordered loops5.
We previously showed that the inhibitor of NFκB, IκBα, accelerates the dissociation of the canonical and most abundant NFκB (RelA-p50) from DNA in vitro in a kinetically-driven process we have termed “molecular stripping”6; 7; 8. Using HDXMS, we showed that free RelA-p50 is surprisingly dynamic, exchanging most of its amides within 5 min. DNA-binding reduces amide exchange within the DBDs, and surprisingly causes increased exchange long distances away from the DNA-binding site9. IκBα binding globally stabilizes RelA-p50 and reduces amide exchange throughout the entire molecule, including the DBDs, which are not in contact with the IκBα9. These data suggested that NFκB dimers may be dynamic, and are consistent with the idea that the relative orientation of the DBDs allosterically respond to binding partners, as we have shown is important for molecular stripping8.
The kinetics of binding of NFκB dimers to a single κB site DNA sequence are rapid, with association rates above 109 M−1s−1 and dissociation rates on the order of 0.5 s−1 as measured by stopped-flow fluorescence7. Intracellular fluorescence recovery after photobleaching experiments give similar rates10. The canonical NFκB (RelA-p50) heterodimer binds single κB-site-containing sequences with affinities in the 3-30 nM range. The RelA homodimer generally binds less tightly to single κB-site-containing sequences (>20 nM), with the difference being the most dramatic for the IgκB promoter sequence (KD of 6.3 nM vs. 7.4 μM)6.
Recent computational analyses of the p50 homodimer, RelA homodimer, and RelA-p50 heterodimer revealed that the heterodimer interface is more frustrated than the homodimer interfaces11 and further suggested that the frustration in the RelA-p50 heterodimer interface might facilitate movement of the DBDs to promote DNA binding. Here we used HDXMS and all-atom molecular dynamics (MD) simulations to investigate this hypothesis. We found that, indeed, the DBDs adopt an open orientation in the RelA-p50 heterodimer and a closed conformation in the RelA homodimer helping to explain the slower DNA binding kinetics observed for the homodimer.
Results
HDXMS comparison of NFκB (RelA-p50) heterodimer vs. NFκB (RelA) homodimer
The HDXMS was optimized to achieve 97% coverage of RelA (Suppl Fig 1) and p50 (Suppl Fig 3). To understand whether RelA dynamics differ when RelA is in the homodimer vs. when it is with p50 in the heterodimer, we compared deuterium uptake into each region of RelA using HDXMS. Both RelA and p50 showed high amide exchange throughout much of the protein within 5 min (Suppl. Fig 2 and Suppl Fig 4) as previously described9. Large differences were observed in the deuterium incorporation into RelA in the context of the RelA-p50 heterodimer as compared to the RelA homodimer, particularly in the DBDs. Within the dimerization domains, residues 195-214 (peptide MH+ 2181.090) and 240-249 (peptide MH+ 1110.565) of RelA, which contribute to the dimer interface12; 13, showed no significant difference in deuterium incorporation between RelA in the p50 heterodimer and RelA in the homodimer (cyan segments and boxed plots, Figure 1). Residues 250-270 (peptide MH+ 2344.248) also showed no difference in exchange. Two loop regions of the dimerization domain of RelA showed slightly lower incorporation of deuterium in the homodimer as compared to the heterodimer. These included residues 215-226, (peptide MH+ 1418.720) and 227-239 (peptide MH+ 1474.675) (green segments and boxed plots). Interestingly, the region from 286 to 309 (peptide MH+ 3143.566) containing the nuclear localization sequence which binds to importin-α14, also exchanged substantially less in RelA homodimer (Suppl Fig 2).
Figure 1.
