

ABSTRACT
During hepatitis B virus (HBV) infections, subviral particles (SVP) consisting only of viral envelope proteins and lipids are secreted. Heterologous expression of the small envelope protein S in mammalian cells is sufficient for SVP generation. S is synthesized as a transmembrane protein with N-terminal (TM1), central (TM2), and hydrophobic C-terminal (HCR) transmembrane domains. The loops between TM1 and TM2 (the cytosolic loop [CL]) and between TM2 and the HCR (the luminal loop [LL]) are located in the cytosol and the endoplasmic reticulum (ER) lumen, respectively. To define the domains of S mediating oligomerization during SVP morphogenesis, S mutants were characterized by expression in transiently transfected cells. Mutation of 12 out of 15 amino acids of TM1 to alanines, as well as the deletion of HCR, still allowed SVP formation, demonstrating that these two domains are not essential for contacts between S proteins. Furthermore, the oligomerization of S was measured with a fluorescence-activated cell sorter (FACS)-based Förster resonance energy transfer (FRET) assay. This approach demonstrated that the CL, TM2, and the LL independently contributed to S oligomerization, while TM1 and the HCR played minor roles. Apparently, intermolecular homo-oligomerization of the CL, TM2, and the LL drives S protein aggregation. Detailed analyses revealed that the point mutation C65S in the CL, the mutation of 13 out of 19 amino acids of TM2 to alanine residues, and the simultaneous replacement of all 8 cysteine residues in the LL by serine residues blocked the abilities of these domains to support S protein interactions. Altogether, specific domains and residues in the HBV S protein that are required for oligomerization and SVP generation were defined.
IMPORTANCE The small hepatitis B virus envelope protein S has the intrinsic ability to direct the morphogenesis of spherical 20-nm subviral lipoprotein particles. Such particles expressed in yeast or mammalian cells represent the antigenic component of current hepatitis B vaccines. Our knowledge about the steps leading from the initial, monomeric, transmembrane translation product of S to SVP is very limited, as is our information on the structure of the complex main epitope of SVP that induces the formation of protective antibodies after vaccination. This study contributes to our understanding of the oligomerization process of S chains during SVP formation and shows that the cytoplasmic loop, one membrane-embedded domain, and the luminal loop of S independently drive S-S oligomerization.
KEYWORDS: hepatitis B virus, membrane proteins, protein oligomerization
INTRODUCTION
The hepatitis B virus (HBV) is the prototype member of the virus family Hepadnaviridae and causes chronic hepatitis in a substantial fraction of infected humans (1, 2). The virus particle consists of an icosahedral capsid with a diameter of 30 nm containing a partially double stranded circular DNA genome and an outer lipid envelope carrying three surface proteins: the large envelope protein L, the medium-sized M protein, and the small S protein (3). During infection of the human liver, virions are secreted from the hepatocytes into the bloodstream, and subviral particles (SVP) are secreted in a ≤104-fold excess over virions. These SVP appear as spheres or filaments with a diameter of 20 nm, consisting solely of lipid and viral envelope proteins (4, 5). The envelopes of virions and SVP express the same antigenicity (hepatitis B surface antigen [HBsAg]). Clearance of the virus and immunity depend on the formation of antibodies against HBsAg.
Heterologous expression of S in eukaryotic cells causes the secretion of spherical SVP consisting of lipid and approximately 100 copies of the S protein (6). Expression of S in Saccharomyces cerevisiae does not lead to SVP secretion. However, lipoprotein complexes of S can be purified from yeast cells (7) and represent the major active hepatitis B vaccine. The structure of the S protein in yeast-derived particles is partially different from the structure in SVP secreted by mammalian cells (8). For example, approximately half of S is N-glycosylated at asparagine 146 in S derived from infected patients or transfected eukaryotic cells, but S from yeast carries no glycan residue. Additionally, disulfide bridges are different between mammalian and yeast-derived S proteins. A detailed structure of S in SVP is still missing. Efforts at crystallization of SVP have been unsuccessful to date, although a relatively low resolution image has been obtained with cryoelectron microscopy (cryo-EM) (9).
The pathway leading from newly translated S protein to secreted SVP is only partially characterized. The S protein is cotranslationally inserted into the endoplasmic reticulum (ER) membrane. This insertion is first initialized by an N-terminal type I signal (containing transmembrane domain 1 [TM1], amino acids [aa] 8 to 22), which directs the N terminus of S into the ER lumen and is not cleaved off (Fig. 1). A central type II translocation signal (containing TM2, aa 80 to 98) initiates a second transmembrane translocation, in this case the translocation of the peptide chain C-terminal of TM2, and anchors the protein in the ER membrane with the N terminus oriented toward the cytoplasm and the C terminus oriented toward the ER lumen (10, 11). The combination of signals I and II generates a cytosolic loop (CL) between TM1 and TM2 and a luminal loop (LL) downstream of TM2. Asparagine 146 in the LL is partially cotranslationally N-glycosylated. A proposed amphipathic helix (aa 156 to 169) may be attached to the inner leaflet of the ER membrane. The location of the hydrophobic C-terminal region (HCR; aa 170 to 226) relative to the membrane is less clear. Since the region between aa 196 and 201 is critical for the envelopment of the cytoplasmic hepatitis D virus nucleoprotein (12), it is conceivable that this part faces the cytoplasm whereas the C terminus of S is oriented toward the ER lumen (10). This suggests that two more transmembrane areas exist between aa 170 and 226.
FIG 1.
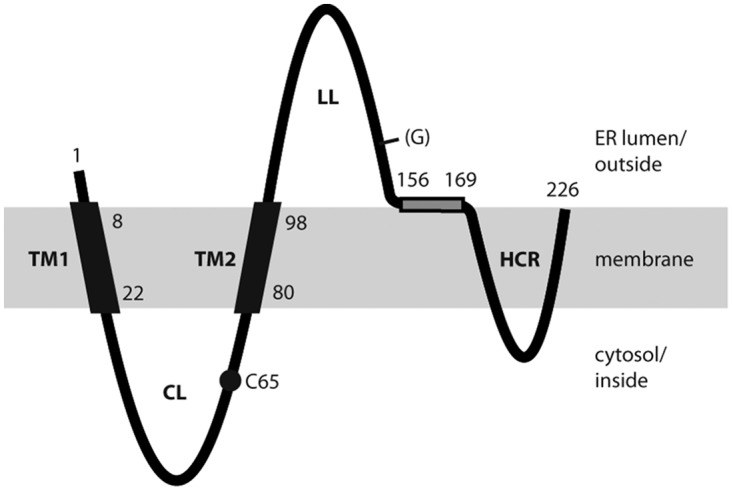
Model for the transmembrane topology of the HBV S protein monomer in the ER membrane (shaded area). TM1 and TM2, transmembrane domains 1 and 2; CL, cytosolic loop; LL, luminal loop; HCR, hydrophobic C-terminal region; (G), facultative glycan N-linked to N146; C65, cysteine residue at position 65. Numbers refer to amino acid positions. The shaded rectangle represents a putative amphipathic helix. Domains in the ER lumen are located on the surfaces of secreted SVP, and domains in the cytosol are located inside SVP.
Shortly after synthesis, the S protein forms disulfide-bridged dimers (13). The N-glycosylation has no influence on dimer formation. Models for the folding of S based on theoretical assumptions have been published (14); however, the number and positions of intra- and intermolecular disulfide bridges in S dimers are not known. Correct disulfide bridges are fundamental for the formation of the main epitope of HBsAg (15).
