

ABSTRACT
Herpes simplex virus 1 (HSV-1) establishes a lifelong latent infection in host peripheral neurons, including the neurons of the trigeminal ganglia (TG). HSV-1 can reactivate from neurons to cause recurrent infection. During latency, the insulator protein CTCF occupies DNA binding sites on the HSV-1 genome, and these sites have been previously characterized as functional enhancer-blocking insulators. Previously, CTCF was found to be dissociated from wild-type virus postreactivation but not in mutants that do not reactivate, indicating that CTCF eviction may also be an important component of reactivation. To further elucidate the role of CTCF in reactivation of HSV-1, we used recombinant adeno-associated virus (rAAV) vectors to deliver a small interfering RNA targeting CTCF to peripheral neurons latent with HSV-1 in rabbit TG. Our data show that CTCF depletion resulted in long-term and persistent shedding of infectious virus in the cornea and increased ICP0 expression in the ganglia, indicating that CTCF depletion facilitates HSV-1 reactivation.
IMPORTANCE Increasing evidence has shown that the insulator protein CTCF regulates gene expression of DNA viruses, including the gammaherpesviruses. While CTCF occupation and insulator function control gene expression in DNA viruses, CTCF eviction has been correlated to increased lytic gene expression and the dissolution of transcriptional domains. Our previous data have shown that in the alphaherpesvirus HSV-1, CTCF was found to be dissociated from the HSV-1 genome postreactivation, further indicating a global role for CTCF eviction in the transition from latency to reactivation in HSV-1 genomes. Using an rAAV8, we targeted HSV-1-infected peripheral neurons for CTCF depletion to show that CTCF depletion precedes the shedding of infectious virus and increased lytic gene expression in vivo, providing the first evidence that CTCF depletion facilitates HSV-1 reactivation.
KEYWORDS: insulator, CTCF, HSV-1 reactivation, epigenetics, chromatin, rabbit ocular, in vivo reactivation, AAV8, gene delivery, HSV-1, in vivo reactivation
INTRODUCTION
Herpes simplex virus 1 (HSV-1) establishes a lifelong latent infection in host peripheral neurons, including the neurons of the trigeminal ganglia (TG) (1). HSV-1 can exit latency and reactivate from TG neurons to cause recurrent infection in the nose, mouth, or eyes, with the latter responsible for herpes keratitis. Herpes keratitis is a global cause of visual impairment and blindness (2); nonetheless, advancements in the development of novel therapeutics to treat herpes recurrence have been scarce due to a poor understanding of mechanisms controlling latency and reactivation.
Following the primary HSV-1 infection in the epithelium where abundant lytic transcription of the virus occurs, HSV-1 enters peripheral neurons via retrograde axonal transport. Here, lytic gene transcription is essentially silenced, while the latency-associated transcript (LAT) is the only transcript abundantly expressed (3). The latent genome persists as a circular episome associated with histones bearing modifications corresponding to the transcriptional status of lytic genes and the LAT. Specifically, the LAT loci is enriched in euchromatic histone marks during latency (4–7), while the juxtaposed immediate-early (IE) lytic genes are enriched in repressive heterochromatic histone marks (8–11). Subsequent to reactivation stimuli, the transcriptional profiles of the LAT and IE change as the LAT region loses euchromatic histone marks, while the IE lytic regions gain euchromatic histone marks (7, 12, 13). The organization of these transcriptional domains within the latent genome and their subsequent dissolution during reactivation indicate that the previously identified chromatin insulators of HSV-1 (14, 15) may control the maintenance of latency in the neuron.
Chromatin insulators are long-range, cis-acting DNA sequence elements and protein complexes that recruit chromatin modifiers and other transcriptional regulatory factors and function to regulate transcriptional domains and gene expression by preventing inappropriate signaling from one transcriptional domain to another (16). Predominantly characterized in eukaryotic cells, chromatin insulators consist of the insulator protein bound to a conserved but complex DNA consensus motif. The most common insulator protein characterized is CCCTC-binding factor (CTCF), an 11 zinc finger nuclear protein that regulates transcriptional domains through a number of functions, including promoter activation, repression, silencing, and barrier activity (17). Further, CTCF-mediated transcriptional repression of lytic phase transcription is an emerging theme in the regulation of DNA viruses. For example, both human cytomegalovirus and human papillomavirus have CTCF sites in their major lytic promoters. When deleted, insulator function is disrupted and lytic transcription increases (18, 19). In the gammaherpesvirus Kaposi's sarcoma-associated herpesvirus (KSHV), CTCF binds to an intergenic site between the open reading frame 73 (ORF73; active during latency) and K14 (active during the lytic infection) to act as an insulator to repress lytic K14 transcription (20). In Epstein-Barr virus genomes, CTCF binds to a site between the viral origin of replication (OriP) and the C promoter (Cp) to repress transcription of Cp (21). These recent works defining the CTCF insulator's role in lytic gene repression of beta- and gammaherpesviruses further supports the possibility that CTCF insulators also regulate alphaherpesvirus latency through lytic gene repression.
Seven conserved reiterated CTCF binding motifs were previously identified in the latent HSV-1 genome (14). Provocatively, these binding domains flank the reactivation critical region of the LAT, as well as each individual IE gene region (14). We previously showed that all CTCF sites were occupied by CTCF during latency, and that three of the sites functioned as enhancer-blocking insulators in rabbit skin cells (14, 22). We also showed that CTCF was evicted from the HSV-1 genome at very early times postreactivation in vivo in mice latent with the wild-type virus 17Syn+, but not in mutants that lacked the ability to reactivate, indicating that CTCF eviction may be an important component of reactivation (22).