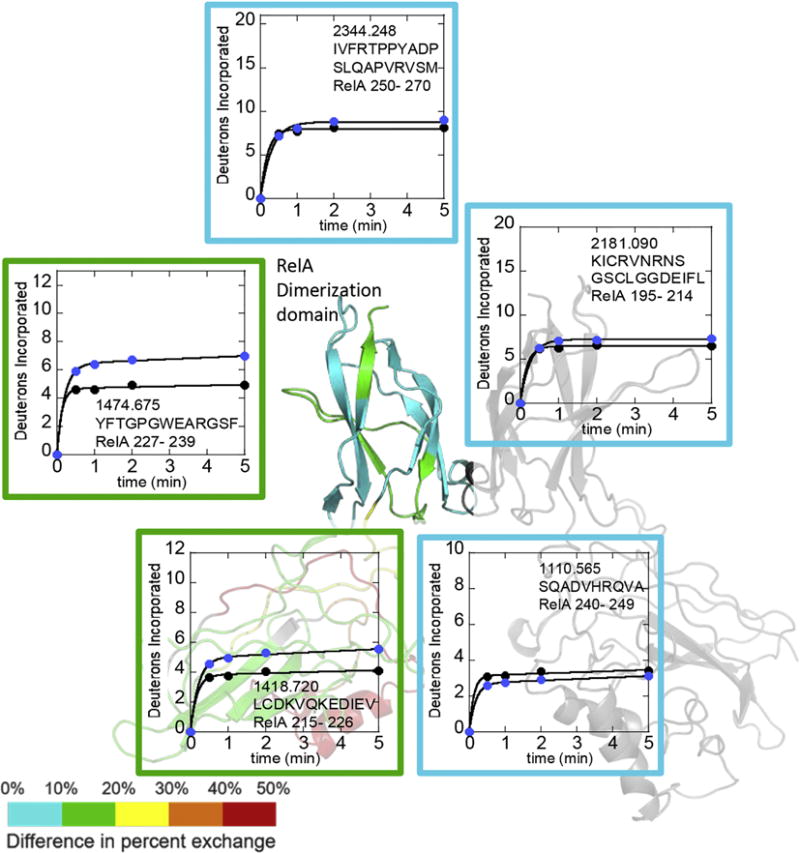
Deuterium uptake plots for regions of RelA from the dimerization domain. The RelA-p50 structure (1LE9 from which DNA was removed) dimerization domain is colored on a rainbow scale representing (uptake of RelA in heterodimer – uptake of RelA in homodimer) with red being the maximum and blue being the minimum. P50 is shown in grey. Uptake of deuterium into RelA is compared for the RelA homodimer (black circles) vs RelA in the heterodimer (blue circles). The plots are boxed according to the color of the region in the structure and are placed near the structural element corresponding to the peptide.
The RelA N-terminal DBD, showed more substantial differences in deuterium incorporation between the homodimer and the heterodimer (Figure 2). Much of the DBD incorporated less deuterium in the context of the homodimer as compared to RelA in the context of the RelA-p50 heterodimer. The N-terminal residues 23-34 (peptide MH+ 1502.826), which span the critical DNA-binding residue R33, incorporated almost 50% less deuterium into the homodimer as compared to the heterodimer (red segment and boxed plot in Figure 2). The core of the DBD including residues 35-76, (peptide MH+ 4505.332), residues 77- 99 (peptide MH+2653.305), residues 100-119 (peptide MH+ 2306.087) and residues 160-176 (peptide MH+1886.032) showed a 10-20% decrease in uptake (green in Figure 2). Residues 120-147, (peptide MH+ 3306.701, red in Figure 2), which form a helix that contains the critical DNA-interacting residues K123K124R125, showed a 50% decrease in deuterium uptake in the homodimer as compared to the heterodimer. Residues 148-156 (peptide MH+ 1051.480, orange in Figure 2) also showed a substantial difference in deuterium incorporation, perhaps because this segment lies underneath the helix corresponding to residues 120-147. Finally, residues 177-194 (peptide MH+ 1991.034, yellow in Figure 2) showed a 30% lower deuterium incorporation in the homodimer as compared to the heterodimer. This region corresponds to the linker which connects the DBD to the dimerization domain that contains the DNA-binding residue R187.
Figure 2.

Deuterium uptake plots for regions of RelA from the N-terminal DNA-binding domain. The RelA-p50 structure is colored as in Figure 1. Comparison of deuterium uptake for some regions of DNA binding domain of RelA-p50 (blue circles) and RelA homodimer (black circles). The plots are boxed in the same manner as in Figure 1.
Molecular dynamics simulations reveal distinct native state ensembles of NFκB dimers
To investigate the possible causes of the difference in deuterium uptake into RelA in the context of the homodimer as compared to the heterodimer, we performed all-atom MD simulations to explore the conformational dynamics of free RelA-p50 heterodimer and RelA homodimer. The simulations started from structures of DNA-bound conformations (PDB entries: 1LE9 and 1RAM) from which the DNA was computationally removed. The initial conformations of the two dimers were superimposable, with root mean square deviation (RMSD) of 2.2 Å. To explore whether the simulations had adequately sampled the final equilibrium state, two independent 400 ns simulations were performed for each system, starting with the same atomic coordinates but different random initial velocities.
Our results showed that, when not bound to DNA, the DBDs of both dimers displayed a wide range of motions by twisting around the linkers connecting the DBD to the dimerization domain (Suppl Figure 3). Rapid large-scale conformational relaxation from the initial DNA-bound conformations was observed as a change in the distance between centers of mass of DBDs over time (Figure 3A) as well as in the all-atom RMSD with respect to the starting conformations (Suppl Figure 4). Despite the high similarity of the RelA-p50 and RelA homodimer in the starting conformations (derived from the DNA-bound structures), they underwent distinct relaxation processes which resulted in significantly different structural ensembles over the time course of the trajectories. While the two DBDs in the RelA-p50 (gold) moved apart from each other and relaxed to an “open” conformational ensemble, those in the RelA homodimer (blue) came together and relaxed to a “closed” conformational ensemble (Figure 3A and Suppl trajectory movies 1-4). We next calculated the probability of the center-of-mass distances between the DBDs after 100 ns of simulation time to reveal the most populated conformational sub-states within the open and closed ensembles (Figure 3B). Whereas the two RelA-p50 trajectories reached the same degree of openness after ~350 ns, the two RelA homodimer simulations did not reach the same final structural ensemble leading to somewhat different average distances between the DBDs (Figure 3). Still, in both trajectories the RelA homodimer consistently closed to structures where the DBDs were in close contact, as opposed to the open conformations sampled by the RelA-p50 heterodimer. Remarkably, in both RelA homodimer trajectories, a new protein-protein interface appeared to form between the two DBDs, although the interfaces that formed in each trajectory were not the same.
