

ABSTRACT
Human noroviruses are the leading cause of acute gastroenteritis in humans. Noroviruses also infect animals, such as cows, mice, cats, and dogs. How noroviruses bind and enter host cells is still incompletely understood. Recently, the type I transmembrane protein CD300lf was identified as the murine norovirus receptor, yet it is unclear how the virus capsid and receptor interact at the molecular level. In this study, we determined the X-ray crystal structure of the soluble CD300lf (sCD300lf) and the murine norovirus capsid protruding domain complex at a 2.05-Å resolution. We found that the sCD300lf-binding site is located on the topside of the protruding domain and involves a network of hydrophilic and hydrophobic interactions. sCD300lf locked nicely into a complementary cavity on the protruding domain that is additionally coordinated with a positive surface charge on sCD300lf and a negative surface charge on the protruding domain. Five of six protruding domain residues interacting with sCD300lf were maintained between different murine norovirus strains, suggesting that sCD300lf was capable of binding to a highly conserved pocket. Moreover, a sequence alignment with other CD300 paralogs showed that the sCD300lf-interacting residues were partially conserved in CD300ld but variable in other CD300 family members, consistent with previously reported infection selectivity. Overall, these data provide insights into how a norovirus engages a protein receptor and will be important for a better understanding of selective recognition and norovirus attachment and entry mechanisms.
IMPORTANCE Noroviruses exhibit exquisite host range specificity due to species-specific interactions between the norovirus capsid protein and host molecules. Given this strict host range restriction, it has been unclear how the viruses are maintained within a species between relatively sporadic epidemics. While much data demonstrate that noroviruses can interact with carbohydrates, recent work has shown that expression of the protein CD300lf is both necessary and sufficient for murine norovirus infection of mice and binding of the virus to permissive cells. Importantly, the expression of this murine protein by human cells renders them fully permissive for murine norovirus infection, indicating that at least in this case, host range restriction is determined by molecular events that control receptor binding and entry. Defining the atomic-resolution interactions between the norovirus capsid protein and its cognate receptor is essential for a molecular understanding of host-range restriction and norovirus tropism.
KEYWORDS: X-ray crystallography, murine norovirus, receptor
INTRODUCTION
Human noroviruses are the leading cause of outbreaks of acute gastroenteritis. Noroviruses belong to the Caliciviridae family and have a single-stranded, positive-sense RNA genome of ∼7.4 to 7.7 kb. Norovirus genomes contain three or four open reading frames (ORFs), with ORF1 encoding the nonstructural proteins, ORF2 encoding the capsid protein, ORF3 encoding a minor structural protein, and murine norovirus (MNV) ORF4 encoding a virulence factor (1). Based on capsid sequences, noroviruses are divided into at least seven genogroups (GI to GVII), with GI, GII, and GIV causing infections in humans and GV infecting mice. Murine noroviruses can be grown routinely in cell culture, can infect laboratory mice, and have been used as a system for defining mechanisms of norovirus replication and pathogenesis (2, 3).
Development of a structural understanding of the norovirus capsid has been enabled by the observation that expression of human norovirus ORF2 in insect cells results in the formation of virus-like particles (VLPs) that are antigenically similar to native virions (4). The virus particles contain 180 copies of the capsid protein that are arranged in a T=3 icosahedral symmetry. The capsid protein is divided into two domains, a shell (S) and a protruding (P) domain, that are connected by a flexible hinge region (5). The S domain forms a roughly spherical structure that surrounds the viral RNA, whereas the P domain, which is further subdivided into P1 and P2 subdomains, protrudes upwards from the S domain and contains the main determinants for cell attachment (5).
The X-ray crystal structures of norovirus P domains belonging to GI, GII, GIII, GIV, and GV indicate that their overall structures are similar and resemble that of the P domain on intact VLPs, having a single α-helix in the P1 subdomain and six antiparallel β-strands in the P2 subdomain (6–13). On the other hand, structural studies of infectious norovirus particles have shown that the P domains can be positioned differently on the S domain. For GI.1 norovirus VLPs, the P domains rest on the S domain (5), while for the GII.10 VLPs and GV virions of murine norovirus, the P dimers are raised off the S domain by ∼15 Å (13, 14). Therefore, the P domains are likely capable of adopting variable conformations with respect to the S domain and the dynamic nature of the P dimers might be an important aspect for the infection process, as previously discussed (13, 15).
Human noroviruses interact with histo-blood group antigens (HBGAs) (6–8, 16–20). Indeed, two recently developed human norovirus cell culture systems have shown that HBGAs and HBGA secretor status are important for virus infection (21, 22). Most human noroviruses bind HBGAs, and GI and GII noroviruses typically bind HBGAs on different regions on the P dimers. On the other hand, murine noroviruses have not been demonstrated to bind HBGAs, although they may bind other glycans as attachment factors (23, 24).