Our previous work demonstrated that CTCF dissociated from the HSV-1 genome postreactivation, in line with changes in the histone marks associated with the LAT and IE regions of HSV-1. However, these earlier studies could not elucidate whether CTCF dissociation was induced by the stimuli to cause HSV-1 reactivation or whether CTCF dissociation was a downstream response to global HSV-1 reactivation. We hypothesized that CTCF knockdown using an small interfering RNA (siRNA) targeted to the protein CTCF would clarify the role of CTCF in the maintenance of latency and reactivation. To accomplish this, we used recombinant adeno-associated virus (rAAV) vectors to deliver an siRNA targeting CTCF to neurons latent with HSV-1 in rabbit TG. rAAV vectors are widely considered to be nonpathogenic, can infect a wide variety of cell types, and are capable of long-term gene expression in nonmitotic, terminally differentiated cells (23, 24). These vectors have a transgene carrying capacity of about 4.5 kb within their single-stranded DNA genomes (25, 26), and we recently developed a method of ocular delivery of rAAV in the rabbit to efficiently transduce >75% of TG neurons (27). Further, we showed in multiple studies that transduction of either the empty rAAV8 vector or the rAAV8-rhodopsin ribozyme vector did not induce ocular pathology, in vivo reactivation, or other ocular delivery-related pathologies (28). Finally, we showed that rAAV vectors colocalized with HSV-1-positive neurons (27), suggesting that rAAV delivery of siRNAs could efficiently knock down host and/or viral proteins in order to define their roles in the establishment, maintenance, and reactivation from latency in vivo in the absence of global pleotropic effects. To explore our hypothesis that CTCF dissociation drives lytic gene activation and subsequent reactivation in HSV-1, we generated an AAV8-siCTCF vector and used ocular delivery of the vector to deplete CTCF in sensory neurons of rabbits following the establishment of HSV-1 latency. In this manuscript, we provide the first evidence that CTCF depletion facilitates HSV-1 reactivation using an in vivo model of latency and reactivation.
RESULTS
An siRNA targeting CTCF efficiently knocks down CTCF in vitro.
Four siRNAs designed to target CTCF were tested for their relative abilities to knock down CTCF following transfection. Figure 1 depicts the normalized quantities of CTCF RNA after transfection of the four siRNAs compared to the negative control (a nontargeting siRNA sequence). The data represent three independent transfections for each siRNA, and the ability of each siRNA to reduce CTCF transcripts is presented as relative quantities of CTCF cDNA normalized to 18S RNA cDNA. At 25 nM (Fig. 1A), one siRNA (CTCF9) showed no significant knockdown relative to the negative control. Further, the knockdown efficiency of CTCF11 was only 34%. The CTCF10 and CTCF12 siRNAs were slightly more efficient, with 48 and 55% reductions in CTCF expression, respectively, although the differences in knockdown by these two were not significant. The positive control, a GAPDH (glyceraldehyde-3-phosphate dehydrogenase)-targeting siRNA, produced a 5-fold knockdown. To determine whether increasing the concentration of siRNAs increased the efficiency of knockdown, the experiment was repeated with 100 nM concentrations of the two most efficient siRNAs from the previous experiment (CTCF10 and CTCF12), as well as both siRNAs at the same time (Fig. 1B). Although the knockdown efficiency increased to ∼75% for both CTCF10 and CTCF12, combining the two siRNAs had little effect on the overall knockdown efficiency. Because there was no statistical difference between CTCF10 and CTCF12 in the in vitro knockdown studies, a transcription cassette to express the CTCF10 siRNA was cloned into a recombinant self-complementary AAV2 vector, which was then pseudotyped and packaged in an AAV8 capsid; the resulting vector AAV8-siCTCF was used in all animal experiments.
FIG 1.
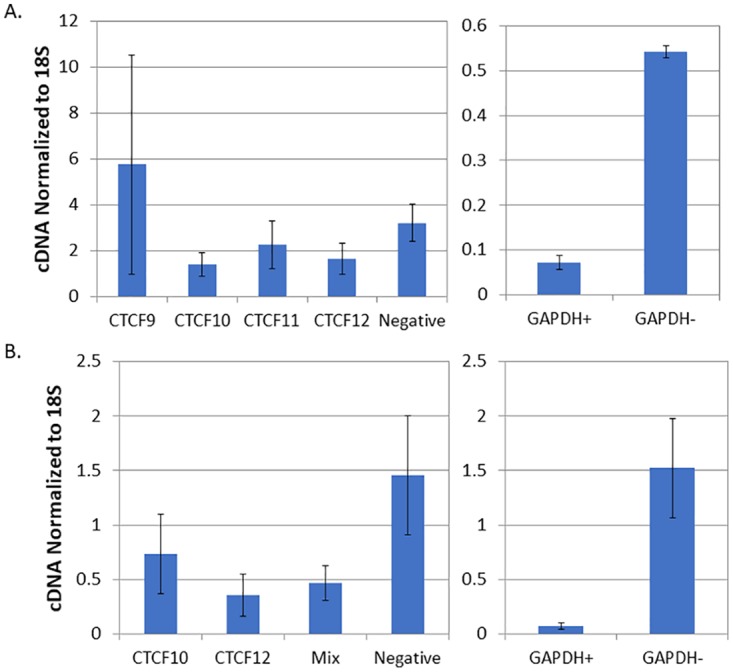
Analysis of siRNAs targeting CTCF for their ability to knock down CTCF following transfection in vitro. Vero cells were transfected with either 25 nM (A) or 100 nM (B) concentrations of four different siRNAs targeting CTCF. After 24 h, RNA was isolated, and the amount of CTCF RNA was determined was determined by real-time PCR on cDNA prepared from the RNA. The right graph represents control experiments with GAPDH-targeting siRNA (GAPDH+) against the negative control (GAPDH−). (A) Normalized quantity of CTCF cDNA following transfection with 25 nM each siRNA; (B) normalized quantity of cDNA using 100 nM CTCF 10 and CTCF 12 siRNAs, as well as a mixture of CTCF10 and CTCF12.
CTCF is depleted in sensory neurons in rabbit TG following ocular delivery of AAV8-siCTCF.
We recently developed an efficient method for the delivery of rAAV8 vectors to sensory neurons in TG of rabbits following ocular delivery with the ability to transduce greater than 75% of neurons, including ones infected with HSV-1 (27). The development of this novel rAAV delivery method, combined with its colocalization with HSV-1 at the site of latency (neuron), provides us with an integral tool to deplete host proteins in a targeted manner in order to study their role in regulating HSV latency in vivo. We delivered AAV8-siCTCF, the vector expressing an siRNA targeting the protein CTCF, to neurons in the rabbit TG via an ocular route. The experimental timeline of the animal study of CTCF depletion is shown in Fig. 2. We infected rabbits via the ocular route with HSV-1 strain 17Syn+ and allowed the virus to establish a latent infection in the TG neurons. Rabbits were considered latent with the absence of viral lesions in the eye (measured by slit lamp microscopy) and the absence of infectious virus in the tear film (at approximately day 24 postinfection [p.i.]). Rabbits were then inoculated with a suspension of AAV8-GFP (control) or AAV8-siCTCF spiked with the AAV8-GFP capsid to allow for immunofluorescence visualization of AAV8-transduced neurons. At the conclusion of the experiment (day 92 p.i.) rabbit TG were removed and analyzed by dual immunofluorescence for CTCF and green fluorescent protein (GFP) to visualize the effect of either AAV8-GFP or AAV8-siCTCF on CTCF protein levels in AAV-transduced neurons (Fig. 3). To quantitate the knockdown efficiencies in vivo, the TG sections were divided into four quadrants, and the neurons were counted to determine the total number of GFP-positive neurons and the total number of CTCF-positive neurons. The ratios of CTCF+ to GFP+ neurons were calculated for both the control and treatment groups. Using this method, we calculated that the knockdown efficiency of CTCF was >70% in neurons transduced with AAV8-GFP (Table 1). Figure 3 shows representative images (one quadrant) for the AAV8-siCTCF and AAV8-GFP control rabbits following dual-labeled immunofluorescence.