For SVP formation, S dimers float horizontally in the cellular membrane and must oligomerize to higher complexes, thereby substantially excluding host proteins, since only HBV envelope proteins have been detected in SVP (5). However, no intermediate oligomers have been observed to date. Particle formation probably takes place at a membrane of the secretory pathway between the ER and the Golgi complex (16). This step may not resemble the process of virion budding and may represent a totally different mechanism (17). While virion budding depends on cellular factors involved in the budding of vesicles from multivesicular bodies, SVP formation does not. During the subsequent passage of the luminal SVP by vesicular transport through the Golgi apparatus, the N-linked glycans are modified from the mannose-rich type to the complex type. Finally, SVP are further transported via vesicles toward the cell membrane and are released from the cell.
Intracellular S is mainly dimeric and carries mannose-rich glycans almost exclusively, whereas secreted SVP carry predominantly complex glycans. This suggests that the interval between the dimerization of S in the ER membrane and sugar modification in the Golgi complex is rather long (hours) compared to the interval between protein translation and dimerization (minutes) and the time span between sugar modification and release (minutes). Possibly, the step leading from transmembrane S to luminal SVP is the time-consuming process.
We intended to define the pathway of SVP formation in more detail and performed a comprehensive study clarifying the contribution of S domains to S oligomerization. The experimental approach was based on the coexpression of S proteins or parts of S linked to yellow fluorescent protein (YFP) and blue fluorescent protein (BFP) in transiently transfected cells. The proximity of a YFP-linked construct to a BFP-linked construct could be measured by Förster resonance energy transfer (FRET).
RESULTS
Experimental strategy.
During the oligomerization phase, the three topologically separated parts of S (the cytosolic, transmembrane, and luminal parts) can directly interact only with parts of other S chains located in the same compartment. Therefore, we could simplify the question “Which parts of S mediate oligomerization?” to “Do cytosolic (transmembrane, luminal) domains of S interact with cytosolic (transmembrane, luminal) domains of other S chains during oligomerization?”
Transmembrane domains.
We started with the membrane compartment of S, consisting of transmembrane domains TM1, TM2, and HCR, and asked whether interactions between these domains could be measured and whether such interactions are required for SVP formation. Previous work suggested that TM2 might be important (18). Because TM2 is part of an autonomous type II translocation/anchor signal, it was possible to assay TM2 separately from other parts of S for its ability to interact with other TM2 domains.
Heterologous expression of TM2.
TM2 and three flanking charged amino acids in the S protein chain (sequence, RRFIIFLFILLLCLIFLLVLLD) were fused to the C terminus of YFP or BFP, and a short hemagglutinin epitope (HA) tag was added at the C terminus of TM2 (Fig. 2A) in the background of a simian virus 40 (SV40) early promoter expression vector (B-TM2 and Y-TM2). As controls, similar chimeras in which of central 13 aa of TM2 were replaced with a polyalanine stretch (sequence, RRFIIA13VLLD; constructs B-2pA and Y-2pA) or in which TM2 was replaced by the type 2 signal sequence of the human transferrin receptor (B-HTR and Y-HTR) were generated. The resulting six chimeras were expected to form transmembrane proteins carrying the fluorescent moiety at the cytoplasmic side of the membrane (Fig. 2B). All six constructs could easily be detected by anti-HA antibodies on Western blots of cell lysates after transient transfection of Huh7 cells with the corresponding plasmids (Fig. 2C). After fractionation of these cells into a soluble and a membranous part, all three YFP-fused proteins were found only in the membrane fraction (Fig. 2D), as was the ER transmembrane protein calnexin, and not in the soluble fraction, as was the ER luminal enzyme protein disulfide isomerase (PDI), suggesting that TM2, its derivative 2pA, and the HTR-derived signal were functional.
FIG 2.

TM2–TM2 interactions. (A) Map of constructs consisting of an N-terminal YFP or BFP moiety fused to either TM2, a derivative of TM2 with alanine substitutions of the central 13 aa (2pA), or the type II signal of the human transferrin receptor (HTR). An influenza virus hemagglutinin tag (HA) was fused to the C terminus. (B) Expected transmembrane topology of the constructs. (C) Western blot from lysates of cells expressing the six constructs stained with anti-HA (top) or with anti-tubulin (bottom) as a loading control. Y, YFP; B, BFP; ab, antibodies. (D) Western blot of cells transfected with the indicated YFP constructs and separated into soluble (s) and membrane (m) fractions. t, unfractionated total cell lysate. Staining was performed with antibodies against the HA tags of the fusion proteins, against calnexin as an ER luminal and soluble protein, against PDI as an ER transmembrane protein, and against tubulin as a cytosolic soluble protein. The YFP constructs appeared exclusively in the membrane fraction. The positions of molecular size markers are given on the left. (E) FACS-FRET analysis. Cells cotransfected with the indicated YFP or BFP pairs were used for FACS-FRET analysis. MEM, palmitoylated 20-aa domain causing membrane attachment of the fusion protein; Y-B, YFP-BFP fusion protein. The percentage of BFP-positive cells giving a positive (pos.) FRET signal is shown along the ordinate. The dashed line indicates the background threshold. TM2 domains specifically interacted with each other. (F) FACS plot of cells cotransfected with the indicated constructs. Gate P3 excluded cells with background FRET signals.
The oligomerization of Y-TM2 with B-TM2 via the TM2 domain was measured by FRET between their fluorescent moieties in living cells (Fig. 2E and F) using fluorescence-activated cell sorter (FACS) analyses (FACS-FRET) (19). To detect FRET signals by flow cytometry, we used a rigorous gating strategy that finally gives the fraction of BFP-positive cells showing a FRET signal. We defined a FRET gate (gate P3) by measuring the background signal using cells that were transfected to coexpress untagged BFP and YFP only (Fig. 2F, Y+B). The FRET gate is set in such a way that <0.5% of these cells exhibit FRET. As a positive control, we utilized cells transfected to express a BFP-YFP fusion protein, which were expected to yield close to 100% FRET-positive cells (Fig. 2F, Y-B). The results of the measurements were expressed as the percentage of cells shifting into the FRET gate, which was set according to the negative and positive controls. Approximately 30% of cells coexpressing Y-TM2 and B-TM2 showed a positive FRET signal (Fig. 2E, lane 7; Fig. 2F, Y/B-TM2) indicating the proximity of the two proteins. Coexpression of Y-TM2 (Fig. 2E, lane 3) or B-TM2 (Fig. 2E, lane 4) with non-membrane-anchored BFP or YFP as a negative control generated no FRET signal. As additional negative controls, we used derivatives of YFP and BFP carrying the 20 N-terminal amino acids of neuromodulin (Y-MEM or B-MEM) at their N termini (20). This short amino acid stretch contains a signal for posttranslational palmitoylation of cysteine residues and causes membrane attachment of the proteins. Coexpression of Y-TM2 (Fig. 2E, lane 5) or B-TM2 (Fig. 2E, lane 6) with B-MEM or Y-MEM, respectively, resulted in no significant FRET, indicating that the FRET signals measured were not induced by overexpression or by unspecific interactions at cellular membranes.