Recently, several different murine noroviruses (termed CW3, CR6, and S7) were found to require the cellular receptor CD300lf for attachment, entry, and replication in both cultured cells and mice (25, 26). CD300 proteins are type I transmembrane proteins containing a single IgV-like extracellular domain that contains two disulfide bonds and a transmembrane helix (reviewed in reference 27). Multiple viruses recognize Ig family proteins as receptors, and structural information deciphering these interactions can offer insights into mechanisms of receptor recognition and antibody neutralization and can facilitate the development of antiviral strategies.
In this study, we demonstrated a direct interaction between the soluble extracellular domain of CD300lf (sCD300lf) and murine norovirus capsid (CR10 strain). We solved the X-ray crystal structure of sCD300lf in complex with the murine norovirus P domain. We found that sCD300lf bound on the topside of the murine norovirus P domain and that five of six protruding domain residues interacting with sCD300lf were highly conserved among different murine norovirus strains. Moreover, the sCD300lf binding site overlaps with the contact areas of two murine norovirus-neutralizing monoclonal antibodies (termed A6.2 and 2D3) and provides a possible structural explanation for the antibody neutralization mechanism (12, 28–30). Surprisingly, the sCD300lf binding site was close to the GI and GII human norovirus HBGA binding pockets. Together, these new findings provide an atomic understanding of receptor selectivity for murine noroviruses.
RESULTS AND DISCUSSION
The biological relevance of the sCD300lf was first evaluated using a murine norovirus plaque replication assay and a viral attachment assay in order to confirm that sCD300lf also interacted with murine norovirus 1 (MNV-1) strain CW1, since several other murine noroviruses (i.e., CW3, CR6, and S7) were previously shown to bind CD300lf (23, 24). The 50% inhibitory concentrations (IC50s) for the plaque and viral attachment assays were 0.2 μg/ml and 0.4 μg/ml, respectively (Fig. 1A). These closely matching IC50s confirmed the neutralizing properties of sCD300lf on the CW1 strain and were in agreement with previous sCD300lf assays using the CW3 strain (25) and indicated the physiological relevance of sCD300lf. We next wanted to test the hypothesis that sCD300lf bound directly to the murine norovirus. However, we did not observe murine norovirus (CR10 P domain or CW1 virion) binding interactions with sCD300lf using a direct enzyme-linked immunosorbent assay (ELISA) (Fig. 1B). These results suggested that sCD300lf might only weakly bind murine norovirus.
FIG 1.

Neutralization properties of sCD300lf against murine norovirus virions. (A) Serially diluted sCD300lf was added to CW1 virions and inhibition of attachment was measured. The IC50s were 0.2 μg/ml and 0.4 μg/ml for the plaque and viral attachment assays, respectively (error bars show SDs; three independent experiments were performed). These findings indicated that the sCD300lf was likely functional and properly folded. (B) A direct ELISA showed that sCD300lf was unable to bind the murine norovirus CR10 P domain or CW1 virions, whereas a positive control murine norovirus-specific nanobody (Nano-45) was able to detect the CR10 P domain and CW1 virions.
Following these results, we proceeded to determine the sCD300lf recognition site on the murine norovirus CR10 P domain using X-ray crystallography. A single crystal of CR10 P domain and sCD300lf complex diffracted to a resolution of 2.05 Å (data statistics are shown in Table 1). The complex consisted of one CR10 P domain dimer and a single sCD300lf molecule. The complex structure indicated that sCD300lf bound to the topside of one CR10 P2 subdomain and interacted with one of the P domains (Fig. 2). Analysis of the alternative sCD300lf-binding pocket on the P domain dimer indicated that binding of a second sCD300lf was not possible because crystal packing sterically hindered this region. In the context of the intact particle, the sCD300lf binding site was located on an easily accessible and exposed region of the P domains (Fig. 2B).
TABLE 1.
Data collection and refinement statistics of murine norovirus CR10 P domain and sCD300lf domain complex structurea
| Parameter | Value(s) for CR10 and sCD300lf (PDB code 5OR7) |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 75.11, 77.46, 140.95 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution range (Å) | 42.82–2.05 (2.12–2.05)b |
| Rmerge | 10.85 (70.51)b |
| I/σI | 11.45 (2.31)b |
| Completeness (%) | 99.50 (97.60)b |
| Redundancy | 5.5 (5.1)b |
| Refinement | |
| Resolution range (Å) | 42.82–2.05 |
| No. of reflections | 52,389 |
| Rwork/Rfree | 18.04/21.68 |
| No. of atoms | 5,836 |
| Protein | 5,371 |
| Ligand/ion | 6 |
| Water | 459 |
| Average B factors (Å2) | |
| Protein | 29.70 |
| Ligand/ion | 35.82 |
| Water | 36.13 |
| RMSD | |
| Bond length (Å) | 0.002 |
| Bond angle (°) | 0.600 |
The data set was collected from a single crystal.