FIG 2.

Experimental timeline for infection of rabbits and the subsequent delivery of either AAV8-GFP or AAV8-siCTCF.
FIG 3.

Analysis of rabbit TG neurons for AAV transduction and CTCF expression. Rabbits were treated with AAV vectors on the eye. Two weeks later, rabbits were sacrificed, and the TG were removed and sectioned for immunofluorescence. The sections were incubated with mouse anti-GFP (primary) and Alexa Fluor 488-conjugated (secondary) antibodies for the visualization of the GFP-expressing AAV8 (AAV8-GFP) and mouse anti-CTCF (primary) and an Alexa Fluor 548-conjugated (secondary) antibodies for the visualization of CTCF. DAPI staining in blue represents nuclei of satellite cells present in the sections. Panel A images are shown at ×10 magnification. Panel B images show only AAV-siCTCF-treated animals at a ×40 magnification.
TABLE 1.
Quantification of knockdown of CTCF
| Treatment group | No. of neurons |
% knockdownb | ||
|---|---|---|---|---|
| CTCF+a | GFP+ | CTCF+/GFP+ | ||
| Control (empty AAV8 vector) | 192 | 192 | 192/192 | 0 |
| AAV8-siCTCF treated | 63 | 212 | 63/212 | 70 |
Only GFP+ neurons were used to calculate CTCF knockdown efficiencies in control and treated groups.
Percent knockdowns were determined by calculating the percentage of CTCF+ neurons and then subtracting that value from 100%.
CTCF-depleted rabbit neurons express the neuronal marker NeuN.
To determine whether CTCF depletion in the TG neurons led to global neuronal death, we performed immunofluorescence analyses on AAV8-siCTCF-treated rabbit TG using the neuronal marker NeuN. NeuN is a neuron-specific nuclear protein found in both the peripheral nervous system (PNS) and the central nervous system of the vertebrate nervous system (29). Immunohistochemically detectable NeuN protein first appears at developmental time points that correspond to the withdrawal of the neuron from the cell cycle and/or with the initiation of terminal differentiation of the neuron. NeuN expression indicates neuron survival, whereas decreased expression of NeuN indicates neuronal death or neuronal incompetence (30–32). We performed dual-immunofluorescence analyses on rabbit TG sections using anti-GFP and anti-NeuN to determine (i) whether there was any impact on NeuN expression by AAV transduction itself (using the control vector AAV8-GFP) and (ii) whether CTCF knockdown resulted in decreased NeuN expression. Using the four-quadrant method described above, we calculated the GFP+/NeuN+ ratios for cells and found no significant reduction in NeuN expression between GFP-positive and GFP-negative neurons in the AAV8-GFP-treated TG or between the control and siCTCF-treated groups. Representative images are shown in Fig. 4. To confirm that NeuN was actively expressed in cells that were actually CTCF depleted, additional dual-immunofluorescence experiments confirmed that NeuN was expressed in cells not expressing detectable CTCF, indicating that CTCF depletion in neurons did not result in marked cell death (Fig. 5).
FIG 4.

Immunofluorescence analysis of rabbit TG for neuronal colocalization of the AAV8 vector and NeuN. The sections were incubated with mouse anti-GFP (primary) and an Alexa Fluor 488-conjugated (secondary) antibodies for the visualization of AAV8-GFP and mouse anti-NeuN (primary) and an Alexa Fluor 548-conjugated (secondary) antibodies for the visualization of NeuN. DAPI staining in blue represents nuclei of satellite cells present in the sections. Panel A images are shown at ×10 magnification. Panel B images show only the AAV-siCTCF-treated animals at a ×40 magnification.
FIG 5.

Immunofluorescence was performed to show neuronal colocalization of the AAV8-GFP, NeuN, and CTCF. The sections were incubated with mouse anti-NeuN (primary) and an Alexa Fluor 488-conjugated (secondary) antibodies for the visualization of the NeuN and mouse anti-CTCF (primary) and Alexa Fluor 548-conjugated (secondary) antibodies for the visualization of CTCF. DAPI staining in blue represents nuclei of satellite cells present in the sections. Images are shown at ×10 magnification.
CTCF depletion in rabbits harboring latent HSV-1 results in persistent shedding of infectious virus and reactivation.
To determine the physiological effect of depletion of CTCF on the maintenance of latency in the rabbit model, we performed ocular swabbing of control and treated rabbits following the delivery of the control AAV8-GFP or the AAV8-siCTCF vector as described in the experimental timeline (see Fig. 2). Following the establishment of latency in our infected rabbits, the animals were evenly divided into two groups. The control group received the AAV8-GFP vector, while the experimental group received the AAV8-siCTCF vector spiked with AAV8-GFP for immunofluorescence visualization. Rabbit eyes were swabbed for 10 consecutive days following the delivery of the vectors to determine whether CTCF depletion resulted in the presence of infectious virus and then remained untouched for another 18 consecutive days. On day 56 p.i., rabbit eyes were again swabbed for an additional 5 days to determine whether infectious virus persisted (Fig. 6). Four of the five rabbits treated with AAV8-siCTCF shed infectious virus, with 16% of the total ocular swabs being positive for virus, compared to only one rabbit in the control group (the control rabbit had only two positive ocular swabs [2%], a finding consistent with spontaneous reactivation) (P < 0.0001) (Fig. 6). Further, in rabbits treated with siCTCF, persistent shedding of infectious virus could be detected through day 61 p.i. (i.e., >30 days after siCTCF delivery) compared to no long-term persistence of infectious virus shedding in the control animals (Fig. 6). These data indicate that CTCF depletion results in in vivo reactivation from latency in the absence of classical reactivation stimuli. To ensure that AAV8-GFP-treated rabbits were capable of reactivating HSV-1, we performed transcorneal iontophoresis of epinephrine (TCIE) on the AAV8-GFP-treated animals, as well as the one AAV8-siCTCF-treated rabbit that did not reactivate. After TCIE, all rabbits reactivated (Table 2). In addition, we observed no significant difference in the number of positive swabs between the AAV8-GFP-treated rabbits subjected to TCIE and the positive swabs from the CTCF-depleted rabbits (no TCIE) (P < 0.165), indicating that CTCF depletion is as efficient a reactivation stimulus as the classical method of TCIE (Table 2).