The positive FRET signal of cells coexpressing Y-HTR and B-HTR indicated that the transmembrane domain of the HTR mediated oligomerization between the two proteins (Fig. 2E, lane 9; Fig. 2F, Y/B-HTR). Moreover, coexpression of the TM2 and HTR constructs (Fig. 2E, lanes 10 and 11) resulted in very low or no detectable FRET signals. This showed that the interaction between TM2 domains was not simply the result of general hydrophobic interactions between transmembrane domains but depended on specific protein-protein contacts. Coexpression of Y-2pA and B-2pA generated no FRET signal (Fig. 2E, lane 8, Fig. 2F, Y/B-2pA) and demonstrated that the central 13 aa of TM2 were important for the interaction.
Requirements of transmembrane domains for SVP formation. (i) TM2.
We tested the importance of TM2–TM2 interactions in the background of a wild type (WT) S protein carrying an HA tag at the C terminus (construct H) (Fig. 3A). Changing the central 13 aa of TM2 to alanine residues (mutant H.2pA) allowed efficient expression (Fig. 3B, lane 3, upper panel) but blocked the appearance of SVP in the culture supernatant (lower panel). This indicates that the polyalanine mutation in TM2, which still supported its function as a translocation and anchor signal (Fig. 2D) but blocked TM2–TM2 interactions (Fig. 2E), also inhibited later steps in SVP formation.
FIG 3.

Influence of mutations in transmembrane domains of S on SVP formation. (A) Map of S derivatives. Filled rectangles, transmembrane domains; shaded box, putative amphipathic helix; hatched box, HA tag. C → S, point mutations of all 8 cysteine residues in the LL to serine residues; 1pA, replacement of the central 11 aa of TM1 with alanine residues; 2pA, replacement of the central 13 aa of TM2 with alanine residues; Δ178 and Δ153, C-terminal deletions (the number indicates the position of the stop codon). (B) Western blots of cell lysates (upper two panels) and culture supernatants (lower two panels) stained with the indicated antibodies (ab). The positions of molecular size markers (in kilodaltons) are shown on the left. The 1pA mutation and the Δ178 truncation were compatible with secretion. (C) SVP formation by secretion-competent mutants. Culture supernatants of cells expressing the indicated constructs were sedimented through a sucrose gradient. For each construct, proteins were detected in gradient fractions by Western blotting using an HA antibody (upper panel) (the positions of molecular size markers [in kilodaltons] are shown on the left), and the sucrose concentrations (percentages [wt/wt]) of the fractions were measured (lower panel).
(ii) TM1.
The type I translocation signal TM1 could not be evaluated in analogy to TM2 (Fig. 2) because it has no membrane anchor function, and testing would result only in the translocation of a fused peptide chain across the ER membrane (21) (at a C-terminal position, TM1 would not be functional at all). Therefore, TM1 (sequence, FLGPLLVLQAGFFLL) was tested in the background construct H by changing the central 11 aa to alanine residues (mutant H.1pA; TM1 sequence, FLA11LL) (Fig. 3A). This rather drastic change was compatible with the release of the mutant into the culture supernatant with slightly lower efficiency than that of construct H (Fig. 3B, lower panels, compare lanes 1 and 4) and with SVP formation, as suggested by rate zonal sedimentation in sucrose gradients (Fig. 3C). This indicates that a potential specific interaction of TM1 with transmembrane sequences of S was probably not essential for SVP morphogenesis.
(iii) HCR.
A C-terminal deletion of H (H.Δ178) (Fig. 3A) eliminating aa 178 to 226 of S removes the putative third and fourth transmembrane domains but keeps the assumed amphipathic helix. This mutant was released from transfected cells (Fig. 3B, lane 6) as SVP (Fig. 3C), indicating that the HCR downstream of aa 177 was not required for particle formation. Particles formed by this mutant sedimented slightly more slowly than SVP consisting of H (or S [see Fig. 4A]), since the peak was shifted two fractions toward the top, and are possibly smaller than 20 nm. Further deletion, including deletion of the amphipathic helix (construct H.Δ153), blocked the appearance of SVP in the culture supernatant (Fig. 3B, lane 7). The expected molecular mass of unglycosylated H.Δ178 was approximately 20 kDa, which fits the apparent molecular mass observed in Western blots. However, H.Δ153 showed an apparent molecular mass of approximately 22 kDa, while the expected value was 17 kDa. We cannot explain this effect.
FIG 4.

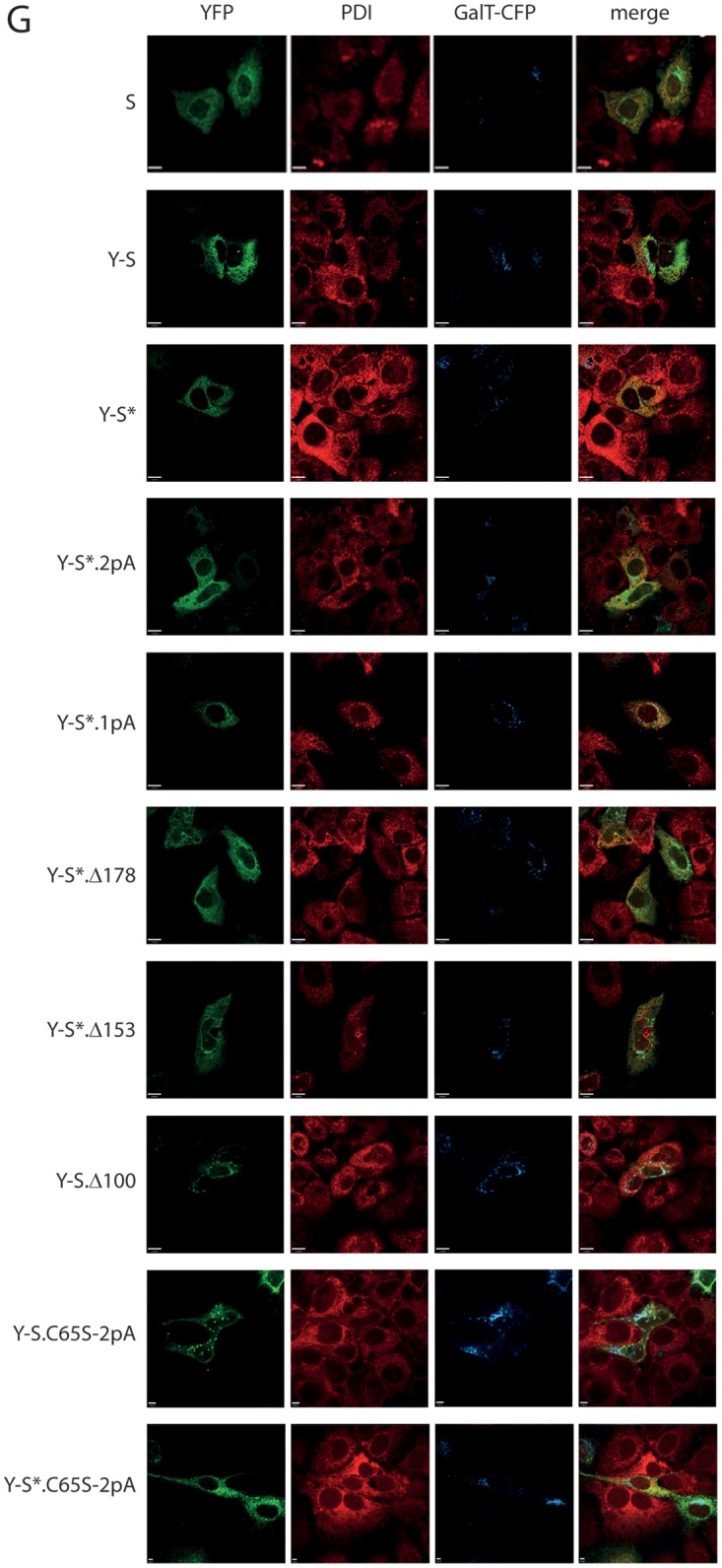
Influence of mutations in S on oligomerization. (A) A fluorescent derivative of S competent for SVP formation. (Top) Map of the fusion protein C-S carrying an N-terminal secretion signal (sig). (Center) Western blot of lysates and culture supernatants of cells expressing WT S or C-S stained with a monoclonal anti-HBs antibody. (Bottom) Western blot of fractions from a sucrose gradient after rate zonal centrifugation. (B) Map of constructs. All constructs carry a YFP or BFP moiety (hatched box) at the N terminus preceded by a secretion signal (small filled box). TM1 and TM2, transmembrane domains; HCR, hydrophobic C-terminal region; shaded box, putative amphipathic helix; pA, substitution of the central 11 aa (in TM1) or the central 13 aa (in TM2) with alanine residues; C → S, substitution of all eight cysteine residues in the LL with serine residues; C65S, replacement of cysteine 65 by serine. The numbers on the right correspond to lanes in panel F. (C) Transmembrane topologies of the Y-S and B-S chimeras. Microsomes from cells expressing the indicated proteins were treated with trypsin in the absence or presence of the mild detergent NP40. The S protein was resistant to trypsin cleavage. The pre-S2 domain of the M protein was cleaved only when microsomes were opened (lane 3), as expected (28). B-S showed a phenotype equivalent to that of M. Y-S was not efficiently cleaved. #Y-S and #B-S are similar to Y-S and B-S, respectively, but lack the N-terminal secretion signal. (D and E) Western blots of lysates from cells expressing the indicated constructs. Blots were stained with anti-GFP (cross-reacting with YFP) (top) or with anti-tubulin as a loading control (bottom). Blots in panel E were developed 5 times longer than those in panel D. The positions of molecular size markers are indicated on the left. (F) FACS-FRET signals from cells expressing the indicated YFP/BFP pairs. Y + B, coexpression of YFP and BFP; Y-B, fused YFP-BFP chimera. The dashed line indicates the background threshold. (G) Intracellular distribution of Y-S derivatives. Cells expressing the indicated constructs (autofluorescence) (green) and a fluorescent version of the Golgi enzyme GalT (GalT-CFP) (blue) were stained with an antibody against the ER-resident protein PDI (red) and were analyzed by immunofluorescence. The WT S protein was stained with polyclonal anti-HBs and a second fluorescent antibody. The bars indicate a distance of 12 μm. All constructs show a diffuse pattern in the ER and a punctate pattern in the Golgi complex, like the WT S protein.