The value in parentheses is for the highest-resolution shell.
FIG 2.

X-ray crystal structure of the CR10 P domain-sCD300lf complex. (A) Asymmetric unit cell containing one P domain dimer and one sCD300lf molecule. The CR10 P domain was subdivided into monomer chains A (dark gray) and B (light gray), while the sCD300lf is shown in light pink. (B) The sCD300lf bound to the topside of the virion, where two sCD300lf molecules (light pink) and one P dimer (blue) were superpositioned onto the murine norovirus virion cryo-EM structure.
Similar to the apo CW1 murine norovirus P domain structure (PDB code 3LQ6), the CR10 P domain could be divided into P1-1 (residues 228 to 277), P2 (residues 278 to 416), and P1-2 (residues 417 to 530) subdomains. The CR10 P1 subdomain comprised β-sheets and one α-helix, while the P2 subdomain contained six antiparallel β-strands that formed a barrel-like structure typical of human noroviruses. The CR10 and CW1 P domain surface-exposed loops were highly equivalent and the two structures had a maximum root mean square deviation (RMSD) of 0.8 Å. This result was not surprising, since the CR10 and CW1 P domains had 95% amino acid sequence identity. Likewise, the sCD300lf in the complex structure was highly reminiscent of the unbound sCD300lf structure (PDB code 5FFL), having a RMSD of 0.3 Å. Overall, the CR10 P dimer and sCD300lf in the complex structure showed no obvious loop movements compared to the apo CW1 P domain and sCD300lf structures.
A network of hydrophilic and hydrophobic interactions held the CR10 P domain and sCD300lf together. The CR10 binding residues were located on or linking the antiparallel β-strands, while for sCD300lf the majority of the binding residues were found on the CC′ and CDR3 loops (Fig. 3A). The P domain bound sCD300lf with five direct hydrogen bonds, which consisted of both side and main chain interactions. The main chain of Gly300 of CR10 (Gly300CR10) held the side chain of Asp2CD300lf. The side chain of Asn364CR10 had two direct hydrogen bonds with the main chain of Arg42CD300lf and side chain of Cys44CD300lf. Additionally, the main chain of Asn364CR10 interacted with the main chain of Arg42CD300lf. The side chain of Asp366CR10 held the main chain of Gly96CD300lf. Three hydrophobic interactions were also observed and involved three residues from CR10 (Val365CR10, Tyr399CR10, and Val304CR10) and two residues from sCD300lf (Pro40CD300lf and Leu97CD300lf) (Fig. 3B). Importantly, mutations in the CC′ loop and/or CDR3 of sCD300lf were previously shown to diminish or abolish murine norovirus infection (25), indicating that these residues were important for the binding of sCD300lf. Moreover, an N-terminal deletion including Asp2CD300lf was also shown to prevent MNV binding (26).
FIG 3.

Binding interactions of the murine norovirus CR10 P domain with sCD300lf. The CR10 P domain and sCD300lf are colored as in Fig. 2. (A) Close-up view of the binding interactions between the CR10 P domain and sCD300lf showing the direct hydrogen bond interactions (black lines) and hydrophobic interactions (orange lines). Residues on CDR3, the CC′ loop, and the N terminus of sCD300lf interact with the CR10 P domain. (B) List of residue interactions, where hydrogen bond distances were between 2.8 and 3.5 Å, while hydrophobic interactions were between 3.9 and 5.3 Å.
Furthermore, a number of P2 subdomain residues that interacted with sCD300lf overlapped the recognition region of two neutralizing murine norovirus monoclonal antibodies, A6.2 and 2D3 (12, 28–30) (Fig. 4). This finding suggested that A6.2 and 2D3 antibodies might compete and/or sterically preclude the sCD300lf-binding pocket on the P2 subdomain, thus providing a possible explanation of the antibody-meditated neutralization.
FIG 4.

Sequence alignment of different murine norovirus capsid proteins. The partial S domain (yellow bar), P1 subdomain (light blue bar), and P2 subdomain (dark blue bar) are indicated on the alignment. The CR10 residues (pink highlight) that interacted with the CD300lf through direct hydrogen bonds and hydrophobic interactions were mostly conserved in different murine norovirus strains. The A6.2 antibody recognition site is indicated by red amino acid letters. Asterisks indicate highly conserved residues.
Interestingly, a HEPES molecule that was previously shown to bind at the surface cleft of sCD300lf (25) was replaced with several CR10 P domain residues that made direct hydrogen bonds with sCD300lf; i.e., Asn364CR10 interacted with Arg42CD300lf and Cys44CD300lf. Also, a sodium atom was held between the P domain and CDR3 of sCD300lf, where Asn364CR10, Asp366CR10, Lys94CD300lf, Gly96CD300lf, and Asp98CD300lf along with a water molecule formed an octahedral-like structure (Fig. 5). Although there is no evidence that the sodium ion is required for binding interactions and it might be replaced with a calcium ion (31), our structural refinements indicated a presence of a sodium atom. Our purification protocol and crystallization condition did not contain calcium. This sodium atom was also further confirmed using the CheckMyMetal web server (https://csgid.org/csgid/metal_sites). The reason for the binding of this ion is currently unknown; however, the ion might help stabilize the interacting residues, since the HEPES molecule that replaced the P domain residues in the apo CD300lf structure also contributed to the binding interactions of this ion (25).