Figure 3.

Distinct conformations of RelA-p50 and RelA homodimer revealed by MD. A) Time evolution of DBD center-of-mass distance showing the relaxation from DNA-bound to free state for RelA-p50 (gold) and RelA homodimer (blue). B) Probability distribution of DBD center-of-mass distance after 100 ns of simulation time reveals conformational sub-states of free RelA-p50 and RelA homodimer. C) Superimposed DNA-bound structures and representative structure for each conformational sub-state. The RelA-p50 heterodimer is shown in gold-grey and the homodimer in blue.
Electrostatic potential calculations predict domain rotations
We decided to investigate whether electrostatic potential differences may be the underlying cause of the opening of the RelA-p50 and closing of the RelA homodimer. Adaptive Poisson-Boltzmann Solver (APBS) electrostatic evaluation of the initial structures after the DNA was computationally removed revealed a much more positive DNA-binding cavity in the RelA-p50 heterodimer as compared to the RelA homodimer. Computation of electrostatic field lines showed the RelA and p50 DBDs repel each other whereas the RelA homodimer displayed an electrostatic attraction between parts of its DBDs (Figure 4). Interestingly, an attractive potential is observed between the p50 DBD and its own dimerization domain, which may explain why the p50 subunit shows a much larger displacement away from the DNA-bound conformation as compared to the RelA subunit.
Figure 4.
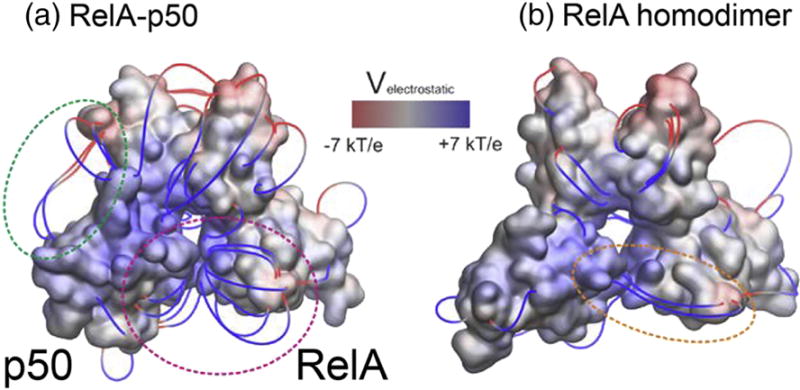
Electrostatics of RelA-p50 heterodimer (A) and of RelA homodimer (B). The electrostatic potential was calculated with APBS and mapped onto the 3D structures of the dimer after computational removal of the DNA and before they underwent any conformational changes in MD simulations. Positive regions are shown in blue and negative regions are shown in red, in a color scale ranging from −7 to +7 kBT/e. We are also displaying electrostatic field lines, filtered according to a gradient magnitude > 8kBT/e/Å. In the heterodimer, several field lines arise from the DBDs near the DNA binding cavity and go in opposite directions (magenta circle), suggesting an electrostatic repulsion. In the homodimer, a few field lines connect the two DBDs, suggesting an electrostatic attraction (orange circle). Field lines in the heterodimer also suggest an electrostatic attraction between the DBD and the dimerization domain of p50 (green circle).
In all simulations, both DBDs of RelA and p50 can rotate around the linker. Figure 5 shows the different orientations of DBDs in the initial and end stages of the simulations. In the initial structures (derived from the DNA-bound conformation), residues responsible for base-specific interaction with the DNA (RelA: R33, R35, Y36, E39 and R187; p50: R54, R56, Y57 E60, H64 and K241) pointed into the DNA-binding cavity. In both RelA-p50 simulations, the RelA DBD rotated and DNA-base-contacting residues pointed away from the binding cavity. The p50 DBD in RelA-p50 rotated to a greater degree than RelA DBD did, making new contacts with a loop of its dimerization domain (Figure 5A).
Figure 5.

Initial and final structures from simulations of heterodimer (RelA in gold, p50 in grey) and homodimer (blue) showing how DBDs rotate around the linkers. Residues responsible for base-specific interactions with the DNA (RelA: R33, R35, Y36, E39, and R187; p50: R54, R56, Y57, E60, H64, and K241) are shown as red spheres. A) In RelA-p50 simulations, the RelA DBD rotated and DNA-binding residues pointed away from the binding cavity. The p50 DBD in RelA-p50 rotated to a greater degree than RelA DBD did. B) In RelA homodimer simulations, the DBDs rotated and formed a new interface with each other. The different directions of the DBD rotation motions in simulation 1 and 2 led to different interacting interfaces and different positions of DNA base-contacting residues in the final structures.