In summary, only TM2–TM2 interactions seem to be crucial for SVP formation in the membrane compartment. The HCR could be deleted, and TM1 could be changed drastically, without blocking SVP formation.
Investigating the contributions of individual parts of S to oligomerization in the background of fluorescent S fusion proteins.
To measure the influence of mutations in S on oligomerization by FRET, we intended to construct fusion proteins of YFP and BFP with S. The phenotypes of these chimeras should resemble the phenotype of WT-S as closely as possible. Prior studies showed that a C-terminal fusion of green fluorescent protein (GFP) to S did not result in a construct capable of efficient SVP formation (22). Moreover, the N-terminal fusion to S allowed only very inefficient SVP formation (22). However, the fusion of such a large domain to the N terminus of S is expected to cause inhibition of the first membrane translocation of the S part (TM1 is nonfunctional at an internal position in a peptide chain). By adding an N-terminal secretion signal to an mCherry-S fusion (23) (Fig. 4A, top), we intended to correct the transmembrane topology of the chimera, which should then have the same topology as the medium-sized HBV envelope protein M. In the M protein, the translocation signal TM1 is able to direct the translocation of the 55-aa N-terminal pre-S2 domain into the ER lumen, because this domain is relatively short (21). By this approach, we were indeed able to generate a fluorescent S derivative that was secreted into the culture supernatant as efficiently as WT S protein (Fig. 4A, center). Rate zonal sedimentation showed that the chimera formed SVP (Fig. 4A, bottom). These particles sedimented slightly faster than 20-nm particles, a finding consistent with their expected larger size.
The mCherry construct, however, was not used in our FRET experiments, because the energy transfer between mCherry and YFP or BFP would be inappropriate. Therefore, we fused the type I secretion signal from the enzyme β-lactamase to the N terminus of YFP-S or BFP-S (constructs Y-S and B-S) (Fig. 4B). For unknown reasons, the Y-S and B-S constructs did not form detectable amounts of subviral particles (data not shown). However, the transmembrane topologies of the Y-S and B-S chimeras appeared as expected, as confirmed by protease protection experiments (Fig. 4C). When microsomes were prepared from cells expressing these constructs by Dounce homogenization and sedimentation, the YFP and BFP moieties were not digested by trypsin (Fig. 4C, lanes 5 and 11) unless the membranes were opened by the addition of detergent (Fig. 4C, lanes 6 and 12). This phenotype indicates translocation of the protected domains into the ER lumen and is similar to the behavior of the pre-S2 domain of the M protein (Fig. 4C, lanes 2 and 3). When the secretion signal sequence was missing at the N terminus (construct #B-S), the BFP moiety was also digested in the absence of detergent, as expected (Fig. 4C, lane 14). The larger apparent molecular size of B-S than of #B-S is caused by covalent modifications of the BFP moiety, such as N-glycosylation, occurring in the ER lumen (24). This supports the notion that the topology of B-S is similar to that of the M protein. These result motivated us to use the Y-S and B-S constructs for FRET analyses to define domains of S involved in S oligomerization.
Mutation of luminal cysteine residues to serine residues.
The LL contains 8 of the 14 cysteine residues of the S protein. This domain very efficiently mediates the oligomerization (probably dimerization) of S (see below), which is stabilized very early after translation by intramolecular disulfide bridges between S chains (13). When all 8 cysteine residues in the LL are changed to serine residues, the LL loses its property of supporting oligomerization (see below). In fact, the resulting construct in the H background (H* [the asterisk indicates the mutation of all luminal cysteine residues in all constructs]) did not form disulfide-linked oligomers, as evident after gel electrophoresis under nonreducing conditions (data not shown), and H* was not able to generate secreted SVP, although its expression level was only slightly lower than that of H (Fig. 3B, lanes 1 and 2). We introduced the 8 cysteine-to-serine mutations in the LLs of the Y-S and B-S constructs, resulting in Y-S* and B-S* (Fig. 4B), respectively, in order to be able to measure the influence of mutations in other parts of S on oligomerization by FRET. The expression levels of the Y/B-S* derivatives were slightly lower than that of Y/B-S (Fig. 4D); however, the FRET signals were comparable (Fig. 4F). All further mutants based on constructs Y-S and B-S (Y/B-S) (Fig. 4B) could be detected in transiently transfected Huh7 cells, although their expression levels were slightly different (Fig. 4D and E). All constructs carrying the N-glycosylation site of the S moiety at N146 appeared as double bands on Western blots, most probably due to partial N-glycosylation at this site (Fig. 4D). All constructs missing N146 showed a single band (Fig. 4E). Mutant Y-S*.Δ153 (Fig. 4D, lane 7; labeled with an asterisk) generated two double bands: one at the expected position, corresponding to a molecular mass of approximately 55 kDa, and an unexpected one at approximately 70 kDa. The Δ153 C-terminal truncation of construct H also showed an abnormal molecular mass after gel electrophoresis (Fig. 3B, lane 7). We have no explanation for this observation so far.