FIG 5.

Sodium atom interactions between the CR10 P domain and sCD300lf. The sodium atom (orange sphere) is held with the CR10 P domain residues (Asn364CR10 and Asp366CR10), CDR3 residues of sCD300lf (Lys94CD300lf, Gly96CD300lf, and Asp98CD300lf), and a water molecule (marine sphere). Interactions are shown as yellow lines. This set of interactions formed an octahedron-like structure, which was also observed in the apo sCD300lf structure (25). The Omit map (mFo-DFc, blue mesh) is countered at 3.0 σ.
In order to determine if other factors facilitated the binding of the sCD300lf to the CR10 P domain, the electrostatic potential of the CR10 P domain and sCD300lf was calculated. The CR10 P domain and sCD300lf binding regions were found to be electrostatically complementary, having negative and positive electrostatic potentials, respectively (Fig. 6A). Moreover, the surface-exposed cleft between the CC′ and CDR3 loops of the sCD300lf molecule elegantly anchored into a complementary cavity on the CR10 P domain (Fig. 6B), with a total interface area of 620 Å2. Correspondingly, previously reported human norovirus P domain and nanobody recognition regions were comparable, having an interface area of ∼700 Å2 (15). Overall, these results showed that the binding pocket on the CR10 P domain was nicely tailored for fitting and binding of sCD300lf.
FIG 6.

Analysis of the CR10 P domain and sCD300lf binding pockets. (A) Binding of sCD300lf was coordinated by a positive (blue) potential on the sCD300lf and a negative (red) potential on the CR10 P domain. Neutral residues were colored white. The scale was from −5 kT/e to 5 kT/e. (B) The cleft between the CC′ loop and CDR3 of sCD300lf (light pink) elegantly fit into the cavity of the CR10 P domain (dark gray). The CR10 P domain and sCD300lf residues contributing the hydrogen bonds are shown in green.
Previously, it was shown that murine norovirus (CW3, CR6, and S7 strains) specifically recognized CD300lf and CD300ld, whereas expression of other CD300 types did not confer susceptibility for infection (25). An amino acid sequence alignment showed that the CD300lf residues interacting with the CR10 P domain were partially conserved in CD300ld but more varied in other CD300 family members (Fig. 7). This observation suggested a possible explanation for murine norovirus selective recognition of CD300lf. Moreover, five of six murine norovirus residues interacting with the receptor were conserved in numerous murine norovirus strains, suggesting that other murine noroviruses would also bind CD300lf at the same pocket (Fig. 4).
FIG 7.

Sequence alignment of CD300 proteins. The sCD300lf residues (pink highlight) that interacted with the CR10 P domain through direct hydrogen bonds and hydrophobic interactions are partially conserved in CD300lf and CD300ld but are more variable in other CD300 types.
In order to investigate how sCD300lf molecules might interact with the P domains on the intact murine norovirus particles, the X-ray structure of the CR10 P domain sCD300lf complex was superpositioned onto the cryo-electron microscopy (cryo-EM) structure of the CW3 virion (Fig. 8) (13). The stoichiometry of CD300lf interaction with VP1 remains unknown. However, the model indicated that sCD300lf molecules were positioned between two P domain dimers. In this model, the three sCD300lf molecules slightly clashed at the intersection between the P dimers. However, since the P domains were raised off the S domain via a flexible hinge region, a certain degree of P dimer movement might occur, which could allow all of the possible binding sites to be occupied with sCD300lf molecules, although direct evidence of this interaction is lacking.
FIG 8.
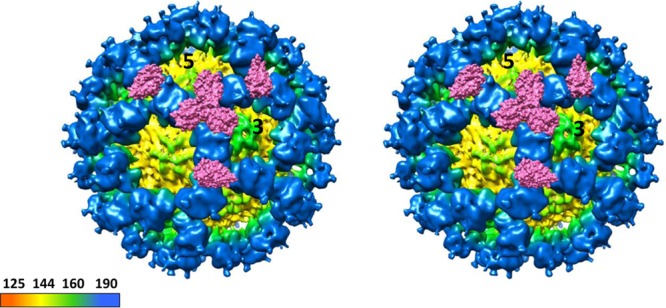
Cryo-EM map of CW1 murine norovirus virion and CR10 P domain sCD300lf complex structures. Shown is a stereo view of the (cryo-EM) CW1 virion structure superpositioned with the X-ray crystal structure of the CR10 P domain sCD300lf complex. The CR10 P domains are removed for clarity, and two molecules of sCD300lf are shown on each P dimer. The virion was colored according to diameter, showing the S domain (yellow) and the P domain (blue). The sCD300lf slightly clashed at the P dimer intersections, suggesting that multiple sCD300lf molecules might not bind simultaneously at this intersection.
