

ABSTRACT
The molecular mechanisms of liver pathology and clinical disease in hepatitis E virus (HEV) infection remain unclear. MicroRNAs (miRNAs) are known to modulate viral pathogenesis either by directly altering viral gene expression or by enhancing cellular antiviral responses. Given the importance of microRNA-122 (miR-122) in liver pathobiology, we investigated possible role of miR-122 in HEV infection. In silico predictions using HEV genotype 1 (HEV-1), HEV-2, HEV-3, and HEV-4 sequences showed that the majority of genomes (203/222) harbor at least one miR-122/microRNA-122-3p (miR-122*) target site. Interestingly, HEV-1 genomes showed a highly (97%) conserved miR-122 target site in the RNA-dependent RNA polymerase (RdRp) region (RdRpc). We analyzed the significance of miR-122 target sites in HEV-1/HEV-3 (HEV-1/3) genomes by using a replicon-based cell culture system. HEV infection did not change the basal levels of miR-122 in hepatoma cells. However, transfection of these cells with miR-122 mimics enhanced HEV-1/3 replication and depletion of miR-122 with inhibitors led to suppression of HEV-1/3 replication. Mutant HEV-1 replicons with an altered target RdRpc sequence (CACTCC) showed a drastic decrease in virus replication, whereas introduction of alternative miR-122 target sites in mutant replicons rescued viral replication. There was enrichment of HEV-1 RNA and miR-122 molecules in RNA-induced silencing complexes in HEV-infected cells. Furthermore, pulldown of miR-122 molecules from HEV-infected cells resulted in pulldown of HEV genomic RNA along with miR-122 molecules. These observations indicate that miR-122 facilitates HEV-1 replication, probably via direct interaction with a target site in the viral genome. The positive role of miR-122 in viral replication presents novel opportunities for antiviral therapy and management of hepatitis E.
IMPORTANCE Hepatitis E is a problem in both developing and developed countries. HEV infection in most patients follows a self-limited course; however, 20% to 30% mortality is seen in infected pregnant women. HEV superinfections in patients with chronic hepatitis B or hepatitis C virus infections are associated with adverse clinical outcomes, and both conditions warrant therapy. Chronic HEV infections in immunocompromised transplant recipients are known to rapidly progress into cirrhosis. Currently, off-label use of ribavirin (RBV) and polyethylene glycol-interferon (PEG-IFN) as antiviral therapy has shown promising results in both acute and chronic hepatitis E patients; however, the teratogenicity of RBV limits its use during pregnancy, while alpha IFN (IFN-α) increases the risk of transplant rejections. Experimental data determined with genotype 1 virus in the current study show that miR-122 facilitates HEV replication. These observations present novel opportunities for antiviral therapy and management of hepatitis E.
KEYWORDS: hepatitis E virus, miR-122, target site, interaction, virus replication, Ago2
INTRODUCTION
Hepatitis E virus (HEV) is the predominant cause of epidemic and sporadic acute hepatitis among adults (1) and is an important public health concern in many developing countries (2). Hepatitis E is generally an acute self-limiting illness (3); however, it can become more severe in pregnant women, with a high incidence of acute liver failure and high (15% to 30%) mortality rates in the third trimester (4, 5). Chronic HEV infections in immunocompromised organ-transplant recipients have also been previously reported (6). Thus, initially considered a less harmful pathogen, HEV poses a new threat for human health globally.
HEV belongs to the family Hepeviridae, and virus strains infecting humans are grouped into species of Orthohepevirus A, in HEV genotype 1 (HEV-1), HEV-2, HEV-3, and HEV-4 (7). Genotype 1 and 2 viruses infect only humans, while genotype 3 and 4 viruses are endemic in pigs and are zoonotically transmitted to humans. HEV is a nonenveloped virus with a single-stranded positive-sense RNA genome of ∼7.2 kb, with a short 5′ noncoding region (5′NCR), 3 open reading frames (ORFs), and a short 3′NCR with a poly(A) tail. Nonstructural and structural proteins are encoded by ORF1 (∼5.1 kb) and ORF2 (∼2.0 kb), respectively. ORF3 (∼342 nucleotides [nt]) overlaps ORF2 and encodes a small phosphoprotein. ORF1 encodes a polyprotein of ∼1,693 amino acids with conserved motifs such as the following: methyltransferase (MeT), papain-like cysteine protease (PCP), RNA helicase (Hel), and RNA-dependent RNA polymerase (RdRp) (8). The recently identified ORF4 (∼478 nt) within ORF1 encodes a protein reported to be essential for optimum functioning of RdRp in HEV-1 viruses (9).
MicroRNA (miRNA) is a 21-to-23-nt small noncoding RNA molecule that functions mainly in posttranscriptional gene regulation (10). Viruses can take advantage of host miRNAs to facilitate their survival and replication (11, 12), while miRNAs can serve as direct antiviral entities for host cells (13, 14). MicroRNA-122 (miR-122) is the most abundant liver-specific miRNA (contributing to ∼70% of the total liver miRNA pool) and is highly conserved among vertebrates (10). It is involved in various pathophysiological processes in liver. miR-122 stimulates hepatitis C virus (HCV) replication via a direct interaction with two binding sites in 5′NCR to increase the stability of the viral genome (15), whereas miR-122 directly binds to a highly conserved hepatitis B virus (HBV) pregenomic RNA sequence and inhibits HBV gene expression and replication (16). miR-122 also inhibits replication of HBV indirectly via the cyclin G1/p53 enhancer pathway in HBV-infected individuals (17). In the present study, we evaluated the relevance of miR-122 in HEV replication using HEV-1/HEV-3 replicons and 3 different human cell lines which are known to support HEV replication.
RESULTS
The HEV-1 genome harbors a highly conserved target site in the RdRp-encoding region.