Furthermore, we compared the intracellular distributions of the Y-S fusion and several selected mutants with that of the WT S protein by immunofluorescence (Fig. 4G). The S protein was detected using a polyclonal anti-HBs antibody and a fluorescent secondary antibody, while the other constructs were visualized by their fluorescence. In addition, the ER was labeled with an antibody against the ER-resident protein PDI, and the Golgi complex was stained by cotransfection of an expression vector for fluorescent galactosyltransferase I (25). All proteins showed diffuse staining of the ER and a more punctate pattern in the Golgi complex, like the WT S protein.
Transmembrane domains.
Replacement of the central 13 aa of TM2 with alanine residues which blocked TM2–TM2 interactions in the Y/B-TM2 background (constructs Y/B-2pA) (Fig. 2) reduced the FRET signal in the Y/B-S* background only from 97% to 78% (Fig. 4F, compare lanes 4 and 5). This indicates that elements other than TM2 contributed to S protein interactions. TM1 was also tested in the context of Y/B-S* by the substituting a polyalanine stretch for the central 11 aa (construct Y/B-S*.1pA) (Fig. 4B). This change reduced the FRET signal from 97% to 60% (Fig. 4F, lane 6), suggesting that TM1 supports the formation or stabilization of S oligomers. The combined alanine substitutions in both TM1 and TM2 (Y/B-S*.12pA) showed a positive FRET signal of 68% (Fig. 4F, lane 7). The differences between the 1pA, 2pA, and 12pA derivatives were not statistically significant.
Removal of the HCR by introduction of the Δ178 truncation into the Y/B-S* background (constructs Y/B-S*.Δ178) resulted in a marked decrease in FRET signals to 42% (Fig. 4F, lane 8). Apparently, the HCR also supports S oligomerization. The additional removal of the putative amphipathic helix in construct Y/B-S*.Δ153 reduced the FRET signal to 35% (Fig. 4F, lane 9). The difference in the FRET signal between Y/B-S*.Δ178 and Y/B-S*.Δ153 was not statistically significant.
Cytosolic loop.
To investigate whether the CL between TM1 and TM2 mediates contacts between S chains during oligomerization, we introduced the point mutation C65S into this region in the Y/B-S* background (mutants Y/B-S*.C65S) (Fig. 4B). Cysteine 65 is not expected to be part of a disulfide bridge, because the redox state of the cytosol generally prevents cysteine formation in this compartment. Previous studies demonstrated that the C65S point mutation completely blocked SVP secretion (26). The C65S mutation also strongly reduced the FRET signal, from 97% to 22%, when introduced into the Y/B-S* pair (Fig. 4F, lane 13). Although a profound effect of the conservative point mutation on the gross folding of the S protein cannot be fully excluded, it seems rather unlikely. A more plausible explanation is that the mutation blocked interactions of S chains via the CL or interactions of this loop with unknown cellular factors supporting the oligomerization of S chains.
In the Y/B-S.12pAΔ100 mutant, transmembrane domains TM1 and TM2 are mutated by polyalanine stretches, and the LL and HCR are deleted. Therefore, the only remaining part of the S protein is the CL. This construct still showed a low but measurable FRET signal (Fig. 4F, lane 14), suggesting homodimer formation by intermolecular interactions of CL domains. The introduction of the C65S point mutation in the CL into this background (constructs Y/B-S.C65S-12pAΔ100), however, completely abolished the FRET signal (lane 15), supporting this notion. The background threshold of the assay (Fig. 4F, dashed line) was defined as the average for at least three negative-control transfections (unfused BFP plus YFP) plus 3 times their standard deviation.
Luminal loop.
The Y/B-S.Δ100 mutant lacks not only the HCR and the putative amphipathic helix but also the LL. This construct showed a FRET signal of 42% (Fig. 4F, lane 10), which is comparable to the FRET signal of Y/B-S*.Δ153 (Fig. 4D, lane 9), carrying, in addition, the LL with the cysteine-to-serine mutations. This result suggests that the LL with the eight cysteine-to-serine point mutations did not contribute to the oligomerization of S chains.
In Y/B-S*.C65S-2pA, we combined the 2pA mutation (replacement of the central 13 aa of TM2 with alanine residues) blocking TM2–TM2 interactions with the C65S mutation in the CL and the cysteine-to-serine mutations in the LL. This mutant generated no detectable FRET signal (Fig. 4F, lane 12). This finding strengthens the conclusion that TM1 and the HCR were not sufficient to establish measurable contacts between S chains. Restoration of all eight cysteine residues of the LL in this mutant (resulting in Y/B-S.C65S-2pA) restored the FRET signal to the WT level (97%) (Fig. 4F, lane 11), indicating that the LL was able to efficiently mediate S oligomerization independently of a functional CL and TM2 when expressed in the context of full-length S protein.
Taken together, our results showed that TM1 and the HCR were not essential for S interactions, whereas the LL, TM2, and the CL independently mediated S oligomerization. In addition, we describe mutations blocking the homo-oligomerization of these three domains.
DISCUSSION
During SVP morphogenesis, approximately 100 HBV transmembrane S proteins assemble with lipids, forming a spherical lipoprotein particle with a diameter of 20 nm. Since host proteins are efficiently excluded during S protein aggregation and SVP morphogenesis, it has to be assumed that specific interactions between the assembling proteins are required for this process. The present study aimed to identify regions in S that were crucial for S protein interactions during this process.
On the one hand, this study was simplified by the fact that the topologically separated areas of S could directly interact only with areas of other S chains in the same compartment (the CL with the CL; the LL with the LL; TM1, TM2, and the HCR with each other). On the other hand, the study was relatively complex, because it turned out that three areas of the S protein (the CL, TM2, and the LL) independently mediated S-S interactions. We were finally able to define the contribution of each domain by FACS-FRET analysis, because we could generate an S mutant carrying relatively subtle mutations in the CL (C65S), TM2 (a polyalanine stretch), and the LL (point mutation of all eight cysteine residues to serine residues) that showed no FRET signal any more (Y/B-S*.C65S-2pA) (Fig. 4D and F, lane 12). This mutant could be used as a background into which to reintroduce individually the WT versions of the CL (Y/B-S*.2pA), TM2 (Y/B-S*.C65S), and the LL (Y/B-S.C65S-2pA). All three variants exhibited strong or medium FRET signals (Fig. 4F, lanes 5, 13, and 11, respectively), suggesting that the CL, TM2, and the LL were each sufficient for generating measurable protein interactions.
The most direct evidence for a specific contribution to S-S interactions was obtained for TM2 (Fig. 2), since this element could be tested in isolation from other parts of S due to the autonomous functionality of the type 2 translocation signal. Previous studies (18) also suggested a role for TM2 in this process, since the replacement of TM2 in S by the type 2 translocation signal of the human transferrin receptor with an identical length or by TM2 from the duck hepatitis B virus S protein resulted in a stable chimera that was unable either (i) to form SVP, (ii) to form mixed particles with WT S, or (iii) to inhibit SVP formation by coexpressed WT S. This phenotype indicated that the chimera and WT S did not form stable, mixed oligomers. Apparently, the changes in TM2 prevented a stable interaction. Also, in the background of a full-length S protein fused to YFP or BFP, the protein contacts were mediated by TM2 (Fig. 4F, compare Y/B-S*.C65S with Y/B-S*.C65S-2pA) (see preceding paragraph). A contribution of transmembrane domains to the assembly of viral envelope proteins has also been reported for other viruses (27).