The putative receptor for human norovirus still remains unknown. A model of the human norovirus P domain and CR10 P domain-sCD300lf complex revealed a close proximity to HBGA binding pockets for GI (similar to the case with GI.1, GI.2, and GI.8) and GII (similar to the case with GII.4, GII.10, GII.12, and GII.17) (Fig. 9). However, this equivalent sCD300lf binding region on the human norovirus P domain is highly variable (14) and different genotypes have P2 subdomain loops of varied lengths (19, 32, 33), which was also evident from a clash on the model (Fig. 9). Moreover, human norovirus-specific monoclonal antibodies and nanobodies that bind nearby the HBGA pockets are typically genotype specific (15, 34, 35), which additionally revealed the variability in this region. Therefore, it is tempting to speculate that the human norovirus receptor-binding site might be different among the numerous genotypes and/or the receptor-binding sites are located away from the known HBGA pockets.
FIG 9.

X-ray crystal structures of human norovirus P domains (GI.1 and GII.10) and CR10 P domain sCD300lf complex. The X-ray crystal structure of CR10 P domain sCD300lf complex was superpositioned on the X-ray crystal structures of the GI.1 P domain (PDB code 2ZL6) (A) and GII.10 P domain (PDB code 3PA1) (B), showing the top and side views. The GI.1 and GII.10 HBGA binding pockets (orange) are in close proximity to the sCD300lf-binding site.
In summary, we showed how the sCD300lf bound on the mouse norovirus P domain. The receptor-binding site was located on the topside of the P2 subdomain and involved hydrophilic and hydrophobic interactions. The sCD300lf bound in a small cavity and was further supported by a positive surface charge on the sCD300lf and negative surface charge on the P domain. The CD300lf residues interacting with the CR10 P domain were partially conserved in CD300ld but not other CD300 family members, which indicated the selective recognition of CD300lf. Defining these atomic-resolution interactions between the capsid protein and receptor can explain in part the host range restriction and murine norovirus tropism.
MATERIALS AND METHODS
Expression and purification of the CR10 P domain.
The CR10 murine norovirus P domain (GenBank accession number ABU55613; residues 226 to 538) was prepared as previously described (15). Briefly, the codon-optimized P domain was cloned into a modified expression vector, pMal-c2X, and transformed into BL21 cells. Transformed cells were grown in LB medium (supplemented with 100 μg/ml of ampicillin) for 4 h at 37°C. Expression was induced with isopropyl-β-d-thiogalactopyranoside (IPTG; 0.7 mM) at an optical density at 600 nm (OD600) of 0.7 for 18 h at 22°C. Cells were harvested by centrifugation and disrupted by sonication on ice. A His-tagged fusion-P domain protein was purified from a nickel-nitrilotriacetic acid (Ni-NTA) column (Qiagen) and digested with HRV-3C protease (Novagen) overnight at 4°C. The cleaved P domain was separated on the Ni-NTA column and dialyzed in gel filtration buffer (GFB; 25 mM Tris-HCl [pH 7.6] and 300 mM NaCl) overnight at 4°C. The P domain was purified by size exclusion chromatography, concentrated to 4 mg/ml in GFB, and stored at 4°C.
Expression and purification of sCD300lf.
The extracellular domain sCD300lf (AAH57864.1; residues 20 to 131, termed 1 to 111 in this report and in reference 25) was cloned into a pHEN6C expression vector as described previously, with slight modifications (15). Expression was induced with IPTG (1 mM) at an OD600 of 0.7 for 18 h at 28°C. sCD300lf was extracted from periplasm and the supernatant collected. sCD300lf was eluted from a Ni-NTA column after a series of washing steps and purified by size exclusion chromatography, concentrated to 3.2 mg/ml in GFB, and stored at 4°C.
Neutralization properties of purified sCD300lf.
Murine norovirus (MNV-1 CW1) was propagated in RAW 264.7 cells as described previously (2). Virus neutralization with sCD300lf was determined using a standard plaque assay (2). Briefly, various concentrations of sCD300lf were preincubated with equal PFU of CW1 murine norovirus for 1 h at room temperature and then added to cell monolayer for 1 h at 37°C and 5% CO2. The culture medium was aspirated and a low-melting-point (LMP) agarose overlay with matching concentrations of sCD300lf was applied. Plates were incubated for 42 h at 37°C and 5% CO2. For inhibition of viral attachment, a modified plaque assay was performed (36). Briefly, CW1 murine norovirus was preincubated with various concentrations of sCD300lf at room temperature (RT) for 1 h and the mixtures were applied to precooled cell monolayer plates for 3 h at 4°C. Unbound virus was removed and plates were washed twice with ice-cold phosphate-buffered saline (PBS) prior to addition of the LMP agarose overlay. Data were analyzed using Prism GraphPad 7. The assays were repeated three times to calculate standard deviation.
sCD300lf reactivity using a direct ELISA.