A protein interface is observed in the RelA homodimer DBDs by both MD and HDXMS
We examined the agreement between representative conformations of the major sub-states observed by MD and the H/D exchange patterns obtained from the HDXMS experiments (Figure 6). Overall, the entire DBD of RelA was more protected in the homodimer than in the heterodimer. A helix (residues 120-136) and the following loop (residues 137-156) exchanged 50% more in the RelA-p50 heterodimer than in the homodimer. This region was completely exposed in the RelA-p50 heterodimer simulations, but was buried at the interface in both MD simulations of the RelA homodimer.
Figure 6.

Representative conformations of RelA homodimer and RelA-p50 heterodimer observed in MD colored according to the HDXMS results on a rainbow scale representing percent uptake with red being the maximum and blue being the minimum. p50 is shown in grey. A) RelA-p50 prefers an open conformation in which RelA residues 120-156 are exposed and show high deuterium exchange (red regions). B) One of the two closed ensemble of conformations from different simulations of RelA homodimer. The loop containing residues 146-156 in one DBD interacts with the loop containing residues 41-56 in the other DBD. C) The other closed ensemble of conformations of RelA homodimer, in which helices from the two DBDs interact with each other.
In both RelA homodimer simulations, the DBDs rotated and formed a new interface with each other. The different directions of the DBD rotation motions in the two simulations led to two different interfaces with different positions of the DNA base-contacting residues and different specific interactions in the final ensemble of structures (Figure 5B). In simulation 1, DNA base-contacting residues from both DBDs were facing the same direction and the loop formed by residues 146-156 in one DBD interacted with the loop formed by residues 41-56 in the other DBD. Residues 148-156 exchanged 40% less in the homodimer, whereas residues 41-56 are found in a large peptide (residues 35-76) in the HDXMS experiment which exchanged 25% less in the homodimer. In simulation 2, a helix-loop structure (residue 123-151) from one DBD interacted with the same helix-loop from the other DBD. They ran antiparallel across the interface so the DNA-base-contacting residues from the two DBDs were pointing in opposite directions. In the HDXMS experiments, residues 120-147 exchanged 50% less in the homodimer as compared to the heterodimer.
To understand the interactions across the DBD interface of RelA homodimer, we analyzed the salt bridges formed during the MD simulations (Figure 7). Five stable inter-DBD salt bridges (all listed with the residue from subunit 1 given first and the residue from subunit 2 given second); R50-E222, D53-R187, D53-K123, K56-D151, and R41-E147; were identified in simulation 1 and three; D151-K123, E127-R124, and R124-E146; in simulation 2. These salt bridges were formed in the later stage of the trajectories as the two DBDs came together (Suppl. Fig 5 and 6). All these salt bridges at the DBD interface last for at least 100 ns with small fluctuations.
Figure 7.
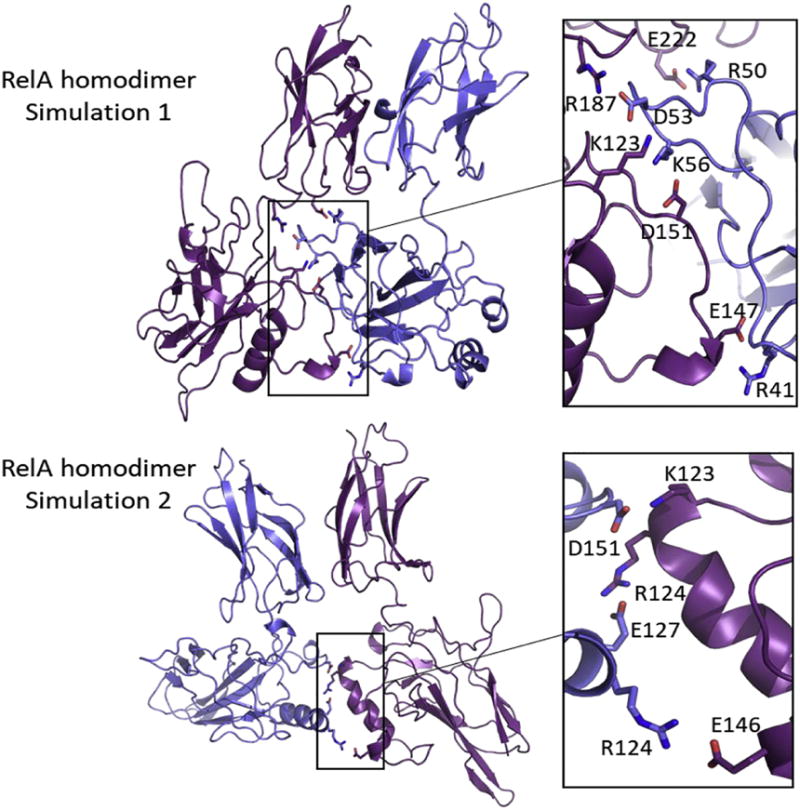
Salt bridges across the interface between the DBDs in RelA homodimer. The two RelA subunits are shown in blue and purple. Oxygen atoms of acidic side chains are shown in red and nitrogen atoms of basic side chains are shown in blue. A) Five stable salt bridges (E222-R50, D53-R187, D53-K123, D151-K56, and E147-R41) were found at the interface in simulation 1. (B) Three stable salt bridges D151-K123, E127-R124, and E146-R124) were found at the interface in simulation 2.