MicroRNA hsa–miR-122 is the most abundant miRNA in human liver. miR-122-5p (miRBase accession identifier [ID] MI0000442), the mature form of miR-122, is encoded by chromosome 18 in humans. Previous reports on the direct interaction between miR-122 and the HCV genome prompted us to look for possible miR-122 target sites in HEV genomes (17, 18). For that, all available human and swine HEV full-genome sequences from GenBank belonging to genotypes 1, 2, 3, and 4 (HEV-1 [n = 32], HEV-2 [n = 2], HEV-3 [n = 107], and HEV-4 [n = 78]) were processed for miRNA target site predictions and phylogenetic analysis. Phylogenetic clusters of these sequences are shown in Fig. S1 in the supplemental material. The results of miRNA target site predictions are summarized in Fig. 1A, and details of these predictions are listed in Tables S1 and S3 in the supplemental material. Genotype-specific prediction analysis was as follows. (i) HEV-1 (n = 32) sequences grouped into 5 different prediction patterns, correlating well with the 5 phylogenetic clusters. Sequences from all 5 clusters depicted the presence of a highly conserved miR-122 target site in the RdRp-encoding region (nucleotides [nt] 4556 to 4577 [nucleotide ranges represent approximations throughout]) (RdRpc). This site was present either alone or in combination with additional miR-122 sites at nt 3930 to 3954 (ORF1) and/or at nt 6256 to 6281 (ORF2) (Fig. 1B) (Table 1). Predictions of miR-122 sites at different locations in 32 HEV-1 genomes were as follows: nt 3930 to 3954 (ORF1), 50% (16/32 sequences); nt 4556 to 4577 (RdRpc), 97% (31/32 sequences); nt 6261 to 6283 (ORF2), 43.75% (14/32 sequences). The miR-122* site at nt 6205 to 6227 (ORF2) was present in 81% (26/32) of the HEV-1 genomes (see Table S1). (ii) HEV-2 (n = 2) sequences showed the presence of the miR-122 site at nt 6231 to 6252 (ORF2) and of the miR-122* site at nt 2301 to 2322 and nt 1788 to 1808 (ORF1). (iii) The HEV-3 (n = 107) genomes clustered into 11 distinct clusters, while 2 genomes remained ungrouped. Unlike the HEV-1 clusters, the HEV-3 clusters (which included both human and pig isolates) revealed no notable patterns, in terms of the presence or absence of as well as the locations of miR-122/miR-122* target sites in viral genomes. (iv) The HEV-4 (n = 78) genomes clustered into 8 distinct clusters with 3 ungrouped genomes and revealed an appreciable correlation with the prediction patterns. However, the human and swine HEV sequences did not segregate.
FIG 1.


(A) Computational prediction of miR-122/miR-122* targets in the HEV genomes. The HEV genomes were screened for putative miR-122/miR-122* target sites using RegRNA, and the prediction patterns were analyzed. The results of the analysis are depicted. (B) Conserved miR-122 target sites in the HEV-1 genome. The replicon developed from the highlighted sequence was used in the present study for experiments. (C) RdRp, a potential target for miR-122 in HEV genotype 1 isolate. (a) Strategy for reverse transcription and amplification of HEV-1 full genome. Full-length SAR55 HEV isolate RNA was amplified in 4 fragments (fragments I, II, III, and IV). The strategy for one of the fragments is shown here. Similar strategies were used for other three fragments. The cDNA fragments were then used as the templates for hybrid PCR using miR-122-specific forward and HEV-specific reverse primers. PCR products were then subjected to TA cloning and sequenced, and the putative target region was identified using BLAST. The hybrid miR-122 primer is shown in the lower left panel of the figure. The RdRp region in the sequenced SAR55 HEV genome, confirmed as the target miR-122 site by hybrid PCR, is shown in the lower right panel of the figure. (b) The HEV genome and potential miR-122 binding site are depicted as follows: 7-methylguanosine (7mG), open reading frame 1 (ORF1), methyltransferase (MT), Y domain, polyproline region (PPR), cysteine protease (P), X domain, helicase (Hel), RNA-dependent RNA polymerase (RdRp), open reading frame 2 (ORF2), open reading frame 3 (ORF3), and open reading frame 4 (ORF4). In the miR-122 binding site, underlined nucleotides indicate the seed region for miR-122 required for binding to the target site.
TABLE 1.
Patterns of target-site interactions of the 3 putative miR-122-5p target sites in HEV-1 isolates

We decided to analyze the significance of the highly conserved miR-122 site at nt 4556 to 4577 (RdRpc) in HEV-1 genomes. This site was absent in 1/32 of the analyzed HEV-1 genomes (AF185822); however, the sequence of the genome in which the site was absent harbored an alternative target site for miR-122. The results indicated that miR-122 probably interacts with the HEV genome, possibly in a favorable way, as all HEV-1 genomes harbored at least one miR-122 site. We examined the HEV-1 SAR55 replicon, which harbored a single miR-122 target site at the highly conserved location RdRpc (Fig. 1B).
Confirmation of potential predicted miR-122 target site by hybrid PCR.
To confirm the presence of the predicted miR-122 target site (RdRpc) in the SAR55 genome, hybrid PCR was carried out using both miR-122-3p and miR-122-5p primers as shown in Fig. 1Ca. There were no predicted miR-122* sites in the SAR55 genome; hence, the miR-122-3p primers served as a negative control. The products (n = 4) obtained with hybrid PCR were of different lengths, and sequence analysis revealed that only one sequence obtained with miR-122-5p matched the HEV genome. This was located in the ORF1 region, flanking the RdRp-encoding site in the SAR55 genome (Fig. 1Cb). These findings supported the prediction of a single miR-122 target site in the SAR55 genome by the use of RegRNA software (Table S1).
Basal levels of miR-122 in different cell lines.