TM1 seems to be less important for stable S oligomer formation than TM2. Indeed, the replacement of 11 aa by alanine residues reduced FRET signals from 97% to 60% (Fig. 4F, lanes 4 and 6); however, this mutation was compatible with SVP formation in the H background (Fig. 3B, lane 4, and C). Furthermore, the replacement of TM1 by the type 1 secretion signal from β-lactamase allowed SVP secretion in the background of the medium-sized HBV envelope protein M carrying an additional 55 aa (pre-S2 domain) at the N terminus of S (18). The large HBV envelope protein L (carrying an additional 119 aa [pre-S1 domain] at the N terminus of M) generates two different transmembrane topologies: one with a cytoplasmic pre-S1 domain (i-pre-S) and the other with a luminal pre-S1 domain (e-pre-S) (28–30). TM1 in the S domain of L chains with an i-pre-S conformation is not expected to traverse the membrane but to be located, like pre-S1, pre-S2, and the LL, on the cytoplasmic side of the ER membrane. The i-pre-S form of L is, however, incorporated into the envelopes of virions and into SVP. This supports the model that TM1 as a transmembrane domain is not required for HBV envelope protein assembly.
The HCR was clearly not required for S protein oligomerization, since its deletion allowed SVP formation (H.Δ178) (Fig. 3B, lane 6, and C). Nevertheless, the deletion caused a reduction of protein-protein interactions as measured by FRET (Fig. 4F, lane 8). Apparently, this interaction was not essential for SVP formation. A similar C-terminal deletion of the S protein at position L176 (31) resulted in a stable protein that did not form SVP but was incorporated into SVP when coexpressed with WT S protein. In this S mutant, a few foreign amino acid residues were fused C-terminally to L176 due to the cloning strategy, which may cause the phenotypic difference from H.Δ178. The formation of mixed particles, however, also indicates that the HCR was not essential for a stable interaction of S protein chains.
The authentic sequence of TM1 and the presence of the HCR were not necessary for SVP formation and therefore were not necessary for S protein assembly. Both domains were also not sufficient for S protein dimerization, since the Y/B-S*.C65S-2pA constructs, containing WT TM1 and the HCR, generated no FRET signal (Fig. 4F, lane 12). This phenotype underlines the minor roles of TM1 and the HCR in S protein oligomerization.
The LL domain had the strongest effect on oligomerization. Changing the eight cysteine-to-serine mutations in the LL of the Y/B-S*.C65S-2pA mutant back to the WT restored the FACS signal from negative to the WT level (Fig. 4F, compare lanes 12 and 11). This might also be explained by the fact that LL-LL interactions are stabilized by disulfide bridges covalently locking the interaction partners.
A positive FRET signal indicates the proximity of coexpressed YFP/BFP pairs. Per se, however, it does not distinguish whether the proximity is created early in the SVP morphogenesis pathway (like dimer formation) or later (like higher-oligomer formation). It is, however, very likely that dimerization is a prerequisite for the formation of higher complexes. Therefore, we propose that the three domains CL, TM2, and LL are involved in dimer formation (Fig. 5). The proposed model does not explain how S dimers form higher oligomers and exclude host protein during assembly. A conceivable possibility is that the proposed amphipathic helix (aa 156 to 169) or C-terminal parts of the luminal loop mediate contacts between S dimers and therefore support the generation of higher oligomers. Alternative approaches will be needed to clarify the requirements for the formation of higher multimers.
FIG 5.

Proposed model for the S homodimer. The TM2 domains of two S proteins interact with each other, as do the two CL domains and the two LL domains. Homodimerization of LL is stabilized by intermolecular disulfide bridges (—S—S—). The authentic sequence of TM1 and the presence of the HCR were not required for SVP formation. Small circles represent cysteine 65. N, N terminus; (G), facultative glycan N-linked to N146.
MATERIALS AND METHODS
Plasmids.
The pSVBX24H plasmid carries the PstI (nucleotide [nt] 21)-to-BglII (nt 1982) fragment of an HBV genotype A genome (32) and directs the expression of the wild-type HBV S gene (nt 153 to 830) under the control of a simian virus 40 early promoter. For easier detection of the S protein on Western blots, a DNA stretch coding for the hemagglutinin-derived epitope YPYDVPDYA (HA) was inserted between the last codon of the S gene and its stop codon, generating the S variant H. By site-directed in vitro mutagenesis, the 8 cysteine codons at positions 107, 121, 124, 137, 138, 139, 147, and 149 of the H gene were mutated to serine codons, generating variant H*. In H, codons 10 to 20 were changed to alanine codons, causing the mutation of the TM1 sequence from FLGPLLVLQAGFFLL to FLA11LL and generating variant H.1pA. Codons 83 to 95 in H were changed to alanine codons, causing the mutation of the TM2 sequence from RRFIIFLFILLLCLIFLLVLLD to RRFIIA13VLLD and generating variant H.2pA. In H.12pA, the polyalanine substitutions in TM1 and TM2 were combined. For the construction of H.Δ178 and H.Δ153, codon 178 or 153, respectively, was mutated in the H background into a stop codon.
For the construction of Y-TM2, a DNA sequence encoding R78 to D99 (TM2) of the S protein followed by the 9 codons for the HA epitope and a stop codon was fused 3′ to the YFP open reading frame in plasmid peYFP-N1 (19) with a (GA)3 linker sequence in between (BsrG1 site → TGTACA AG GGA GCA GGT GCA GGA GCA CGG ← R78 of S). For B-TM2, the same TM2-HA coding sequence was fused to the XhoI site of plasmid pmTagBFP-C1 (33) (XhoI site → CTCGAG CT CGG ← R78 of S). For Y-HTR, a DNA sequence coding for the type II signal CSGSICYGTIAVIVFFLIGFMIGYLGY from the human transferrin receptor (34) fused to the HA coding sequence was ligated 3′ to the YFP open reading frame of plasmid peYFP-N1 (linker, BsrG1 site → TGTACA AG GGA GCA GGA GCA TGT ← N-terminal cysteine codon of the transmembrane domain of the HTR). For B-HTR, the HTR-HA sequence was fused 3′ to the BFP open reading frame in plasmid pmTagBFP-C1 (linker, XhoI site → CTCGAG CT TGT ← N-terminal cysteine codon of the transmembrane domain of the HTR). B/Y-MEM were constructed by fusing the N-terminal 20 aa of neuromodulin (20) to the N termini of YFP and BFP using plasmids peYFP-N1 and pmTagBFP-C1, respectively. The neuromodulin fragment contains a signal for posttranslational palmitoylation of cysteines 3 and 4 that targets the fusion proteins to cellular membranes.