The sCD300lf reactivities against murine norovirus virions and the CR10 P domain were determined using a direct ELISA (15). Microtiter plates (Maxisorp) were first coated with 100 μl (10 μg/ml) of murine norovirus virions or 100 μl (15 to 20 μg/ml) of the CR10 P domain. Wells were washed three times with PBS (pH 7.4) containing 0.1% Tween 20 (PBS-T) and then blocked with 300 μl of PBS containing 5% skim milk (PBS-SM) for 1 h at room temperature. After washing, 100 μl of serially diluted sCD300lf (starting from 200 μg/ml) was added to each well. The wells were washed, and then 100 μl of a 1:3,000 dilution of horseradish peroxidase (HRP)-conjugated anti-His IgG (Sigma) was added to the wells for 1 h at 37°C. After washing, 100 μl of 50 mM phosphate-citrate buffer (pH 5.0) containing the substrate o-phenylenediamine dihydrochloride (OPD) and H2O2 was added to wells and the plates were left in the dark for 30 min at room temperature. The reaction was stopped with the addition of 50 μl of 3 N HCl, and the absorbance was measured at 490 nm (OD490). A murine norovirus-specific nanobody (termed Nano-45) that was produced against the MNV1 murine norovirus P domain (developed as previously described [15]) was used as a positive control. Data were analyzed using Prism GraphPad 7, and experiments were repeated three times to calculate standard deviations.
Crystallization of the CR10 P domain and sCD300lf.
The CR10 domain and sCD300lf were mixed in a 1:1.2 molar ratio and incubated overnight at 4°C. The complex was purified by size exclusion chromatography using a Superdex-200 column. The CR10 P domain and sCD300lf were eluted as two separate peaks at their corresponding molecular weights, rather than as a complex. The two peaks were mixed and concentrated to 5.1 mg/ml in GFB. The mixture was crystallized in a mother solution containing 25% (wt/vol) polyethylene glycol 3000 (PEG 3000) and 0.1 M sodium acetate (pH 4.6). Prior to flash freezing, crystals were transferred to a cryoprotectant solution containing the same mother liquor with an addition of 40% PEG 3000.
Data collection, structure solution, and refinement.
A single crystal diffracted to a resolution of 2.05 Å, and the structure was solved as described earlier (15). Briefly, X-ray diffraction data were collected at the European Synchrotron Radiation Facility, France, at beamline ID29 and processed with XDS (37). The structure was solved using molecular replacement in PHASER (38) using the previously solved murine norovirus P domain (PDB code 3LQ6) and sCD300lf (PDB code 5FFL) as search models. The CR10 P domain-sCD300lf complex formed crystals in space group P212121 with one P dimer and one sCD300lf per asymmetric unit. Possible interfaces derived from crystal packing were evaluated using the PDBePISA server (http://www.ebi.ac.uk/pdbe/pisa). The biologically relevant interaction that involves the variable CDR region and has the highest total area of interface was selected. The complex structure was refined in multiple rounds of manual model building in COOT (39), with subsequent refinement with PHENIX (40). The structure was validated with COOT and Molprobity (41). The binding interactions were analyzed using Accelrys Discovery Studio, with hydrogen bonding interaction distances between 2.8 and 3.5 Å and hydrophobic interaction distances between 3.9 and 5.3 Å. Figures and protein contact potentials were generated using PyMOL and Chimera.
Sequence alignments.
Amino acid sequences of CD300 and murine norovirus VP1 were aligned using ClustalX (Genetyx). CD300 sequences used included (GenBank accession numbers are in parentheses) mouse CD300lf (AAH57864.1), mouse CD300ld (NP_663412.1), mouse CD300lh (XP_017169688.1), mouse CD300la (NP_739564.1), mouse CD300lb (AAI07353.1), mouse CD300lc (NP_954695.1), mouse CD300le (NP_742047.1), mouse CD300lg (NP_001154183.1), human CD300f (NP_620587.2), and human CD300d (NP_001108624.1). Murine norovirus VP1 sequences used included those from strains CR10 (ABU55613.1), CR6 (JQ237823.1), MNV-1 (AAO63099.2), CW1 (DQ285629.1), CW3 (EF014462.1), and S7 (AB435515).
Accession number(s).
Atomic coordinates and structure factors of the complex were deposited in the Protein Data Bank with accession code 5OR7.
ACKNOWLEDGMENTS
We acknowledge the protein crystallization platform within the excellence cluster CellNetworks of the University of Heidelberg for crystal screening. We thank the staff of the European Synchrotron Radiation Facility (ESRF) and EMBL-Grenoble for assistance and support in using beamline ID29. We thank Herbert W. Virgin for the murine norovirus virions.