We needed an appropriate cell culture system for studying the effect of miR-122 on HEV replication. For that purpose, hepatoma cells (S10-3 and HepG2/C3A cells) and lung carcinoma cells (A549 cells) were considered, as those cell lines are used as model systems for studying HEV-1 replication. S10-3 cells are known to be the most permissive for HEV-1 among these 3 cell lines (19, 20). Previous reports have revealed the presence of higher basal levels of miR-122 in Huh7 cells and lower levels in HepG2 cells, though both are human hepatocarcinoma cell lines (21). The Huh-7 cell line is the parental cell line of the S10-3 clonal cell line, while the HepG2 cell line is the parental cell line of HepG2/C3A clonal cells. In agreement with the previously reported miR-122 levels in parental cell lines, quantitative analysis of miR-122 in the current study also showed lower levels in HepG2/C3A cells (∼104 copies/106 cells) and higher levels in S10-3 cells (∼108 copies/106 cells). Nonhepatic A549 cells had low levels of miR-122 (<100 copies/106 cells) (Fig. 2).
FIG 2.
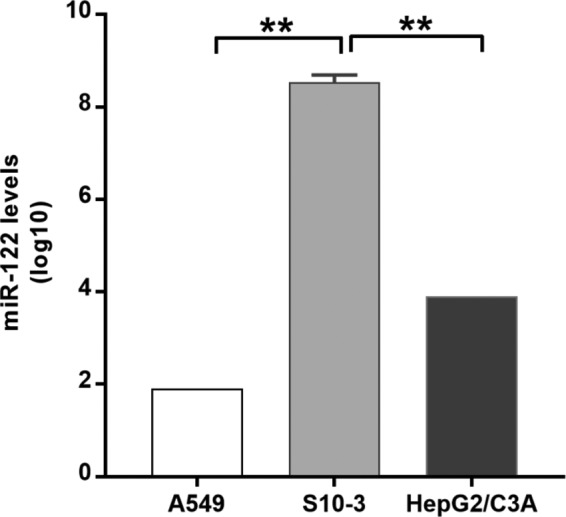
Differential basal expression of miR-122 in the cell lines used. Cells were harvested, and RNA was isolated using a mirVana miRNA isolation kit. miRNA was reverse transcribed using has–miR-122-5p- and U6 snRNA-specific stem-loop primers. miR-122 levels were measured using a TaqMan-based quantitative PCR (qPCR) assay. U6 snRNA served as an endogenous control for normalization. *, P < 0.05 (considered to represent significance).
Effect of HEV replication on miR-122 levels in hepatoma cells.
The presence of a highly conserved target site (RdRpc) in HEV-1 genomes prompted us to examine whether HEV has the ability to modulate miR-122 expression in HepG2/C3A and S10-3 cells. For that purpose, HEV infection was initiated by transfecting cells with SAR55 full-genome replicon (pSK-HEV-2) and virus replication was confirmed by negative-strand RNA detection and immunofluorescence assay as described previously (22) (Fig. 3Ai and ii). In both cell lines, miR-122 levels remained similar in both HEV-infected and uninfected cells, indicating that HEV has no influence on miR-122 biogenesis (Fig. 3B).
FIG 3.

(A) Confirmation of HEV-1 replication in S10-3 cells. (i) Negative-strand HEV RNA detection by tagged primer-based PCR. S10-3 cells were transfected with capped pSK-HEV-2 RNA transcripts (SAR55 full-genome clone) and harvested at the indicated time points. Total RNA was isolated and processed for negative-strand RNA PCR. A 1% agarose gel is shown with lanes indicated as follows: lane M, 100-bp DNA ladder; lane 1, mock-transfected cells; lane 2, cells at 48 h posttransfection; lane 3, cells at 96 h posttransfection. (ii) Immunofluorescence staining (IFA) for detection of HEV ORF2 protein. The left panel shows S10-3 cells transfected with capped pSK-HEV-2 RNA transcripts after 6 days, and the right panel shows mock-transfected S10-3 cells as a negative control. Cells were stained with monoclonal antibodies developed against ORF2 protein. (B) HEV-1 has no effect on miR-122 expression levels in human hepatoma cells. S10-3 and HepG2/C3A cells were transfected with capped pSK-HEV-2 RNA transcripts (2 μg/well), and miR-122 expression levels were determined by qPCR at 48 and 96 h. miR-122 levels were determined per million cells. The data represent the log values of means ± standard deviations (SD) of results from three independent triplicate sets of experiments.
Experimental alterations of miR-122 levels in cells and effect on HEV replication. (i) Overexpression of miR-122 enhanced HEV replication in cells.
To determine the possible influence of miR-122 on HEV replication, S10-3, HepG2/C3A, and A549 cells were transfected with 50 nM miR-122 mimics as described previously (16). Both hepatoma cells lines showed a significant increase in HEV replication as follows: for S10-3 cells, ∼3.4-, 3.1-, and 3.8-fold at 48, 72, and 96 h, respectively; for HepG2/C3A cells, ∼1.9-, 2.8-, and 3.2-fold at 48, 72, and 96 h, respectively (Fig. 4A and Bi). However, A549 cells exhibited an initial ∼4.4-fold increase at 48 h followed by a significant decrease at further time points (Fig. 4Bii). This indicated that although increased miR-122 levels boosted HEV replication in A549 cells initially, activation of cellular antiviral responses probably restricted virus replication at later time points. Similarly, although higher miR-122 levels improved HEV replication in HepG2/C3A cells, the increase was not as great as that seen in S10-3 cells. We previously reported that A549 and HepG2/C3A cells have intact cellular antiviral pathways and restrict HEV replication, while S10-3 cells have mutations in both Toll-like receptor-3 (TLR3) and RIG-I genes (double-stranded RNA [dsRNA] sensors) and generate poor antiviral responses after HEV infection. Hence, S10-3 cells are more permissive for HEV than A549 and HepG2/C3A cells (19, 20).
FIG 4.