The construction of the expression plasmid for the fusion protein between the fluorescent protein mCherry carrying an N-terminal secretion signal and the HBV S protein (construct C-S), which is competent for subviral particle secretion, has been described elsewhere (23). For the construction of Y-S and B-S, the mCherry-derived part of C-S was replaced by open reading frames coding for YFP and BFP, respectively. #B-S and #Y-S were constructed by digesting the plasmids for the expression of Y-S and B-S with BamHI, uniquely cutting in the region encoding the N-terminal signal sequence, and with XmaI, cutting downstream of the HBV-derived insertion. The corresponding fragment was inserted into BamHI-XmaI-digested pSVBX24H. Using Y/B-S, cysteine codons 107, 121, 124, 137, 138, 139, 147, and 149 of the S gene were mutated to serine codons, resulting in constructs Y/B-S*. The derivatives Y/B-S*.1pA, Y/B-S*.2pA, Y/B-S*.12pA, Y/B-S*.Δ178, and Y/B-S*.Δ153 were constructed in the same way as the corresponding H variants (see above). For Y/B-S.Δ100, a stop codon was introduced at codon 100 of Y/B-S*. For Y/B-S*.C65S, cysteine codon 65 in the S open reading frame in Y/B-S* was changed to a serine codon. In Y/B-S.12pAΔ100, Y/B-S.C65S-12pAΔ100, and Y/B-S*.C65S-2pA, the corresponding mutations were combined. Y/B-S.C65S-2pA corresponds to Y/B-S*.C65S-2pA, but codons 107, 121, 124, 137, 138, 139, 147, and 149 of the S gene code for cysteines as in the wild type.
Cell culture, transfection, and harvest.
Huh7 cells grown in 12-well dishes were transiently transfected with a total amount of 0.5 μg of plasmid using Fugene HD (Promega) or X-tremeGene (Roche) according to the instructions of the manufacturer. For cotransfections, an equal molar ratio of plasmids was used. After transfection, the culture supernatant was removed, the cells were washed twice with 1 ml of phosphate-buffered saline (PBS), and 1 ml of fresh medium was added to the cells. Five days posttransfection, the culture supernatant was harvested and was centrifuged for 10 min at 13,000 rpm. From the supernatant, 40 μl was used for Western blotting. The cells on the dish were washed with 1 ml of PBS and were lysed by adding 0.25 ml of lysis buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 20 mM EDTA, 0.5% [vol/vol] Nonidet P-40) and incubating on ice for 10 min. The cell lysate was collected and was spun at 13,000 rpm for 10 min. Ten microliters of the supernatant was used for Western blotting.
Cell fractionation.
Three days posttransfection, cells transiently transfected in 6-well dishes were washed twice with 1 ml of ice-cold PBS and were incubated for 45 min with 0.4 ml of 10 mM Tris-Cl (pH 7.4), 150 mM NaCl, and 1% (vol/vol) Triton X-114 on ice (35). The cell lysate was collected and was spun for 45 min at 12,000 × g and 4°C. Ten microliters of the supernatant was used for Western blotting (total lysate). From the supernatant, 350 μl was layered on top of 350 μl of a cushion of 6% (wt/vol) sucrose, 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.06% (vol/vol) Triton X-114 and was incubated for 5 min at 30°C. After centrifugation for 3 min at 12,000 × g and room temperature (RT), 20 μl of the upper phase was used for Western blotting (aqueous phase). The remaining part of the aqueous phase was removed. Twenty microliters of the detergent phase was used for Western blotting (membrane fraction).
Western blotting.
Cleared cell culture supernatants or lysates of transfected Huh7 cells were denatured, reduced with sodium dodecyl sulfate and dithiothreitol, and used for electrophoresis through 12% polyacrylamide gels. The proteins in the gel were transferred to nitrocellulose membranes (Amersham Protran 0.45 NC; GE Healthcare) by wet blotting. Proteins were detected with a rabbit antibody against the HA epitope (Sigma-Aldrich Chemie GmbH, Schnelldorf, Germany) for all constructs carrying this tag and with a cross-reacting rabbit anti-GFP antibody (D5.1XP; New England BioLabs, Ipswich, MA, USA) for YFP fusion proteins. As a loading control, Western blots were stained with rabbit anti-β-tubulin (New England BioLabs, Ipswich, MA, USA). The HBV S protein was detected with the monoclonal anti-HBs antibody HB1 (provided by courtesy of Aurelia Zvirbliene, Vilnius University, Vilnius, Lithuania). Rabbit antibodies against calnexin and PDI were from New England BioLabs, Ipswich, MA, USA. Peroxidase-labeled antibodies against rabbit and mouse immunoglobulins were from Dianova (Hamburg, Germany).
FACS-FRET.
FRET analysis was carried out as described by Banning and colleagues (19). One day after transfection in 12-well dishes, Huh7 cells were washed twice with PBS, detached from the dish with 100 μl of trypsin solution, and suspended in 800 μl of 1% (vol/vol) fetal calf serum (FCS) in PBS. Cells were then sedimented by centrifugation at 1,300 rpm and 4°C for 5 min, and the cell pellet was suspended in 200 μl of 1% (vol/vol) FCS in PBS and applied to the FACS machine. BFP was excited with a laser beam at 405 nm, and its emission was measured at 450 nm. YFP was excited with a laser light at 488 nm, and its emission was measured at 529 nm.
Rate zonal sedimentation.
A sucrose gradient consisting of 0.5 ml of 60% (wt/wt) sucrose in 50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 0.1 mM EDTA, and 1 ml each of corresponding 45%, 35%, 25%, and 15% sucrose solutions was prepared in centrifugation tubes and was left for 4 h at 4°C. Then 0.5 ml of the culture supernatant was layered on top, and the gradient was spun for 16 h at 50,000 rpm and 10°C in a Beckman SW55 rotor. The gradient was harvested from the top (15 0.33-ml fractions). The sucrose concentrations of the fractions were measured by refractometry, and the distribution of proteins was analyzed by Western blotting using the monoclonal anti-HBs antibody HB1.
Protease protection experiment.
Microsomes were prepared, and protease protection experiments were carried out, as described elsewhere (28), except that the cells were not labeled with radioactive methionine and the samples were not used for immunoprecipitation prior to polyacrylamide gel electrophoresis. Instead, 10 μl of each sample was denatured and was loaded directly onto the gel.
Immunofluorescence.
Cells were seeded on coverslips and were transfected the next day. Two days posttransfection, cells were washed twice with PBS and were fixed by incubation with 4% paraformaldehyde for 15 min at room temperature. Cells were again washed three times with PBS for 5 min each time with shaking and were incubated with ice-cold methanol for 10 min at −20°C. After being washed again with PBS, cells were incubated for 1 h in 5% (vol/vol) goat serum–0.3% (vol/vol) Triton X-100 in PBS. Cells were then incubated with a rabbit anti-PDI antibody (New England BioLabs) diluted 1:200 in 1% (wt/vol) bovine serum albumin–0.3% (vol/vol) Triton X-100 in PBS (antibody dilution buffer) or with the monoclonal anti-HBs antibody HB1 diluted 1:100 in the same buffer overnight at room temperature in a humid box. After three washes with PBS for 5 min with shaking, cells were incubated with an Alexa Fluor 555-labeled goat anti-rabbit antibody (Life Technologies) diluted 1:2,000 in antibody dilution buffer or with an Alexa Fluor 488-labeled goat anti-mouse antibody (Life Technologies) diluted 1:2,000 in the same buffer for 2 h at room temperature in the dark. After being washed three times with PBS for 5 min each time, cells were fixed with Mowiol (Sigma-Aldrich) and were stored at 4°C until examination with a fluorescence microscope.
ACKNOWLEDGMENTS
This work was funded by institutional support to V.B. from the Helmholtz Zentrum München. A.O. was supported by the Deutscher Akademischer Austauschdienst (DAAD) in cooperation with the Egyptian Ministry of Higher Education and Scientific Research.