Kinetics of DNA binding
Both the MD and the HDXMS data indicated that the DBDs in the free RelA homodimer form a new interacting interface. To test whether this interface may impede DNA binding, we carried out stopped-flow experiments to measure the DNA binding rate for the RelA-p50 heterodimer and RelA homodimer. We had previously measured DNA binding kinetics by monitoring the fluorescence intensity increase of a pyrene-labeled DNA hairpin corresponding to the IFN κB site upon rapid mixing with the NFκB dimer7. As reported previously, the fluorescence increases upon RelA-p50 binding to labeled DNA with bi-exponential kinetics, however the second phase was slow with a weak dependence on protein concentration7. We therefore compared the fast phases of the two association curves. The association rate constant for labeled DNA binding was 2.7 ± 0.06 × 108 M−1 s−1 for binding to RelA-p50 and 0.75 ± 0.02 × 108 M−1 s−1 for binding to RelA homodimer. Thus, association of DNA to the RelA homodimer was approximately 3.6 times slower than association to the RelA-p50 (Figure 8).
Figure 8.
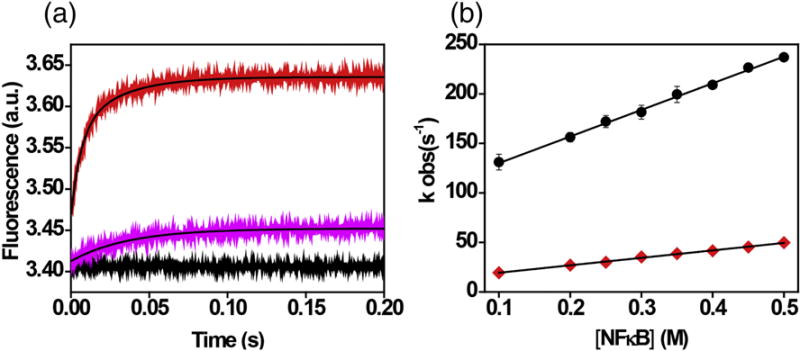
Stopped-flow fluorescence kinetics of RelA-p50 and RelA homodimer binding to pyrene-labeled DNA at various protein concentrations. A) Representative kinetic traces of association of RelA-p50 (red line) and RelA homodimer (pink) with DNA (Final Concentrations of DNA and RelA-p50 and RelA homodimer are 0.1 μM and 0.2 μM respectively). The black line represents the DNA baseline. B) Plot of the concentration dependence of NFκB association with DNA; RelA-p50 (black circles, slope = 2.7 ± 0.06 × 108 M−1 s−1) and RelA homodimer (red diamonds, slope = 0.75 ± 0.02 × 108 M−1s−1).
Discussion
The combined HDXMS and all-atom MD simulations provide the first evidence of the solution conformation(s) of the free NFκB RelA-p50 heterodimer and RelA homodimer. Although the DNA-bound conformations are highly similar, both the HDXMS and all-atom MD revealed significant differences in the RelA amide exchange depending on whether it was in the homodimer or in the RelA-p50 heterodimer. We observed slightly higher amide exchange in the dimerization domain of the heterodimer consistent with recent coarse-grained simulations that predicted higher local frustration in the heterodimer dimerization interface11. The regions with the largest difference in HDX were in the RelA DBD where the heterodimer showed high amide exchange and the homodimer showed much lower exchange. There were two possible explanations for this. On the one hand, the RelA DBD might be more folded into a compact domain in the homodimer while being somewhat disordered in the heterodimer. On the other hand, the DBDs could be forming an interface in the homodimer. To explore these possible explanations, we performed all-atom MD simulations. During the simulations, the heterodimer DBDs rapidly moved farther apart from each other whereas the homodimer DBDs moved closer together. These conformational relaxation processes led to an “open” ensemble for the heterodimer and an “closed” ensemble for the homodimer.
In all of the simulations, the DBDs reoriented relative to the dimerization domain in both RelA and p50, which preferred somewhat different relative domain orientations. The p50 DBD flipped up and appeared to form contacts between its positively charged loop (residues 74-77, KNKK) and a negatively charged loop in the dimerization domain (residues 284-287, EEEE). This reorientation of the p50 DBD opened the DNA-binding cavity exposing it to higher amide exchange. In all simulations, the RelA DBDs rotated around the DNA-binding position, but did not move much away from their original position. In the homodimer simulations, the rotation of the DBDs appeared to allow the formation of inter-domain contacts.