Altered miR-122 levels and HEV replication. miR-122 facilitates HEV replication in human hepatoma cells. (A) S10-3 cells. (B) (i) HepG2/C3A cells. (ii) Nonhepatoma cells. A549 cells were transfected with 50 nM miR-122 mimic. After 24 h, cells were cotransfected with HEV Rluc RNA and firefly luciferase plasmid DNA to normalize cell transfection efficiency. Cell-associated Renilla luciferase activity was determined to monitor HEV replication. (C) Toxicity of miR-122 LNA in S10-3 cells. Toxicity was checked by cotransfecting cells with 5 nM LNA and plasmid harboring the HBV replicon (500 ng/well). HBV replication was monitored by measuring HBsAg and HBV DNA levels using ELISA and qRT-PCR, respectively, at 48 and 72 h. (D) Effect of anti–miR-122 LNA on HEV replication in S10-3 cells. Cells were transfected with 5 nM miR-122 LNA. After 24 h, cells were cotransfected with HEV Rluc RNA and firefly luciferase plasmid DNA to normalize cell transfection efficiency. Cell-associated Renilla luciferase activity was determined to monitor HEV replication. For all the graphs representing the experiments described above, the data represent means ± SD of results from three independent triplicate sets of experiments. **, P < 0.01; ***, P < 0.001. Statistical comparisons between mock-transfected cells and cells transfected with HEV-Rluc RNA were done by one-way analysis of variance (ANOVA).
(ii) Decreased levels of miR-122 resulted in poor HEV replication.
To study effect of miR-122 depletion, we used miR-122-LNA (miR-122–locked nucleic acid), which is a potent miR-122 inhibitor used in loss-of-function studies. miR-122-LNA toxicity was checked in all 3 cell lines used in the study by exposing cells to different concentrations (1, 5 and 10 nM) of LNA. There was no effect of any of these concentrations of LNA on the viability and morphology of these cells for up to 7 days (data not shown). To see whether LNA-treated cells still supported virus replication, we checked replication of another hepatotropic virus, HBV, by cotransfecting S10-3 cells with 5 nM LNA plus plasmid harboring a genotype D HBV replicon (23). Replication of the virus was monitored by measuring HBsAg levels in the culture supernatants and HBV DNA levels in the cell pellets. As expected, LNA-treated cells showed an increase in HBsAg secretion as well as increased numbers of HBV DNA copies compared to cells transfected with HBV DNA alone (16) (Fig. 4C), confirming that miR-122-LNA was not affecting normal cellular functions essential for virus replication.
To study effects of miR-122-LNA on HEV replication, we preferred to use S10-3 cells since miR-122 basal levels were high in these cells and there was no reason for decreasing miR-122 levels further in HepG2/C3A cells. In addition, S10-3 cells were known to be more permissive for HEV. Interestingly, S10-3 cells treated with miR-122-LNA showed drastic decreases in HEV replication at 72 h (P < 0.01) and 96 h (P < 0.001) (Fig. 4D).
Effect of miR-122 supplementation/depletion on HEV-3 replication.
In order to further determine whether the phenomena observed for HEV-1 apply to other HEV genotypes, we used a p6 HEV-3 full-genome clone (24). This clone harbored a single miR-122 target site at ∼1903 to 1924 nucleotides in the genome. To see the effect of miR-122 on virus replication, S10-3 cells were transfected with either miR-122 mimics or LNA prior to HEV RNA and virus replication was monitored using real-time PCR. There was an ∼2.0-fold increase in virus replication with miR-122 mimics (supplementation) compared to HEV-3 alone, whereas LNA-treated cells (depletion) showed a significant inhibition of HEV-3 replication (Fig. 5).
FIG 5.

Altering miR-122 levels in S10-3 cells affects HEV-3 replication. Cells were transfected with 50 nM miR-122 mimic (mim). After 24 h, cells were cotransfected with HEV-3 RNA. For all graphs, the data represent means ± SD of results from three independent triplicate sets of experiments. ****, P < 0.0001. Statistical comparisons between mock-transfected cells and cells transfected with HEV RNA were done by one-way analysis of variance (ANOVA).
Taken together, both HEV-1 and HEV-3 replicons, each harboring a single miR-122 target site, appeared to be influenced similarly by cellular miR-122 levels.
HEV replication, miR-122, and cellular antiviral responses.
Several studies have documented influential effects of miR-122 on interferon (IFN) signaling. miR-122 is known to target SOCS1, a negative regulator of the JAK-STAT pathway, and to enhance type I IFN signaling (25, 26). Interestingly, miR-122 expression is downregulated by type I interferons (14, 27, 28). To see the effect of increased levels of miR-122 on cellular antiviral responses, we used HepG2/C3A cells, since these cells have intact antiviral pathways (20). Cells were transfected with HEV RNA either alone or along with miR-122 mimics as follows: set 1, miR-122 mimics; set 2, miR-122 mimics plus SAR55 RNA; set 3, SAR55 RNA. Transcript levels of selective antiviral pathway genes were analyzed. There was a moderate increase in expression of the cytosolic dsRNA sensor RIG-I gene initially (∼2-fold at 12 h and 48 h) in all 3 sets of the cells, with a further increase seen at later time points (Table 2). Levels of expression of TLR3 (endosomal dsRNA sensor) remained similar in all 3 sets. There was no change in the transcript levels of IFN-α in any of the sets. Interferon-stimulated genes (ISGs) such as PKR and ISG56 showed higher induction in response to HEV RNA. Higher levels of ISG56 (due to RNA) were evident in both set 2 cells and set 3 cells at 12 h. These levels decreased in set 3 but remained higher in set 2 at 96 h due to the cumulative effect of miR-122 plus HEV RNA. Similarly, higher expression of PKR was noted at 12 h in all 3 sets. From these observations, it was concluded that the enhancement in HEV replication due to increased miR-122 levels was not due to altered cellular antiviral responses in HepG2/C3A cells.
TABLE 2.
Effect of miR-122 on the expression of innate immune response genes
| Gene | Fold expression change |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mimics (set 1) |
Mimics + HEV (set 2) |
HEV (set 3) |
|||||||
| 12 h | 48 h | 96 h | 12 h | 48 h | 96 h | 12 h | 48 h | 96 h | |
| RIG-I | 1.3 | 1.09 | 111 | 2.21 | 1.73 | 116 | 2.23 | 2.12 | 78.5 |
| TLR3 | 1.53 | 1.22 | 0.54 | 2.1 | 1.47 | 0.61 | 9.03 | 1.7 | 0.6 |
| IFN-α | 1.37 | 0.6 | 0.81 | 0.59 | 0.61 | 0.79 | 4.92 | 0.99 | 0.91 |
| PKR | 4.33 | 1.04 | 0.32 | 6.5 | 3.67 | 0.81 | 2.61 | 0.46 | 0.35 |
| ISG56 | 2.09 | 0.05 | 1 | 199 | 31.8 | 33.2 | 164 | 33.7 | 5.44 |
The intact miR-122 target site in the HEV genome favored viral genome replication.