G.S.H. was funded by the CHS foundation, the Helmholtz-Chinese Academy of Sciences (HCJRG-202), the BMBF VIP+ (Federal Ministry of Education and Research) (NATION, 03VP00912), and Deutsche Forschungsgemeinschaft (DFG, FOR2327).
REFERENCES
- 1.McFadden N, Bailey D, Carrara G, Benson A, Chaudhry Y, Shortland A, Heeney J, Yarovinsky F, Simmonds P, Macdonald A, Goodfellow I. 2011. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog 7:e1002413. doi: 10.1371/journal.ppat.1002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. 2004. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol 2:e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW IV. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. doi: 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X, Wang M, Graham DY, Estes MK. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J Virol 66:6527–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290. doi: 10.1126/science.286.5438.287. [DOI] [PubMed] [Google Scholar]
- 6.Hansman GS, Biertumpfel C, Georgiev I, McLellan JS, Chen L, Zhou T, Katayama K, Kwong PD. 2011. Crystal structures of GII.10 and GII.12 norovirus protruding domains in complex with histo-blood group antigens reveal details for a potential site of vulnerability. J Virol 85:6687–6701. doi: 10.1128/JVI.00246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi JM, Hutson AM, Estes MK, Prasad BV. 2008. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc Natl Acad Sci U S A 105:9175–9180. doi: 10.1073/pnas.0803275105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao S, Lou Z, Tan M, Chen Y, Liu Y, Zhang Z, Zhang XC, Jiang X, Li X, Rao Z. 2007. Structural basis for the recognition of blood group trisaccharides by norovirus. J Virol 81:5949–5957. doi: 10.1128/JVI.00219-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bu W, Mamedova A, Tan M, Xia M, Jiang X, Hegde RS. 2008. Structural basis for the receptor binding specificity of Norwalk virus. J Virol 82:5340–5347. doi: 10.1128/JVI.00135-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh BK, Koromyslova A, Hansman GS. 2016. Structural analysis of bovine norovirus protruding domain. Virology 487:296–301. doi: 10.1016/j.virol.2015.10.022. [DOI] [PubMed] [Google Scholar]
- 11.Singh BK, Glatt S, Ferrer JL, Koromyslova AD, Leuthold MM, Dunder J, Hansman GS. 2015. Structural analysis of a feline norovirus protruding domain. Virology 474:181–185. doi: 10.1016/j.virol.2014.10.028. [DOI] [PubMed] [Google Scholar]
- 12.Taube S, Rubin JR, Katpally U, Smith TJ, Kendall A, Stuckey JA, Wobus CE. 2010. High-resolution X-ray structure and functional analysis of the murine norovirus 1 capsid protein protruding domain. J Virol 84:5695–5705. doi: 10.1128/JVI.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katpally U, Voss NR, Cavazza T, Taube S, Rubin JR, Young VL, Stuckey J, Ward VK, Virgin HW IV, Wobus CE, Smith TJ. 2010. High-resolution cryo-electron microscopy structures of murine norovirus 1 and rabbit hemorrhagic disease virus reveal marked flexibility in the receptor binding domains. J Virol 84:5836–5841. doi: 10.1128/JVI.00314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansman GS, Taylor DW, McLellan JS, Smith TJ, Georgiev I, Tame JR, Park SY, Yamazaki M, Gondaira F, Miki M, Katayama K, Murata K, Kwong PD. 2012. Structural basis for broad detection of genogroup II noroviruses by a monoclonal antibody that binds to a site occluded in the viral particle. J Virol 86:3635–3646. doi: 10.1128/JVI.06868-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koromyslova AD, Hansman GS. 2015. Nanobody binding to a conserved epitope promotes norovirus particle disassembly. J Virol 89:2718–2730. doi: 10.1128/JVI.03176-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Chen Y, Jiang X, Xia M, Yang Y, Tan M, Li X, Rao Z. 2015. A unique human norovirus lineage with a distinct HBGA binding interface. PLoS Pathog 11:e1005025. doi: 10.1371/journal.ppat.1005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hao N, Chen Y, Xia M, Tan M, Liu W, Guan X, Jiang X, Li X, Rao Z. 2015. Crystal structures of GI.8 Boxer virus P dimers in complex with HBGAs, a novel evolutionary path selected by the Lewis epitope. Protein Cell 6:101–116. doi: 10.1007/s13238-014-0126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koromyslova AD, Hansman GS. 2017. Nanobodies targeting norovirus capsid reveal functional epitopes and potential mechanisms of neutralization. PLoS Pathog 13:e1006636. doi: 10.1371/journal.ppat.1006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh BK, Leuthold MM, Hansman GS. 2016. Structural constraints on human norovirus binding to histo-blood group antigens. mSphere 