An interface formed between the DBDs in the simulations of the homodimer. In one simulation, the interface formed between loops and in the other simulation it was formed between helices. Both the loops and the helices were the regions of RelA that showed the most dramatic difference in exchange suggesting that the interfaces formed in the MD simulations may both be contributing to the lower observed exchange in the homodimer. These different interfaces suggested that instead of forming a unique strong inter-domain interaction, the DBDs could interact from different orientations in the homodimer. Both interfaces were stabilized by salt bridges, which could break and allow the DBD to rotate before they interact again and form a new interface. Such breaking and reforming of salt bridges is not expected to be observed within the 400 ns MD simulations. Taken together, the HDXMS and MD simulations are most consistent with the formation of an interface between the RelA DBDs in the homodimer.
We had previously reported that the RelA homodimer binds DNA more weakly than the heterodimer, and this is true for a number of different κB promoter sites including those for MIP2, RANTES, IgκB, IFN, and urokinase6. For most of the promoters, the homodimer bound 3-5 fold more weakly than the heterodimer. The weaker binding observed in the homodimer could be due to either lower association rate, or faster dissociation rate, or both. If, indeed, the free homodimer occluded the DNA binding cavity due to the formation of an interface between the DBDs, the association rate should be decreased. Using stopped-flow fluorescence, we were able to show that for the IFN promoter sequence, all of the difference in binding affinity could be accounted for by a decreased association rate. Taken together, our results strongly support the formation of a DBD interface in the homodimer that occludes DNA-binding. We considered testing the propensity for DBD domain reorientation and homodimer DBD interface formation using site-directed mutagenesis, but the electrostatic potential calculations suggest that all three parts of the proteins, the dimerization domain, the linker, and the DBD contribute to the electrostatic interaction which apparently drives the RelA homodimers toward each other and the p50 subunit away. Disruption of one charged residue among all of them is not likely to have a very large effect. In the homodimer, two different DBD-DBD interfaces were observed in the simulations suggesting that while an interface can form, it is not particularly stable or unique, and therefore again a single point mutation would not likely disrupt this “fuzzy” interaction completely. Our results provide a mechanistic explanation for why the RelA homodimer binds DNA more weakly, an observation that was not understood from the analysis of static structures of the DNA-bound dimers.
Materials and Methods
Protein expression and purification
Murine, N-terminal hexahistidine-p5039–350/RelA19–321 heterodimer (full-length NFκB) was co-expressed as described previously15 RelA-p50 was purified using nickel affinity chromatography, followed by cation exchange chromatography (MonoS; GE healthcare) and finally size exclusion chromatography (Superdex S200; GE healthcare). It is important to note that by using a His-tagged p50, every RelA molecule in the preparation is present as a RelA-p50 heterodimer. Most of the experiments were performed with fresh proteins preparations. RelA homodimer was purified using cation exchange chromatography (SP sepharose Fast Flow; GE healthcare). Further, protein was purified using cation exchange chromatography (MonoS; GE healthcare) and finally size exclusion chromatography (Superdex S200; GE healthcare). Protein concentration of RelA-p50 (ε = 43760 M/cm) and RelA (ε = 40800 M/cm) were determined using their molar extinction coefficients.
Labeled DNA preparation
A hairpin DNA sequence corresponding to the ten-nucleotide IFN-κB site (underlined) 5′-AmMC6/GGGAAATTCCTCCCCCAGGAATTTCCC-3′ (IDT Technologies) was labeled with pyrene succinimide (Sigma) and purified as described7.
Hydrogen-deuterium exchange mass spectrometry
Hydrogen/deuterium exchange mass spectrometry (HDXMS) was performed using a Waters Synapt G2Si equipped with nanoACQUITY UPLC system with H/DX technology and a LEAP autosampler. Individual proteins were purified by size exclusion chromatography in 25 mM Tris pH 7.5, 150 mM NaCl, 1 mM DTT, 0.5 mM EDTA immediately prior to analysis. The final concentrations of proteins in each sample were 5 μM. For each deuteration time, 4 μL complex was equilibrated to 25 °C for 5 min and then mixed with 56 μL D2O buffer (25 mM Tris pH 7.5, 150 mM NaCl, 1 mM DTT, 0.5 mM EDTA in D2O) for 0, 0.5, 1, 2, or 5 min. The exchange was quenched with an equal volume of quench solution (3 M guanidine, 0.1% formic acid, pH 2.66).
The quenched sample (50 μL) was injected into the sample loop, followed by digestion on an in-line pepsin column (immobilized pepsin, Pierce, Inc.) at 15°C. The resulting peptides were captured on a BEH C18 Vanguard pre-column, separated by analytical chromatography (Acquity UPLC BEH C18, 1.7 μM, 1.0 × 50 mm, Waters Corporation) using a 7-85% acetonitrile in 0.1% formic acid over 7.5 min, and electrosprayed into the Waters SYNAPT G2Si quadrupole time-of-flight mass spectrometer. The mass spectrometer was set to collect data in the Mobility, ESI+ mode; mass acquisition range of 200–2,000 (m/z); scan time 0.4 s. Continuous lock mass correction was accomplished with infusion of leu-enkephalin (m/z = 556.277) every 30 s (mass accuracy of 1 ppm for calibration standard). For peptide identification, the mass spectrometer was set to collect data in MSE, ESI+ mode instead.