FIG 6.

Measurement of the effect of CTCF knockdown on HSV-1 reactivation in the rabbit eye. The eyes of rabbits latently infected with HSV-1 were swabbed for 10 consecutive days following treatment with either the AAV8-GFP (control) or AAV8-siCTCF, starting at day 28 p.i. Swabs were assayed by plaque assays for infectious virus. Additional swabs were obtained and plaque assayed starting on day 56 p.i. to determine whether rabbits continued to shed infectious virus in the siCTCF-treated group long term. An exact version of chi-square analysis for all pairwise comparisons with control (AAV8-GFP) was performed and indicated a significant increase in infectious virus present in the AAV8-siCTCF-treated animals versus the control group (P < 0.0001).
TABLE 2.
Rabbits positive for infectious virus following TCIE at 71 to 81 days p.i.
| Treatment group | No. of positive swabs/total no. of swabs (%) | No. of rabbits shedding infectious virus/total no. of rabbits |
|---|---|---|
| Control (empty AAV8 vector) | 22/100 (22) | 5/5 |
| AAV8-siCTCF treateda | 16/100 (16) | 4/5 |
An AAV8-siCTCF-treated rabbit that did not shed infectious virus prior to TCIE was subjected to the reactivation protocol. After TCIE, we detected infectious virus in the ocular swabs. These swabs were excluded from the table.
CTCF depletion does not alter genome loads in rabbits.
We performed quantitative real-time PCR using a primer/probe set targeted to the viral gene HSV-1 DNA polymerase to determine whether CTCF depletion resulted in increased HSV-1 genomes in rabbit TG. The relative HSV-1 DNA polymerase gene copies were normalized to the host gene rabbit GAPDH gene following PCR (Fig. 7). Our results show that depletion of CTCF in the neuron did not result in increased viral genomes in vivo, which is consistent with our previous observations showing that viral loads in the ganglia do not increase significantly in the TG following reactivation (22). These data indicate that CTCF depletion does not alter the number of viral genomes in the TG but instead may affect the function of CTCF insulators in HSV-1.
FIG 7.

The relative quantity of HSV-1 latent DNA in the TG of each treatment group was determined by quantitative real-time PCR. The relative quantities of the HSV-1 DNA polymerase gene and rabbit GAPDH gene were determined by qPCR using five rabbit TG per group. TG were harvested at day 92 p.i. Data are presented as an HSV-1 DNA polymerase/GAPDH ratio. One-way analysis of variance (ANOVA) showed no significant difference in HSV-1 copies between the treatment groups.
CTCF depletion in sensory neurons leads to increased ICP0 expression in vivo.
We previously showed that three of the seven CTCF sites in the HSV-1 genome were functional enhancer-blocking insulators in epithelial cells, namely, the CTRL2 site positioned between the LAT enhancer and ICP0 loci, the CTa'm site upstream the ICP0 promoter, and the CTRS3 site located upstream of the ICP4 promoter (14, 22). We hypothesized that CTCF depletion in the neurons might result in the inability of these sites to function as enhancer-blocking insulators, resulting in altered viral transcript expression. To determine whether changes in immediate-early (IE), early (E), and late (L) gene expression could be detected following CTCF depletion, we performed quantitative reverse transcriptase PCR (qRT-PCR) on TG from siCTCF-treated and control animals using primer/probe sequences targeted to the LAT intron, ICP0, HSV-1 DNA polymerase, and VP16. All relative expressions were normalized to the expression of GAPDH for comparisons between the groups. Strikingly, we observed a significant increase in the expression of ICP0 in the AAV8-siCTCF-treated group relative to the control group. It is important to note that the ICP0 region is flanked by two CTCF binding motifs, the CTRL2 and the CTa'm sites, both of which have been found to be enhancer-blocking insulators to the LAT enhancer (see Fig. 9 for the locations of the CTCF binding motifs in the HSV-1 genome) (14). In contrast, no changes in the transcript abundance of the LAT intron, the early genes, or the late genes assessed were observed (Fig. 8). Importantly, there are no CTCF binding motifs located near the E or L genes in HSV-1. Of importance, we also assayed the expression of ICP27 following depletion due to the fact that it lies near the CTRL1 binding motif in HSV-1. Again, we found only baseline levels of ICP27 (or no ICP27) expressed in either the AAV8-siCTCF or control groups (data not shown). These findings consistently support the hypothesis that insulator function might be altered to increase the leakiness and allow activation of ICP0 following CTCF depletion in vivo.
FIG 9.

Model for the initiation of reactivation through CTCF dissociation. During latency, each CTCF binding domain in HSV-1 is occupied by the protein CTCF, and the CTRL2 site has been characterized as a functional enhancer-blocking insulator (14, 22). We propose that during latency the CTRL2 blocks the LAT enhancer from activating the ICP0 promoter to control lytic transcription. CTCF dissociation results in a loss of insulator function and activation of ICP0 by the LAT enhancer to ultimately cause the production of infectious virus and HSV-1 reactivation in epithelial cells.
FIG 8.

LAT and lytic transcript relative abundance was determined from TG harvested at day 92 p.i. RNA was isolated and analyzed by qRT-PCR using primers and probes specific for the LAT intron, ICP0, HSV-1 DNA polymerase, and VP16 (see Table 3). The relative quantities were determined for viral genes and then normalized to the relative quantities of rabbit GAPDH to account for variations between samples. The average normalized values for each gene region are presented, along with the standard deviations. All samples have an n = 5 TG. One-way ANOVA showed that a significant increase in ICP0 transcription was found in siCTCF-treated rabbits versus the control animals.
DISCUSSION
The hypothesis that CTCF insulators control lytic transcription during HSV-1 latency is provocative and timely, especially given that insulator control of lytic gene transcription is an emerging theme in DNA viruses. Previously, we showed that CTCF occupied the HSV-1 genome at all seven CTCF binding motifs during latency and was dissociated at very early times postreactivation, but these data did not discern whether CTCF dissociation was a facilitator of HSV-1 reactivation or merely resulted from the reactivation process as IE transcription occurred. To answer this important question, we combined in vivo delivery of AAV8 vectors to knock down CTCF in neurons. Our data showed a reactivation phenotype following CTCF depletion, marking the first time AAV8-siRNAs have been applied to the cornea, transduced neurons in latently infected animals and produced an in vivo reactivation phenotype.