It is known that miR-122 enhances replication via direct binding to the 5′ untranslated region (5′UTR) and increasing stability of the HCV genome (29). It was evident from the miR-122 target search analysis that HEV-1 genomes contain at least one miR-122 target site and that depletion of miR-122 molecules results in a drastic decrease in HEV replication. These observations indicated that miR-122 possibly interacts with the HEV genome.
The seed sequence of miR-122 involved in binding is GUGAGG, i.e., CACTCC in the conserved (RdRpc) target site. To determine whether enhanced viral replication involved a direct interaction between miR-122 and the target site in the HEV genome, we generated pSK-HEV-2-Rluc replicon mutants (mut) by altering the RdRpc target site as follows: for mutant mut1, AACTCC (H to N) (where the underlining and boldface indicate an altered nucleotide); for mutant mut2, CACTAC (S-Y); for mutant mut3, AACTAC (H-N, S-Y); for mutant mut4, CATTCC (H-H); for mutant mut5, CACTCT (S-S). After the alterations were performed, we assessed the replication of the mutants in S10-3 cells. Surprisingly, mutants mut1, mut2, and mut3 showed almost no replication, indicating a critical role of those nucleotides in the HEV genome. Further analysis showed that these changes were nonsynonymous and caused amino acid substitutions in the catalytic site of RdRp which probably resulted in loss of polymerase function. Changes in mut4 and mut5 replicons were of the synonymous type, and these mutants successfully replicated, albeit with >95% and ∼55% decreases in the replication efficiency, respectively (Fig. 6Ai). These results suggested that the HEV-2-Rluc genome harbored a functional miR-122 target site that conferred miR-122-mediated enhancement of virus replication, possibly via direct interaction.
FIG 6.

(A) miR-122 interacts with the HEV genome to facilitate its replication. S10-3 cells were transfected with either wild-type HEV-Rluc RNA or HEV Rluc–miR-122 target site mutant replicons (2 μg/well). (i) mut1, AACTCC (where the underlining and boldface represent an altered nucleotide); mut2, CACTAC; mut3, AACTAC; mut4, CATTCC; mut5, CACTCT. (ii) mut4, ORF2 miR-122-rescue mutant (mut6), and RdRp miR-122-rescue mutant (mut7). (iii) HEV Rluc and mut7 along with miR-122/mut4 mimics. (iv) mut4 along with miR-122/mut4 mimics. In all experiments, cells were cotransfected with firefly luciferase plasmid DNA along with replicon RNA and were harvested after different time intervals. Cell-associated Renilla luciferase activity was determined to monitor HEV replication. The data represent means ± SD of results from three independent triplicate sets of experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Statistical comparisons between mock-transfected cells and cells transfected with HEV-Rluc RNA were done by one-way analysis of variance (ANOVA). (B) miR-122 directly binds to the HEV-1 genome. (i) S10-3 cells were transfected with HEV-1 full-genome replicon RNA and were harvested after 48 h. Cell lysates were processed for immunoprecipitation with Ago2-specific monoclonal antibodies or unrelated IgG2a isotype control antibodies. RNA was isolated from immunoprecipitates using a viral RNA extraction kit, and HEV RNA levels were determined by qRT-PCR. (ii) Relative levels of miR-122 in the immunoprecipitates were analyzed by qRT-PCR. (iii) S10-3 cells were cotransfected with HEV RNA and biotinylated control miRNA/miR-122 mimics. After 48 h, cell lysates were incubated with streptavidin beads, and complexes were pulled down. RNA was isolated from these complexes using a viral RNA extraction kit, and HEV RNA expression was analyzed by qRT-PCR. The data represent means ± SD of results from three independent triplicate sets of experiments. **, P < 0.01; ****, P < 0.0001.
Introduction of an alternative miR-122 target site rescued HEV replication.
A significant decrease in the replication efficiency of the virus containing target site mutations suggested a crucial function of miR-122 target sites during HEV-1 replication. With these results, we anticipated that it might be possible to rescue replication of the mutant virus by introducing an alternative miR-122 target site(s) in the viral genome. For that purpose, we selected a mut4 construct with a synonymous mutation in the RdRpc target site (exhibiting >95% inhibition) and generated two rescue mutants. Alternative target sites were introduced at nt 6261 to 6283 (ORF2 miR-122-rescue mutant [mut]; mut6) and nt 3932 to 3956 (RdRp miR-122-rescue mutant; mut7) without changing the amino acid sequences of these two proteins. These two target sites were present in several HEV-1 genomes (Fig. 1B) but absent in pSK-HEV-Rluc. The replication efficiency of both the rescue mutants was significantly improved compared to that seen with the mut4 replicon. However, the mut7 mutant, the rescue mutant generated by introducing the site at nt 3932 to 3956 (RdRp), showed a significant increase in replication compared to the rescue mutant corresponding to nt 6261 to 6283 (ORF2), i.e., the mut6 mutant (Fig. 6Aii). These results indicated that the target site in the HEV-1 genome is required for miR-122-mediated enhancement.
We further confirmed this finding by examining the effect of a mut4 sequence-matched mutant mimic on the wild-type (WT) HEV and mutant mut7 and mut4 replication efficiencies. There was no effect of mutant mimics on the replication of the wild-type and mut7 replicons (Fig. 6Aiii), whereas there was a marginal rescue of mut4 replication seen with the mut4 mimics (Fig. 6Aiv).
miR-122 possibly binds to the RdRp region of the HEV-1 genome.