The peptides were identified from triplicate MSE analyses of 10 μM NFκB, and data were analyzed using PLGS 2.5 (Waters Corporation). Peptide masses were identified using a minimum number of 250 ion counts for low energy peptides and 50 ion counts for their fragment ions. The peptides identified in PLGS were then analyzed in DynamX 3.0 (Waters Corporation) and the deuterium uptake was corrected for back-exchange as previously described9. The RelA peptides identified from the RelA homodimer were the same as those identified from the RelA in the RelA-p50 heterodimer because we ensured that the pepsin digestion went to completion. The relative deuterium uptake for each peptide was calculated by comparing the centroids of the mass envelopes of the deuterated samples vs. the undeuterated controls following previously published methods16. The experiments were performed in triplicate, and independent replicates of the triplicate experiment were performed to verify the results.
Molecular Dynamics Simulations
The atomic coordinates of mus musculus NFκB dimers (RelA-p50 and RelA homodimer) were obtained from crystal structures of NFκB-DNA complexes (PDB entries: 1LE9 and 1RAM), in which RelA ranges from residue 19 to 291, missing the NLS region (292-321), and p50 ranges from residue 39 to 350. DNA molecules were computationally removed for simulating free NFκB. Prior to simulations, the two NFκB dimers were aligned. The RMSD of the aligned structures was 2.2 Å as calculated by Lovoalign17. The protonation states of ionic residues were assigned at pH 7 using the H++ server18. Proteins were solvated with TIP3P water19 in a rectangular box with a 15 Å buffer region using tleap in the AMBER package20. Chloride ions were added to keep the systems electrically neutral. The total numbers of atoms are approximately 150000 for RelA-p50 and 160000 for RelA homodimer. To allow the bonds and angles to relax and eliminate possible bad contacts, the complete and solvated model was submitted to the following refinement procedure using AMBER 1420 and the ff14SB forcefield21: i) 1000 steps of energy minimization of the water molecules with the protein atoms restrained and ii) 2500 steps of energy minimization with no position restraints. Each stage was performed using conjugate gradient method for 1000 and 2500 cycles.
Molecular dynamics simulations were performed with periodic boundary conditions and an integration time step of 2 fs. A Langevin thermostat was used for temperature control. The SHAKE algorithm was used to constrain hydrogen atoms during the simulation. The particle-mesh Ewald method22 was used to handle long-range electrostatic interactions. A cutoff of 8 Å was used for short-range nonbonded interactions. Equilibration of the systems was performed in two stages. First, the system was heated up from 100 K to 300 K with position restraints on solutes with a force constant of 10 kcal/mol for 20 ps. Next, the entire system was equilibrated at 300 K and 1 bar for 1 ns to equilibrate the density. After equilibration, two independent 400-ns production runs were performed for each system. In the NVT regime, atomic coordinates were recorded every 2 ps and the resulting trajectories were visualized using Visual Molecular Dynamics (VMD)23. CPPTRAJ24 in the AMBER package was used to perform RMSD and distance analysis. The distance between the two DBDs in the NFκB dimers was defined as the distance between their centers of mass. VMD was used to performed salt bridge analysis. A salt bridge is defined as an acidic-basic residue pair with a distance between their O and N atoms less than 3.2 Å.
Electrostatics analysis with APBS
Electrostatic calculations were performed with the Adaptative Poisson-Boltzmann Solver (APBS1.3)25, for the initial relaxed and minimized NFκB dimer structures described above (RelA-p50 and RelA homodimer). The structures were prepared for calculations with the pdb2pqr software26, with the same force field parameters and protonation states as used in the MD simulations. Calculations were performed at 298 K, with the traditional Linearized Poisson Boltzmann Equation (LPBE), with a grid size of ~ 0.7 Å. using dielectric constants of 2 and 78 for the protein and water environments, respectively. We used an internal dielectric constant of 2.0 to represent the non-polar environment of the protein and an external constant of 78.0 to represent the polar aqueous environment of the solvent. The electrostatic potentials resulting from these calculations were then mapped onto the 3D structures of the NFκB dimers. The field lines represent the electrostatic pathways along which a positive point charge is most likely to travel. Since they represent the electrostatic potential before the systems underwent any of the conformational changes observed in MD simulations, they help us understand the causal role of electrostatics.
Stopped flow kinetics experiments
The stopped flow experiments were carried out on an Applied Photophysics SX-20 apparatus at 25 °C. The mixing ratio of the apparatus is 1:1. The final concentrations of the DNA and protein samples after mixing were 0.1 μM and 0.2 μM. Different concentrations (after mixing) of proteins used were 0.1, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45 and 0.5 μM. The pyrene-labeled DNA was excited by 343 nm wavelength light and total fluorescence was collected using 350 nm longpass filter. The data was collected for the 0.5 seconds with 5000-time points. The multiple traces were collected and averaged for better signal to noise ratio. The obtained data was fitted using bi-exponential equation for RelA-p50 and mono-exponential equation for RelA homodimer.