We also explored the possibility that two different siRNAs could be combined to complement each other, resulting in a higher knockdown efficiency both in vitro and in vivo. We did not observe an increase in knockdown efficiency by combining the CTCF10 and CTCF12 constructs. However, it is important to note that our pool of siRNAs to CTCF was limited to only four in this study and that our findings do not preclude the fact that other combinations of siRNAs in general may still be more effective if the correct combinations of siRNAs were established.
CTCF is a fundamental protein involved in cellular gene regulation and, as such, it is possible that CTCF depletion could cause off-target effects in the rabbit. It is important to note that while we did observe the in vivo reactivation phenotype in response to CTCF depletion, we did not observe any other gross adverse effects in the AAV8-siCTCF-treated rabbits over the entire course of this study. On a molecular level, CTCF depletion did not result in global neuronal death; we observed no change in the NeuN expression in neurons treated with AAV8-siCTCF compared to the AAV8-GFP controls. These findings are consistent with other reports in eukaryotic gene expression that showed that CTCF depletion in neurons resulted in changes to chromatin architecture and/or gene expression and not global neuronal death (33, 34). Nonetheless, while we did not observe changes in the numbers of neurons that expressed NeuN in treated versus untreated rabbits, we cannot rule out the possibility that CTCF depletion resulted in the activation of an unidentified cellular stress response that ultimately triggered HSV-1 reactivation. Although this remains a possibility, other investigators have also established the role of CTCF and CTCF binding in latency and reactivation. Specifically, Lee et al. recently reported that one of the CTCF binding sites in HSV-1, namely, the CTRL2 site positioned between the LAT enhancer and ICP0, functions as a heterochromatic barrier to keep the viral genome latent. Conversely, deletion of the CTRL2 site attenuates HSV-1 reactivation following explant and results in heterochromatin spread to the LAT region of the genome (35). These findings, together with our previous reports showing CTCF eviction on the viral genome can be detected at early times postreactivation, argue that CTCF depletion results in changes to the chromatin architecture and could drive the process of reactivation.
The timeline for the detection of infectious virus in the tear film of rabbits at 3 days postdelivery of AAV8-siCTCF was consistent with other reports showing that AAV8 delivery of an siRNA targeted to calcineurin induced behavioral changes by 10 days in vivo (36). We also found that the percentages of positive swabs found in the AAV8-siCTCF-treated rabbits were consistent with the percentages of positive swabs in the control group following a classical reactivation stimulus, TCIE. This finding suggested that CTCF knockdown, even at just 60% knockdown efficiency, was enough to efficiently drive the production of infectious virus at the periphery (cornea). Furthermore, rabbits treated with AAV8-siCTCF shed virus for over 2 months, further confirming that long-term knockdown of CTCF can be achieved with AAV8 delivery of the siRNA to CTCF.
Our data also suggested that the reestablishment of latency was disrupted by CTCF depletion. Rabbits that received the AAV8-siCTCF continuously shed infectious virus in the tear film for over 2 months. In contrast, control rabbits that were subjected to TCIE shed virus for less than 10 days and, at the termination of the experiment, were no longer shedding infectious virus at the periphery, an observation indicative of latency. This was confirmed with qRT-PCR data that showed no lytic transcription in the control animals. These findings suggest that the ability of HSV-1 to establish and maintain latency is dependent on CTCF occupation of the binding domains. This hypothesis is currently being explored and will be the subject of an additional manuscript.
The fact that we did not detect changes in virus copies in the ganglia of the AAV8-siCTCF-treated rabbits compared to control animals was not surprising. HSV-1 is a virus that has the ability of establishing a lifelong infection in the host, and host survival is critical to the viral life cycle program. Production of high titers of infectious virus in the neuron would likely result in neuronal death and not be beneficial for HSV-1. Therefore, we speculate that CTCF dissociation initiates reactivation on a molecular level in the neuron, spurred by a loss of CTCF insulator function that poises the genome for the expression of lytic genes, possibly through a mechanism that changes the chromatin profiles of genes critical to HSV-1 reactivation, including ICP0 and ICP4. Previous studies have already quantified the chromatin changes (in parallel to increased IE expression) of HSV-1 in rabbits latently infected with HSV-1 at the LAT, ICP0, and ICP4 loci (12). These changes happen at very early times postreactivation, and it is feasible that the changes in chromatin are facilitated by the dissociation of CTCF.
Finally, we recognize that using the AAV8-siCTCF vector to deplete CTCF in latently infected neurons could result in the global dissociation of CTCF on the HSV-1 viral genome. However, we hypothesize that global dissociation of CTCF is not required for reactivation of HSV-1. For example, it has been shown in other studies, particularly in KSHV, that global eviction of CTCF does not drive lytic gene expression. CTCF is dynamically evicted from some sites in KSHV and is maintained or gradually reduced at other CTCF sites after reactivation (37). Importantly, we have experimental evidence that CTCF is dynamically evicted from some sites in HSV-1, but not others (40), suggesting that a similar mechanism of transcriptional control during latency is conserved among herpesviruses. We know that the CTRL2 site (downstream LAT enhancer) and CTa'm site (upstream ICP0) are both functional enhancer blocker insulators. Both of these sites lose CTCF occupation at early times after reactivation is initiated (22). The fact that CTCF depletion results in an increase in ICP0 transcription, but not ICP27 transcription, supports this hypothesis and suggests the dominant mechanism for controlling IE transcription during latency may be through CTCF preventing the LAT enhancer from activating ICP0 transcription due to its enhancer blocking activity. During reactivation, CTCF is dissociated from this site, and the LAT enhancer would then be able to activate the ICP0 promoter, initiating the process of reactivation (Fig. 9). In the present study, we found that ICP0 expression was significantly higher in AAV8-siCTCF-treated rabbit ganglia compared to the AAV8-GFP controls. These findings are consistent with a loss of CTRL2 insulator function to allow the LAT enhancer to activate the ICP0 promoter to initiate reactivation. We speculate that this response leads to a change in chromatin profiles, thereby allowing the process of reactivation to occur in wild-type virus (17Syn+). This hypothesis is supported by the recent findings by Lee et al. that identified the CTRL2 site in HSV-1 (strain KOS) as a heterochromatic barrier element that prevented the spread of heterochromatin onto the LAT region to maintain latency in vivo (35). Collectively, these studies indicate that CTCF plays a key role in contributing to the maintenance of HSV-1 latency, while a loss of CTCF binding facilitates reactivation.
MATERIALS AND METHODS
Cells and viruses.