REFERENCES
- 1.Lavanchy D. 2004. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 2.Seeger C, Mason WS. 2015. Molecular biology of hepatitis B virus infection. Virology 479–480:672–686. doi: 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruss V. 2007. Hepatitis B virus morphogenesis. World J Gastroenterol 13:65–73. doi: 10.3748/wjg.v13.i1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganem D, Varmus HE. 1987. The molecular biology of the hepatitis B viruses. Annu Rev Biochem 56:651–693. doi: 10.1146/annurev.bi.56.070187.003251. [DOI] [PubMed] [Google Scholar]
- 5.Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. 1984. Large surface proteins of hepatitis B virus containing the pre-S sequence. J Virol 52:396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laub O, Rall LB, Truett M, Shaul Y, Standring DN, Valenzuela P, Rutter WJ. 1983. Synthesis of hepatitis B surface antigen in mammalian cells: expression of the entire gene and the coding region. J Virol 48:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valenzuela P, Medina A, Rutter WJ, Ammerer G, Hall BD. 1982. Synthesis and assembly of hepatitis B virus surface antigen particles in yeast. Nature 298:347–350. doi: 10.1038/298347a0. [DOI] [PubMed] [Google Scholar]
- 8.Diminsky D, Schirmbeck R, Reimann J, Barenholz Y. 1997. Comparison between hepatitis B surface antigen (HBsAg) particles derived from mammalian cells (CHO) and yeast cells (Hansenula polymorpha): composition, structure and immunogenicity. Vaccine 15:637–647. doi: 10.1016/S0264-410X(96)00239-3. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert RJ, Beales L, Blond D, Simon MN, Lin BY, Chisari FV, Stuart DI, Rowlands DJ. 2005. Hepatitis B small surface antigen particles are octahedral. Proc Natl Acad Sci U S A 102:14783–14788. doi: 10.1073/pnas.0505062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eble BE, Lingappa VR, Ganem D. 1986. Hepatitis B surface antigen: an unusual secreted protein initially synthesized as a transmembrane polypeptide. Mol Cell Biol 6:1454–1463. doi: 10.1128/MCB.6.5.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eble BE, MacRae DR, Lingappa VR, Ganem D. 1987. Multiple topogenic sequences determine the transmembrane orientation of the hepatitis B surface antigen. Mol Cell Biol 7:3591–3601. doi: 10.1128/MCB.7.10.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komla-Soukha I, Sureau C. 2006. A tryptophan-rich motif in the carboxyl terminus of the small envelope protein of hepatitis B virus is central to the assembly of hepatitis delta virus particles. J Virol 80:4648–4655. doi: 10.1128/JVI.80.10.4648-4655.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wunderlich G, Bruss V. 1996. Characterization of early hepatitis B virus surface protein oligomers. Arch Virol 141:1191–1205. doi: 10.1007/BF01718824. [DOI] [PubMed] [Google Scholar]
- 14.Berting A, Hahnen J, Kroger M, Gerlich WH. 1995. Computer-aided studies on the spatial structure of the small hepatitis B surface protein. Intervirology 38:8–15. doi: 10.1159/000150409. [DOI] [PubMed] [Google Scholar]
- 15.Mangold CM, Unckell F, Werr M, Streeck RE. 1995. Secretion and antigenicity of hepatitis B virus small envelope proteins lacking cysteines in the major antigenic region. Virology 211:535–543. doi: 10.1006/viro.1995.1435. [DOI] [PubMed] [Google Scholar]
- 16.Huovila AP, Eder AM, Fuller SD. 1992. Hepatitis B surface antigen assembles in a post-ER, pre-Golgi compartment. J Cell Biol 118:1305–1320. doi: 10.1083/jcb.118.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prange R. 2012. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med Microbiol Immunol 201:449–461. doi: 10.1007/s00430-012-0267-9. [DOI] [PubMed] [Google Scholar]
- 18.Siegler VD, Bruss V. 2013. Role of transmembrane domains of hepatitis B virus small surface proteins in subviral-particle biogenesis. J Virol 87:1491–1496. doi: 10.1128/JVI.02500-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. 2010. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One 5:e9344. doi: 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skene JH, Virag I. 1989. Posttranslational membrane attachment and dynamic fatty acylation of a neuronal growth cone protein, GAP-43. J Cell Biol 108:613–624. doi: 10.1083/jcb.108.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eble BE, Lingappa VR, Ganem D. 1990. The N-terminal (pre-S2) domain of a hepatitis B virus surface glycoprotein is translocated across membranes by downstream signal sequences. J Virol 64:1414–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert C, Thome N, Kluck CJ, Prange R. 2004. Functional incorporation of green fluorescent protein into hepatitis B virus envelope particles. Virology 330:158–167. doi: 10.1016/j.virol.2004.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayer K, Banning C, Bruss V, Wiltzer-Bach L, Schindler M. 2016. Hepatitis C virus is released via a noncanonical secretory route. J Virol 90:10558–10573. doi: 10.1128/JVI.01615-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costantini LM, Subach OM, Jaureguiberry-bravo M, Verkhusha VV, Snapp EL. 2013. Cysteineless non-glycosylated monomeric blue fluorescent protein, secBFP2, for studies in the eukaryotic secretory pathway. Biochem Biophys Res Commun 430:1114–1119. doi: 10.1016/j.bbrc.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rocks O, Gerauer M, Vartak N, Koch S, Huang ZP, Pechlivanis M, Kuhlmann J, Brunsveld L, Chandra A, Ellinger B, Waldmann H, Bastiaens PI. 2010. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141:458–471. doi: 10.1016/j.cell.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Mangold CM, Streeck RE. 1993. Mutational analysis of the cysteine residues in the hepatitis B virus small envelope protein. J Virol 67:4588–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.da Silva DV, Nordholm J, Madjo U, Pfeiffer A, Daniels R. 2013. Assembly of subtype 1 influenza neuraminidase is driven by both the transmembrane and head domains. J Biol Chem 288:644–653. doi: 10.1074/jbc.M112.424150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruss V, Lu X, Thomssen R, Gerlich WH. 1994. Post-translational alterations in transmembrane topology of the hepatitis B virus large envelope protein. EMBO J 13:2273–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prange R, Streeck RE. 1995. Novel transmembrane topology of the hepatitis B virus envelope proteins. EMBO J 14:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ostapchuk P, Hearing P, Ganem D. 1994. A dramatic shift in the transmembrane topology of a viral envelope glycoprotein accompanies hepatitis B viral morphogenesis. EMBO J 13:1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruss V, Ganem D. 1991. Mutational analysis of hepatitis B surface antigen particle assembly and secretion. J Virol 65:3813–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valenzuela P, Quiroga M, Zaldivar J, Gray R, Rutter W. 1980. The nucleotide sequence of the hepatitis B viral genome and the identification of the major viral genes. UCLA Symp Mol Cell Biol 18:57–70. [Google Scholar]
- 33.Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, Gruenwald D, Souslova EA, Chudakov DM, Verkhusha VV. 2008. Conversion of red fluorescent protein into a bright blue probe. Chem Biol 15:1116–1124. doi: 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McClelland A, Kuhn LC, Ruddle FH. 1984. The human transferrin receptor gene: genomic organization, and the complete primary structure of the receptor deduced from a cDNA sequence. Cell 39:267–274. doi: 10.1016/0092-8674(84)90004-7. [DOI] [PubMed] [Google Scholar]
- 35.Bordier C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem 256:1604–1607. [PubMed] [Google Scholar]