To analyze whether there is a direct interaction between HEV-1 RNA and miR-122, we used the following two approaches.
(i) Argonaute2 (Ago2) immunoprecipitation.
For this, S10-3 cells were transfected with a pSK-HEV-Rluc/mut4/mut6/mut7 replicon and harvested after 96 h. Cell lysates were processed for immunoprecipitation using Ago2 monoclonal antibody (MAb) or an unrelated isotype control antibody. Analysis of RNA isolated from an immunoprecipitate obtained with Ago2 antibodies showed the following fold changes in numbers of HEV RNA copies: for the wild type, 555.8-fold-higher expression; for the mut4 mutant, 0.78-fold-higher expression; for the mut6 mutant, 8.17-fold-higher expression; for the mut7 mutant, 15.40-fold-higher expression (compared to the respective nonspecific antibody controls). Similarly, the following fold changes were seen for the numbers of miR-122 copies in the corresponding complexes: for the wild type, 590.81-fold-higher expression; for the mut4 mutant, 0.02-fold-higher expression; for the mut6 mutant, 16.84-fold-higher expression; for the mut7 mutant, 77.74-fold-higher expression (compared to the respective nonspecific antibody controls) (Fig. 6Bi and ii). Effective pulldown of both miR-122 and HEV RNA in RNA silencing complexes with Ago2 antibodies indicated that miR-122 binds directly to the HEV-1 genome.
(ii) miR-122-RNA pulldown assay.
Further confirmation of miR-122 binding was performed by using biotin-labeled miR-122 mimics and a nonspecific miRNA as the control. For that purpose, S10-3 cells were cotransfected with HEV-Rluc RNA and biotin-labeled control miRNA or miR-122 mimics and harvested after 48 h. Cell lysates were incubated with streptavidin beads, and viral RNA isolated from beads was analyzed by quantitative reverse transcription-PCR (qRT-PCR). HEV RNA levels were found to be ∼3-fold higher in S10-3 cells cotransfected with miR-122 mimics than control miRNA levels (Fig. 6Biii). These results further confirmed interaction of miR-122 with the HEV genome, since miR-122 was found to be associated with HEV RNA.
Taken together, these results indicated a possible interaction between miR-122 and the HEV genome.
DISCUSSION
miR-122 plays a central role in liver development, differentiation, and homeostasis (10). Viruses infecting liver, namely, hepatitis A virus (HAV), HBV, HCV, and HEV, are taxonomically unrelated viruses. HEV has a positive-sense RNA genome and initiates viral protein synthesis immediately after entry into host cells. This increases the possibility of its being directly modulated by cellular miRNAs. We checked for the presence of an miR-122 target site(s) in HEV genomes and, to our surprise, observed that the majority (203/222) had at least one miR-122 and/or miR-122* target site (see Table S1 in the supplemental material), suggesting the possibility of an miR-122-mediated influence on the pathogenesis of HEV.
There were larger variations in the locations of these sites in HEV-3 and HEV-4 sequences; however, the existence and the locations of these sites were comparatively consistent in HEV-1 genomes. The driving force behind generating heterogeneity in target sites was possibly exerted by host cells. Given that they are restricted to humans, there is a limited scope for HEV-1 evolution compared to that of HEV-3 and HEV-4 viruses, as they have a broader host range.
HEV-1 genomes harbored at least one miR-122 target site. Considering the conventional inhibitory role of miRNAs, the presence of a conserved miR-122-5p target site in HEV-1 genomes was unexpected, as it indicated a possible inhibition of HEV replication by miR-122. It was unclear why HEV, a virus with a highly error-prone RNA polymerase, would retain such a sequence unless it was under selection pressure to do so.
Downregulation of miR-122 expression by HBV has been reported by Wang et al. (17), whereas HCV has been shown to interfere with the RNA interference (RNAi) machinery (30). We observed that HEV does not interfere in the biogenesis of miR-122. However, HEV replication efficiencies in hepatoma cells showed a direct correlation with miR-122 levels. Exogenous overexpression of miR-122 facilitated HEV replication in both S10-3 and HepG2/C3A cells, though the level was higher in S10-3 cells. Similarly, mimics also showed enhanced replication in nonhepatic A549 cells which returned to basal levels at later time points. Also, altering miR-122 levels in S10-3 cells resulted in a similar effect on HEV-3 replication (Fig. 5]. We have previously shown that both A549 cells and HepG2/C3A cells exhibit higher levels of antiviral responses than S10-3 cells (20). It was determined from the present observations that increasing miR-122 levels alone was not sufficient to enhance HEV replication and that cellular antiviral responses played a key role in determining the permissiveness of cells for the virus. S10-3 cells proved to be a better model for visualizing the direct effect of miR-122 levels on HEV replication. Sequestration of endogenous miR-122 molecules in S10-3 cells resulted in a drastic decrease in the level of HEV replication (Fig. 4D]. These results suggested a probable positive regulatory role of miR-122 during HEV replication, similarly to HCV (30).
The next issue concerned understanding the mechanism of the miR-122-mediated enhancement in HEV replication. The possibilities included enhancement in translation of HEV RNA, direct interaction with the HEV genome, and modulation of antiviral responses. The increase in luciferase activity seen upon transfection of cells with miR-122 mimics indirectly indicated enhancement in translation. During replication, ORF1 is immediately translated from the viral genome to generate viral replicative enzymes. RdRp then synthesizes negative-sense RNA and utilizes it as the template to synthesize subgenomic RNA. The luciferase gene, located in frame with ORF2, is expressed via subgenomic RNA. The presence of the miR-122 target site in the RdRp region probably enhanced ORF1 translation. However, this postulate needs experimental confirmation. Current HEV-1 cell culture systems are not useful for such studies, and this issue will remain unresolved until we devise an efficient cell culture system. Though microRNAs are translational repressors, they can also enhance translation in rare situations, depending on the complementarity between miRNA and the target sequence or cell cycle phase (31). Positive regulation of HEV ORF1 by miR-122 could be among those rare miRNA functions.