3T3 mouse fibroblasts were obtained from the American Type Culture Collection and passaged in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum (Atlanta Biological) and 250 U of penicillin, 250 μg of streptomycin, and 292 μg of l-glutamine/ml (Life Technologies). A low-passage-number stock of HSV-1 strain 17Syn+ was obtained from J. Stevens and recently sequenced. 17Syn+ was grown and titrated on rabbit skin cells using Eagle minimal essential medium (Life Technologies) supplemented with 5% calf serum, 250 U of penicillin/ml, 250 μg of streptomycin/ml, and 292 μg of l-glutamine/ml (Life Technologies).
siRNAs.
Four different siRNAs targeting CTCF were obtained from Dharmacon: CTCF9 (GUUCAAAUUUGGAUCGUCA in the ORF), CTCF10 (CUGUGUUUCAUGAGCGAUA in the ORF), CTCF11 (GCUAAUAAAUCAUAACGGA in the untranslated region [UTR]), and CTCF12 (CCAAACAUACUGAGAACGA in the UTR). For a negative control, a nontargeting siRNA sequence was used, and for a positive control, siRNA targeting GAPDH was used (both obtained from Dharmacon).
Transfections.
For transfection, confluent 3T3 mouse fibroblasts were trypsinized and diluted to a concentration of 5 × 103 cells/ml using a hemocytometer. Then, 500 μl was pipetted into each well of a 24-well plate. Transfections was carried out in triplicate using the transfecting reagent Dharmafect1 (Dharmacon) in accordance with the manufacturer's recommendations. Cells were treated with the siRNAs for 24 to 48 h; the transfecting reagent was replaced with complete DMEM at 24 h.
RNA isolation and cDNA production (in vitro experiments).
RNA from transfected cells was isolated using TRIzol according to the manufacturer's protocol. A Turbo DNA-free kit (Ambion, Inc.) was used to eliminate contaminating DNA. cDNA was synthesized using an Omniscript RT kit (Qiagen).
Quantification of cDNA.
To determine the amount of cDNA present compared to the negative, real-time quantitative PCR (RT-qPCR from Applied Biosystems) was used. A primer/probe combination for CTCF was used on every sample transfected with CTCF-targeting siRNA, as well as the negative control. For the positive control, a GAPDH primer/probe was used for the samples transfected by GAPDH targeting siRNA, as well as the negative. This is to ensure that the small amount of RNA detected for CTCF was due to a successful transfection. The cDNA quantity of 18S rRNA was measured using an 18S primer/probe. The normalized quantity of DNA is calculated as the quantity of cDNA in question divided by the quantity of 18S.
AAV8-GFP.
The plasmid scAAV-GFP was obtained from the UF Powell Gene Therapy Core. This plasmid has a SalI site at 2,108 bp and a cytomegalovirus promoter/enhancer within the two terminal repeat sequences (AAV2). This plasmid was packaged by the UF Powell Gene Therapy Core and pseudotyped into AAV8 capsids.
AAV8-siCTCF.
A pIDT SMART plasmid with ampicillin resistance was designed by IDT to contain the CTCF10 siRNA cassette flanked by SalI cleavage sites to facilitate subcloning of the siRNA cassette into an AAV vector, the H1 promoter region for the recruitment of polymerase III, the sense and antisense sequences that will bind to the mRNA, and a hairpin loop in between. This cassette was then subcloned into the plasmid scAAV-GFP; the plasmid was packaged by the UF Powell Gene Therapy Core and pseudotyped into AAV8 capsids.
Rabbit ocular infections.
New Zealand White rabbits were anesthetized using intramuscular injections of ketamine (30 to 45 mg/kg [body weight]) and xylazine (7.5 to 11.5 mg/kg [body weight]). Using a blunt-tip 27-gauge needle, a 3-by-3 crosshatch pattern was made on the corneal surface. HSV-1 strain 17Syn+ was applied to each eye in a volume of 20 μl (200,000 PFU/eye). Rabbits were monitored, and slit lamp exams were performed on postinfection days 3, 5, 7, 10, and 14 to assess ocular lesions during the acute infection. After the establishment of latency (at 24 days p.i.), either the empty capsid AAV8-GFP (control) or AAV8-siCTCF spiked with AAV8-GFP was applied to the corneal surface of rabbits following corneal abrasion at an initial inoculum of 1010 viral genome equivalents/eye for each AAV vector in a total volume of 30 μl. All rabbits were monitored for signs of ocular pathology until the termination of the experiment.
Ocular swabs.
Rabbit eyes were swabbed with sterile cotton-tipped applicators that were rubbed across the corneal surface and under the eyelids. Each applicator was immediately placed in a tube containing 1 ml of sterile DMEM supplemented with 1% fetal bovine serum and 2% penicillin-streptomycin. The tube containing the applicator was placed on a rocker at room temperature for 1 h. A 24-well plate was plated with Vero cells 24 h earlier. The normal growth medium was removed from the cells, and the medium from each tube was then transferred to a single well in the plate. The plate was placed in the incubator at 37°C and 5% CO2. Plates were monitored under the microscope for plaques and/or cytopathic effects for 7 days.
HSV-1 reactivation.
Rabbit corneas were subjected to TCIE using previously reported methods (38, 39). Briefly, latently infected rabbits were anesthetized using IM ketamine/xylazine and subjected TCIE using a 0.015% epinephrine solution in water at 0.8 mA for 8 min per eye. Rabbits were swabbed for 12 consecutive days following TCIE. At the end of the experiment, rabbits were euthanized, and the TG were rapidly removed and either placed in 10% buffered formalin (histology) or placed in RNAlater (Ambion, Inc., Austin, TX) for RNA and DNA extraction.
Immunofluorescence.