Supplementary Material
Highlights.
Structures of RelA-containing NFκB dimers have not been examined to date because all crystal structures have the NFκB dimer in complex with either DNA or inhibitor and the free dimers do not crystallize. To investigate the dynamics of free Rel-A-containing NFκB dimers in solution, we performed amide exchange experiments and molecular dynamics simulations.
NFκB dimers appear very dynamic in the absence of DNA. In particular, the DNA-binding domains (DBDs) reorient due to the flexible linker that connects them to the dimerization domain.
Amide exchange (HDXMS) shows much less exchange in the RelA homodimer DBDs than in the RelA-p50 DBDs.
The RelA-p50 heterodimer DBDs move away from each other during the MD simulations whereas the RelA homodimer DBDs move towards each other. The MD results are consistent with the HDXMS and provide an explanation for the dramatically reduced exchange in the homodimer – the homodimer DBDs actually form an interface that is protected from exchange.
The observation of such different DNA binding cavities in the two RelA-containing NFκBs suggested that DNA may associate more slowly with the RelA homodimer, and stopped-flow measurements show significantly slower association with the RelA homodimer. Taken together the results help explain a long-standing puzzle in the NFκB field, why DNA binds more weakly to the RelA homodimer than to the RelA-p50 heterodimer.
Acknowledgments
The authors thank J. Andrew McCammon for helpful discussions. This work was supported by NIH grant P01GM071862 to EAK. WC acknowledges fellowship support from the Ministry of Education of Taiwan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 4.Lee CH, Jeon YT, Kim SH, Song YS. NF-kappaB as a potential molecular target for cancer therapy. Biofactors. 2007;29:19–35. doi: 10.1002/biof.5520290103. [DOI] [PubMed] [Google Scholar]
- 5.Huang DB, Huxford T, Chen YQ, Ghosh G. The role of DNA in the mechanism of NFkappaB dimer formation: crystal structures of the dimerization domains of the p50 and p65 subunits. Structure. 1997;5:1427–36. doi: 10.1016/s0969-2126(97)00293-1. [DOI] [PubMed] [Google Scholar]
- 6.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-kappaB•DNA dissociation by IkappaBalpha. Proc Nat Acad Sci USA. 2009;106:19328–19333. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alverdi V, Hetrick B, Joseph S, Komives EA. Direct observation of a transient ternary complex during IkBa-mediated dissociation of NFkB from DNA. Proc Natl Acad Sci U S A. 2014;111:225–230. doi: 10.1073/pnas.1318115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potoyan DA, Zheng W, Komives EA, Wolynes PG. Molecular stripping in the NF-κB/IκB/DNA genetic regulatory network. Proc Natl Acad Sci U S A. 2016;113:110–115. doi: 10.1073/pnas.1520483112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsey KM, Dembinski HE, Chen W, Ricci CG, Komives EA. DNA and IκBα Both Induce Long-Range Conformational Changes in NFκB. J Mol Biol. 2017;429:999–1008. doi: 10.1016/j.jmb.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosisio D, Marazzi I, Agresti A, Shimizu N, Bianchi ME, Natoli G. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-kappaB-dependent gene activity. EMBO J. 2006;25:798–810. doi: 10.1038/sj.emboj.7600977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potoyan DA, Bueno C, Zheng W, Komives EA, Wolynes PG. Resolving the NFκB Heterodimer Binding Paradox: Strain and Frustration Guide the Binding of Dimeric Transcription Factors. J Am Chem Soc. 2017;139:18558–18566. doi: 10.1021/jacs.7b08741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 13.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 14.Fagerlund R, Kinnunen L, Köhler M, Julkunen I, Melén K. NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J Biol Chem. 2005;280:15942–15951. doi: 10.1074/jbc.M500814200. [DOI] [PubMed] [Google Scholar]
- 15.Sue SC, Cervantes C, Komives EA, Dyson HJ. Transfer of Flexibility between Ankyrin Repeats in IkappaBalpha upon Formation of the NF-kappaB Complex. J Mol Biol. 2008;380:917–931. doi: 10.1016/j.jmb.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martínez L, Andreani R, Martínes JM. Convergent Algorithms for Protein Structural Alignment. BMD Bioinformatics. 2007;8:306. doi: 10.1186/1471-2105-8-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. H++: a server for estimating p K as and adding missing hydrogens to macromolecules. Nuc Acids Res. 2005;33:368–371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jorgensen WL, Chandrasekhar J, Madura JD. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 20.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, Darden TA, I, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- 21.Maier JA, Carmenza Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling CL. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darden T, York D, Pedersen L. Particle mesh Ewald: An N- log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 23.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 24.Roe DR, Cheatham TE., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 25.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10042. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nuc Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.