Complementarity between the miRNA “seed region” (positions 2 to 8 from the 5′ end) and the mRNA target sequence is crucial for the interaction and the final effects (32). miRNAs bind to target sequences via partial base pairing and regulate both mRNA stability and translation. Emerging evidence suggests that cellular miR-122 is a crucial regulator of replication for hepatitis viruses. To learn whether the interaction between miR-122 and RdRp in HEV-1 has any enhancing effect on virus replication, we generated RdRpc target site mutants. Experiments performed with mutants mut1, mut2, and mut3, with nonsynonymous mutations in the catalytic domain of RdRp, resulted in complete inhibition of HEV replication. It was observed that though amino acids in the catalytic domains of RdRp are highly conserved across HEV genotypes 1 to 4, there are third-base variations. Hence, we generated 2 additional mutants, mut4 and mut5, with synonymous changes and saw that there were significant decreases (>95% and ∼55%, respectively) in HEV replication (Fig. 6Ai). This indicated the importance of complementarity between viral and host sequences. Interestingly, introduction of alternative miR-122 target sites in the HEV genome rescued HEV replication (Fig. 6Aii). There was enrichment of both HEV-1 and miR-122 RNAs in RNA-induced silencing complexes in HEV-infected cells. A possible direct interaction between the HEV genome and miR-122 was also indicated by the selective pulldown of HEV RNA along with miR-122 molecules. The replication efficiencies of the HEV replicons varied with respect to miR-122 enrichment in the complexes. It was clearly evident with the low level of pulldown of miR-122 molecules with mutant mut4 and the improved enrichment with the rescue mutants (Fig. 6Bi and ii). Together, these results confirmed a requirement of at least one miR-122 target sequence in the HEV-1 genome for establishing efficient replication in hepatic cells.
In conclusion, we demonstrate for the first time that miR-122 facilitates replication of HEV in human hepatoma as well as nonhepatoma cells. Inhibition of miR-122 functional molecules drastically reduces HEV replication. These findings suggest that regulating miR-122 levels could help in the management of HEV infection in humans. One miR-122 inhibitor, miravirsen (SPC3649), has undergone successful phase II clinical trials for HCV (33) and could also be used for the treatment and management of hepatitis E.
MATERIALS AND METHODS
miR-122 target site prediction.
miR-122 target sites were predicted in full-length HEV genome sequences representing different HEV genotypes as follows: 32 from HEV-1, 2 from HEV-2, 107 from HEV-3, and 78 from HEV-4 (from GenBank; http://www.ncbi.nlm.nih.gov/GenBank). Target sites were predicted using RegRNA computational software (http://regrna.mbc.nctu.edu.tw). To examine the extent of conservation of predicted target sites, sequences were aligned using MEGA6 (34) and phylogenetic analysis was done. Details of the sequences analyzed are given in Table S1 in the supplemental material.
Cell lines.
Human lung carcinoma cells (A549) and subclonal hepatoma cells (HepG2/C3A) were obtained from ATCC (USA), while Huh-7-derived clonal S10-3 cells were a kind gift from S. Emerson, NIH, USA. Cells were grown at 37°C with 5% CO2 in either Dulbecco's modified Eagle's medium (DMEM) or minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. All cells were maintained at 34.5°C after transfection with HEV replicons.
HEV and HBV replicon plasmids.
The HEV-1 subgenomic replicon (pSK-HEV-Rluc) encoding the Renilla luciferase (Rluc) gene was a kind gift from X. J. Meng (Virginia Tech, Blacksburg, VA, USA). This clone was developed from the original pSK-HEV-2 (HEV-1) infectious cDNA clone (GenBank accession no. AF444002 [23]). pSK-HEV-2 and p6 HEV-3 (GenBank accession no. JQ679013) (24) infectious cDNA clones were a kind gift from Suzanne Emerson, NIAID, NIH, Bethesda, MD, USA. Five miR-122 target site mutants (mutant 1 [mut1], AACTCC; mutant 2 [mut2], CACTAC; mutant 3 [mut3], AACTAC; mutant 4 [mut4], CATTCC; mutant 5 [mut5], CACTCT) were generated by site-directed mutagenesis using plasmid pSK-HEV-Rluc as the template. Two rescue mutants were generated by introducing new miR-122 target sites by introducing synonymous mutations in the ORF2 region or the RdRp-encoding region using mutant mut4 as the template and mutant mut6 (ORF2 miR-122-rescue mut [nt 6261 to 6283]) and mutant mut7 (RdRp miR-122-rescue mut [nt 3932 to 3956]). The HBV 1.3-mer WT genotype D replicon (GenBank accession no. V01460) (35) was a kind gift from Wang-Shick Ryu.
miR-122 mimics and inhibitors.
miR-122 levels in cells were increased using miR-122 mimics (hsa–miR-122-5p) (Dharmacon, USA), while functional miR-122 levels were depleted by using miR-122 inhibitor (miRCURY LNA; hsa–miR-122-5p inhibitor) (Exiqon, Germany). A nonspecific miRCURY LNA miRNA inhibitor control (Exiqon; catalog no. YI00199006) was used in experiments with LNA. For confirming the functional relevance of the seed region, we used an miR-122 mimic with a mut4 seed sequence (mut4 mimic). Sequences of the miR-122 mimics and the inhibitor used are given in Table S2.
Hybrid PCR for confirmation of the predicted miR-122 target sites.