Immediately upon removal, the TG were placed in 10% neutral buffered formalin, stored at 4°C overnight, and then transferred to 70% ethanol. All samples were then paraffin embedded and serial sectioned to 10 μm in thickness with three to four sections per each slide. Slides containing serial sections of rabbit TG were deparaffinized, hydrated, and peroxidase blocked through a series of washes that included (i) two xylene washes for 5 min each, (ii) two 100% ethanol washes for 2 min each, (iii) one 3% hydrogen peroxide wash (30% hydrogen peroxide was diluted 1:10 in 100% methanol) for 10 min, (iv) one 95% ethanol wash for 3 min, (v) one 70% ethanol wash for 1 min, and (vi) one double-distilled H2O wash for 1 min. Epitope retrieval was done using epitope retrieval solution by steaming the slides in buffer using a Coplin jar for 45 min on high. The slides were removed from the buffer, rinsed in water, and washed in Tris-buffered saline/Tween. A normal serum block was performed using normal serum from the species in which the secondary antibody was raised. Sections were then incubated with primary antibody (a 1:200 dilution of either mouse anti-GFP or mouse anti-NeuN or a 1:500 dilution of mouse anti-CTCF) overnight at 4°C. Secondary antibody incubation was then performed using a 1:1,000 dilution of either goat anti-mouse Alexa Fluor 548 (Invitrogen) or goat anti-mouse Alexa Fluor 488 (Invitrogen) for 2 h at room temperature. The slides were mounted using ProLong gold mounting media (Invitrogen). All immunofluorescence images were captured with a Leica DM RA2 digital microscope equipped with the appropriate filters, excitation sources, and a motorized z-stage controlled by Slidebook 5.0 software. Images were deconvolved (no neighbors). Negative controls included either naive rabbit TG subjected to the full immunohistochemistry protocol described above with antibody incubation or sections incubated without the primary antibody.
qRT-PCR analysis (in vivo experiments).
Rabbit TG were isolated and placed in RNAlater and stored according to the manufacturer's specifications. RNA was extracted by removing RNAlater from samples and adding TRIzol reagent (Sigma-Aldrich) to each sample. Briefly, each TG was homogenized in 1.2 ml of TRIzol and, following the addition of 0.2 volume of chloroform, the samples were centrifuged for phase separation. RNA was precipitated from the aqueous phase using 0.7 volume of isopropanol, followed by DNase treatment using DNA-free (Ambion), according to the manufacturer's directions. Reverse transcription using random primers was performed with a high-capacity cDNA reverse transcription kit (ABI), according to the manufacturer's instructions. Briefly, 20-μl reaction mixtures contained DNase-treated RNA, manufacturer-supplied buffer, deoxynucleoside triphosphate mix, a 1 μM concentration of random hexamer primer, 1 U of RNase inhibitor (Ambion), and MultiScribe reverse transcriptase. In the case of ICP0, a 10-μl aliquot of purified RNA was used with the strand-specific primer for the ICP0 transcript (LAT I-1, GACACGGATTGGCTGGTGTAGTGGG; nucleotides 120797 to 120820) (13). Real-time PCRs were performed on cDNA according to the above-described procedures and protocols using the primers and probes listed in Table 3.
TABLE 3.
Primers and probes
| Target | Sequence (5′-3′) | GenBank accession no. (nucleotide range) |
|---|---|---|
| Rabbit GAPDH | ||
| Primer, forward | GCA CCA CCA ACT GCT TAG C | |
| Primer, reverse | CCT CCA CAA TGC CGA AGT G | |
| Probe | CTG GCC AAG GTC ATC C | |
| HSV-1 ICP0 | NC_001806 (121385–121453) | |
| Primer, forward | GGC CGA GGG AGG TTT CC | |
| Primer, reverse | CCG CTT CCG CCT CCT C | |
| Probe | CTC CCA GGG CAC CGA C | |
| HSV-1 VP16 | NC_001806 (103946–104191) | |
| Primer, forward | CCT CGA TGG TAG ACC CGT AA | |
| Primer, reverse | ACA TTC GCG AGC ACC TTA AC | |
| Probe | CAT AAA GTA CCC AGA GGC | |
| HSV-1 DNA polymerase | NC_001806 (65880–65953) | |
| Primer, forward | AGA GGG ACA TCC AGG ACT TTG T | |
| Primer, reverse | CAG GCG CTT GTT GGT GTA C | |
| Probe | ACC GCC GAA CTG AGC A | |
| HSV-1 LAT intron | ||
| Primer, forward | ACC CAC GTA CTC CAA GAA GGC | NC_001806 (119721–119795) |
| Primer, reverse | TAA GAC CCA AGC ATA GAG AGC CA | |
| Probe | TCC CAC CCC GCC TGT GTT TTT |
qPCR for genome copies/ganglia.
Real-time primer/probe sets specific to HSV-1 DNA polymerase were used to determine HSV-1 genome copies per ganglia. Real-time PCR was performed in duplicate as described above. The relative HSV-1 copies for each sample were further normalized to the relative copies of the host control rabbit GAPDH.
Statistical analysis.
Statistical analyses were performed using SigmaPlot 12.3 for Windows XP.
ACKNOWLEDGMENTS
This study was supported in part by grants R56 AI101174 (D.M.N.), COBRE P30 GM106392 (D.M.N.), and R01AI048633 (D.C.B.) from the National Institutes of Health; an unrestricted grant from Research to Prevent Blindness; and the Department of Pharmacology, Louisiana State University Health Sciences Center, New Orleans, LA.