1:e00049-. doi: 10.1128/mSphere.00049-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh BK, Leuthold MM, Hansman GS. 2015. Human noroviruses' fondness for histo-blood group antigens. J Virol 89:2024–2040. doi: 10.1128/JVI.02968-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, Kou B, Opekun AR, Burrin D, Graham DY, Ramani S, Atmar RL, Estes MK. 2016. Replication of human noroviruses in stem cell-derived human enteroids. Science 353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones MK, Grau KR, Costantini V, Kolawole AO, de Graaf M, Freiden P, Graves CL, Koopmans M, Wallet SM, Tibbetts SA, Schultz-Cherry S, Wobus CE, Vinje J, Karst SM. 2015. Human norovirus culture in B cells. Nat Protoc 10:1939–1947. doi: 10.1038/nprot.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taube S, Perry JW, McGreevy E, Yetming K, Perkins C, Henderson K, Wobus CE. 2012. Murine noroviruses bind glycolipid and glycoprotein attachment receptors in a strain-dependent manner. J Virol 86:5584–5593. doi: 10.1128/JVI.06854-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taube S, Perry JW, Yetming K, Patel SP, Auble H, Shu L, Nawar HF, Lee CH, Connell TD, Shayman JA, Wobus CE. 2009. Ganglioside-linked terminal sialic acid moieties on murine macrophages function as attachment receptors for murine noroviruses. J Virol 83:4092–4101. doi: 10.1128/JVI.02245-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, Virgin HW. 2016. Discovery of a proteinaceous cellular receptor for a norovirus. Science 353:933–936. doi: 10.1126/science.aaf1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haga K, Fujimoto A, Takai-Todaka R, Miki M, Doan YH, Murakami K, Yokoyama M, Murata K, Nakanishi A, Katayama K. 2016. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc Natl Acad Sci U S A 113:E6248–E6255. doi: 10.1073/pnas.1605575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borrego F. 2013. The CD300 molecules: an emerging family of regulators of the immune system. Blood 121:1951–1960. doi: 10.1182/blood-2012-09-435057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolawole AO, Li M, Xia C, Fischer AE, Giacobbi NS, Rippinger CM, Proescher JB, Wu SK, Bessling SL, Gamez M, Yu C, Zhang R, Mehoke TS, Pipas JM, Wolfe JT, Lin JS, Feldman AB, Smith TJ, Wobus CE. 2014. Flexibility in surface-exposed loops in a virus capsid mediates escape from antibody neutralization. J Virol 88:4543–4557. doi: 10.1128/JVI.03685-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katpally U, Wobus CE, Dryden K, Virgin HW IV, Smith TJ. 2008. Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J Virol 82:2079–2088. doi: 10.1128/JVI.02200-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolawole AO, Smith HQ, Svoboda SA, Lewis MS, Sherman MB, Lynch GC, Pettitt BM, Smith TJ, Wobus CE. 2017. Norovirus escape from broadly neutralizing antibodies is limited to allostery-like mechanisms. mSphere 2(5):e00334-. doi: 10.1128/mSphere.00334-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi SC, Simhadri VR, Tian L, Gil-Krzewska A, Krzewski K, Borrego F, Coligan JE. 2011. Cutting edge: mouse CD300f (CMRF-35-like molecule-1) recognizes outer membrane-exposed phosphatidylserine and can promote phagocytosis. J Immunol 187:3483–3487. doi: 10.4049/jimmunol.1101549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weichert S, Koromyslova A, Singh BK, Hansman S, Jennewein S, Schroten H, Hansman GS. 2016. Structural basis for norovirus inhibition by human milk oligosaccharides. J Virol 90:4843–4848. doi: 10.1128/JVI.03223-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh BK, Koromyslova A, Hefele L, Gurth C, Hansman GS. 2015. Structural evolution of the emerging 2014/15 GII.17 noroviruses. J Virol 90:2710–2715. doi: 10.1128/JVI.03119-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shanker S, Czako R, Sapparapu G, Alvarado G, Viskovska M, Sankaran B, Atmar RL, Crowe JE Jr, Estes MK, Prasad BV. 2016. Structural basis for norovirus neutralization by an HBGA blocking human IgA antibody. Proc Natl Acad Sci U S A 113:E5830–E5837. doi: 10.1073/pnas.1609990113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koromyslova A, Tripathi S, Morozov V, Schroten H, Hansman GS. 2017. Human norovirus inhibition by a human milk oligosaccharide. Virology 508:81–89. doi: 10.1016/j.virol.2017.04.032. [DOI] [PubMed] [Google Scholar]
- 36.Tai CJ, Li CL, Tai CJ, Wang CK, Lin LT. 2015. Early viral entry assays for the identification and evaluation of antiviral compounds. J Vis Exp 2015(105):e53124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26:795–800. doi: 10.1107/S0021889893005588. [DOI] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]