REFERENCES
- 1.Croen KD, Ostrove JM, Dragovic LJ, Smialek JE, Straus SE. 1987. Latent herpes simplex virus in human trigeminal ganglia. Detection of an immediate early gene “anti-sense” transcript by in situ hybridization. N Engl J Med 317:1427–1432. [DOI] [PubMed] [Google Scholar]
- 2.Austin A, Lietman T, Rose-Nussbaumer J. 2017. Update on the management of infectious keratitis. Ophthalmology 124:1678–1689. doi: 10.1016/j.ophtha.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 4.Deshmane SL, Fraser NW. 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol 63:943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kubat NJ, Amelio AL, Giordani NV, Bloom DC. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol 78:12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubat NJ, Tran RK, McAnany P, Bloom DC. 2004. Specific histone tail modification and not DNA methylation is a determinant of HSV-1 latent gene expression. J Virol 78:1139–1149. doi: 10.1128/JVI.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM. 2007. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol 81:13248–13253. doi: 10.1128/JVI.01569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A 102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristie TM, Liang Y, Vogel JL. 2010. Control of alpha-herpesvirus IE gene expression by HCF-1 coupled chromatin modification activities. Biochim Biophys Acta 1799:257–265. doi: 10.1016/j.bbagrm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Creech CC, Neumann DM. 2010. Changes to euchromatin on LAT and ICP4 following reactivation are more prevalent in an efficiently reactivating strain of HSV-1. PLoS One 5:e15416. doi: 10.1371/journal.pone.0015416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amelio AL, Giordani NV, Kubat NJ, O'Neil J, Bloom DC. 2006. Deacetylation of the herpes simples virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J Virol 80:2063–2068. doi: 10.1128/JVI.80.4.2063-2068.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amelio AL, McAnany PK, Bloom DC. 2006. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol 80:2358–2368. doi: 10.1128/JVI.80.5.2358-2368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Q, Lin L, Smith S, Huang J, Berger SL, Zhou J. 2007. CTCF-dependent chromatin boundary element between the latency-associated transcript and ICP0 promoters in the herpes simplex virus type 1 genome. J Virol 81:5192–5201. doi: 10.1128/JVI.02447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.West AG, Gaszner M, Felsenfeld G. 2002. Insulators: many functions, many mechanisms. Genes Dev 16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 17.Ghirlando R, Felsenfeld G. 2016. CTCF: making the right connections. Genes Dev 30:881–891. doi: 10.1101/gad.277863.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paris C, Pentland I, Groves I, Roberts DC, Powis SJ, Coleman N, Roberts S, Parish JL. 2015. CCCTC-binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J Virol 89:4770–4785. doi: 10.1128/JVI.00097-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez FP, Cruz R, Lu F, Plasschaert R, Deng Z, Rivera-Molina YA, Bartolomei MS, Lieberman PM, Tang Q. 2014. CTCF binding to the first intron of the major immediate early (MIE) gene of human cytomegalovirus (HCMV) negatively regulates MIE gene expression and HCMV replication. J Virol 88:7389–7401. doi: 10.1128/JVI.00845-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang H, Lieberman PM. 2009. Cell cycle control of Kaposi's sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J Virol 83:6199–6210. doi: 10.1128/JVI.00052-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tempera I, Klichinsky M, Lieberman PM. 2011. EBV latency types adopt alternative chromatin conformations. PLoS Pathog 7:e1002180. doi: 10.1371/journal.ppat.1002180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ertel MK, Cammarata AL, Hron RJ, Neumann DM. 2012. CTCF occupation of the HSV-1 genome is disrupted at early times post-reactivation in a transcription-dependent manner. J Virol 86:12741–12759. doi: 10.1128/JVI.01655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirata RK, Russell DW. 2000. Design and packaging of adeno-associated virus gene targeting vectors. J Virol 74:4612–4620. doi: 10.1128/JVI.74.10.4612-4620.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovric J, Mano M, Zentilin L, Eulalio A, Zacchigna S, Giacca M. 2012. Terminal differentiation of cardiac and skeletal myocytes induces permissivity to AAV transduction by relieving inhibition imposed by DNA damage response proteins. Mol Ther 20:2087–2097. doi: 10.1038/mt.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. 2003. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther 10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 26.McCarty DM, Monahan PE, Samulski RJ. 2001. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther 8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- 27.Watson ZL, Ertel MK, Lewin AS, Tuli SS, Schultz GS, Neumann DM, Bloom DC. 2016. Adeno-associated virus vectors efficiently transduce mouse and rabbit sensory neurons coinfected with herpes simplex virus 1 following peripheral inoculation. J Virol 90:7894–7901. doi: 10.1128/JVI.01028-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bloom DC, Lewin AS, Neumann DM, Watson ZL, Tuli SS, Schultz GS. 2017. Method for delivering RNA to neurons to treat herpes infections. USWO Patent 2017/223248 A1.
- 29.Kim KK, Adelstein RS, Kawamoto S. 2009. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J Biol Chem 284:31052–31061. doi: 10.1074/jbc.M109.052969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Estrada FS, Hernandez VS, Medina MP, Corona-Morales AA, Gonzalez-Perez O, Vega-Gonzalez A, Zhang L. 2009. Astrogliosis is temporally correlated with enhanced neurogenesis in adult rat hippocampus following a glucoprivic insult. Neurosci Lett 459:109–114. doi: 10.1016/j.neulet.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 31.Ahn HC, Yoo KY, Hwang IK, Cho JH, Lee CH, Choi JH, Li H, Cho BR, Kim YM, Won MH. 2009. Ischemia-related changes in naive and mutant forms of ubiquitin and neuroprotective effects of ubiquitin in the hippocampus following experimental transient ischemic damage. Exp Neurol 220:120–132. doi: 10.1016/j.expneurol.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 32.Goodman T, Trouche S, Massou I, Verret L, Zerwas M, Roullet P, Rampon C. 2010. Young hippocampal neurons are critical for recent and remote spatial memory in adult mice. Neuroscience 171:769–778. doi: 10.1016/j.neuroscience.2010.09.047. [DOI] [PubMed] [Google Scholar]
- 33.Sams DS, Nardone S, Getselter D, Raz D, Tal M, Rayi PR, Kaphzan H, Hakim O, Elliott E. 2016. Neuronal CTCF is necessary for basal and experience-dependent gene regulation, memory formation, and genomic structure of BDNF and Arc. Cell Rep 17:2418–2430. doi: 10.1016/j.celrep.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 34.Marsman J, O'Neill AC, Kao BR-Y, Rhodes JM, Meier M, Antony J, Mönnich M, Horsfield JA. 2014. Cohesin and CTCF differentially regulate spatiotemporal runx1 expression during zebrafish development. Biochim Biophys Acta 1839:50–61. doi: 10.1016/j.bbagrm.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Lee JS, Raja P, Pan D, Pesola JM, Coen DM, Knipe DM. 2018. CCCTC-binding factor acts as a heterochromatin barrier on herpes simplex viral latent chromatin and contributes to poised latent infection. mBio 9:e02372-. doi: 10.1128/mBio.02372-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mineur YS, Taylor SR, Picciotto MR. 2014. Calcineurin downregulation in the amygdala is sufficient to induce anxiety-like and depression-like behaviors in C57BL/6J male mice. Biol Psychiatry 75:991–998. doi: 10.1016/j.biopsych.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li DJ, Verma D, Mosbruger T, Swaminathan S. 2014. CTCF and Rad21 act as host cell restriction factors for Kaposi's sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog 10:e1003880. doi: 10.1371/journal.ppat.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hill JM, O'Callaghan RJ, Hobden JA. 1993. Ocular iontophoresis, p 331–354. In Mitra AK. (ed), Ophthalmic drug delivery systems. Marcel Dekker, Inc, New York, NY. [Google Scholar]
- 39.Hill JM, Shimomura Y, Kwon BS, Gangarosa LP Sr. 1985. Iontophoresis of epinephrine isomers to rabbit eyes induced HSV-1 ocular shedding. Invest Ophthalmol Vis Sci 26:1299–1303. [PubMed] [Google Scholar]
- 40.Washington SD, Musarrat F, Ertel MK, Backes GL, Neumann DM. 2018. CTCF binding sites in the herpes simplex virus 1 genome display site-specific CTCF occupation, protein recruitment, and insulator function. J Virol 92:e00156-18. doi: 10.1128/JVI.00156-18. [DOI] [PMC free article] [PubMed] [Google Scholar]