Hybrid PCR was carried out as described previously (36). miR-122-specific primers for both the 5p and 3p forms were designed using reverse complementary sequences of hsa–miR-122 and hsa–miR-122*, respectively. Complementary seed regions were located in the 3′ ends of the miRNA-specific primers. Since G:U pairs are acceptable in miRNA:mRNA duplexes, primers were synthesized as compatible primers where “R” in the primer represents either adenine (A) or guanine (G). Four sets of HEV-specific nested reverse primers, i.e., set I, set II, set III, and set IV, were designed for amplifying the HEV-1 genome in 4 overlapping fragments (Table S2). Combinations of miR-122/miR-122*-specific forward primers and HEV-specific reverse primers were used to perform hybrid PCR (an example for one cDNA fragment is shown in Fig. 1B). Briefly, full-genome HEV-1 RNA was converted into 4 overlapping cDNA fragments, which were used as the templates to identify putative binding sites of miR-122/122* by the use of heminested PCR (a miRNA-specific primer was used in both rounds of PCR). A low annealing temperature (37°C) was used in the first round of amplification to allow hybridization of the miRNA primer with putative target sequences under a condition similar to core body temperature. A round of nested PCR performed at a higher annealing temperature (55°C) was performed for further specific amplification of sequences from putative target RNA sites. To acquire the actual sequences of the miR-122 putative target sites, all PCR products were subjected to thymine and adenine (TA) cloning into pGEM T-Easy vector and were sequenced.
Cell transfections with mimics and inhibitor.
Cells were transfected with a miR-122 mimic, i.e., the mut4 mimic (50 nM) (16), or with an inhibitor using HiPerFect transfection reagent (Qiagen, Hilden, Germany) 24 h prior to transfection with HEV RNA, wherever required. To determine the toxicity of LNA inhibitor, cells were transfected with 1, 5, and 10 nM LNA and monitored for up to 120 h.
Determination of miR-122 levels.
miR-122 levels were determined using TaqMan assays. Briefly, miRNAs were isolated from cells using a mirVana miRNA isolation kit (Ambion, Life Technologies, USA). RNA was reverse transcribed using a TaqMan miRNA reverse transcription kit and hsa–miR-122-5p- and U6 snRNA-specific stem-loop primers (Applied Biosystems, CA, USA). Absolute quantification was done using synthetic miR-122 mimics (Integrated DNA Technologies, IA). Data were normalized across samples using the median normalization method with U6 snRNA as the endogenous control (37).
Cotransfection of cells with miR-122 inhibitor plus HBV replicon plasmid and monitoring of virus replication.
S10-3 cells were cotransfected with 5 nM LNA and 500 ng of HBV replicon plasmid/well of a 24-well plate using Lipofectamine 3000 transfection reagent (Invitrogen, USA) per the manufacturer's instructions. Cells were harvested at the stipulated time points, and HBV replication was monitored by quantitating HBsAg and HBV DNA levels in the culture supernatants and cell pellets, respectively. Briefly, HBsAg levels were determined using an enzyme-linked immunosorbent assay (ELISA) kit (J. Mitra, India) per the manufacturer's instructions. HBV DNA levels were quantitated by real-time PCR as described previously (38).
Transfection of cells with HEV replicon RNA and monitoring of HEV replication.
The pSK-HEV-2/pSK-HEV-Rluc (HEV-1) plasmids were linearized with BglII and p6 HEV-3 plasmid with MluI and were used as the templates to generate capped transcripts using an in vitro transcription kit (mMESSAGE mMachine T7 Ultra kit; Ambion, USA). Plasmid DNA was removed by adding 5 μl (1 unit/μl) of RQ1 RNase-free DNase I (Promega, USA), and the reaction mixture was incubated at 37°C for 30 min. RNA was precipitated using 7.5 M LiCl. The RNA pellet was washed with 70% ethanol, air dried, and dissolved. The concentration of RNA was determined spectrophotometrically using a NanoDrop ND-1000 spectrophotometer at 260 nm; RNA integrity was checked by denaturing agarose gel electrophoresis. RNA was diluted appropriately in Opti-MEM, and cells were transfected (2 μg/well of a 24-well culture plate) using 1,2-dimyristyl Rosenthal inhibitor ether (DMRIE-C) reagent (Invitrogen). For measuring the replication efficiency of the pSK-HEV-RLUC replicon, cells were cotransfected with firefly luciferase (FLuc) plasmid DNA (pGL-3 promoter vector; 100 ng/well) along with HEV Rluc RNA to normalize cell transfection efficiency and Renilla luciferase signal. After 4 h of incubation at 34.5°C, the transfection mixture was replaced with DMEM containing 10% FBS and cells were incubated further. All transfections were carried out in triplicate, and each set of experiments was repeated twice. For mimic and inhibitor assays, cells were transfected a day prior with miRNA and then with HEV RNA.
Reporter gene assay.
Cells were harvested at different time intervals after transfection with pSK-HEV-Rluc RNA, lysed in 100 μl of 1× passive lysis buffer, and stored at −80°C until use. To measure Rluc activity, which indicates the efficiency of HEV replication, a dual-luciferase assay kit (Promega, USA) was used. Briefly, samples were thawed and centrifuged at 5,000 rpm for 2 min, and 20 μl of supernatants was used for measuring dual-luciferase activities (Rluc and firefly luciferase [FLuc]) as described earlier (19).
Negative-strand-specific RT-PCR.
Total cellular RNA was extracted from cells using a Ribopure RNA extraction kit (Ambion, Life Technologies, USA), and detection of negative-sense RNA (replicative intermediate) was done by using a tagged-primer-based reverse-transcription PCR protocol as described previously (22).
Immunofluorescence assay.
Cells were plated in Lab-Tek chamber slides 24 h prior to the stipulated time point (Nunc, Thermo Scientific, USA). The next day, cells were fixed with acetone for 30 min followed by blocking with superblock reagent (Pierce Biotechnology, Thermo Fisher Scientific, USA) for 30 min and were incubated with anti-HEV MAb (generated against a partial ORF2 protein [458 to 607 amino acids]) at room temperature for 30 min. After 3 washes, cells were incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG antibodies (Invitrogen, USA) for 30 min and viewed on a FLoid cell imaging station (Life Technologies, CA, USA).
qRT-PCR for monitoring HEV RNA levels.
HEV-1 RNA levels were determined by using a TaqMan MGB probe and primers as described previously (39). For measuring HEV-3 RNA levels, we used a SYBR green-based qRT-PCR assay. Briefly, in vitro-transcribed HEV-3 RNA was serially diluted from 109 to 101 copies/μl and used as a standard. RNA was isolated from transfected cells using a viral RNA extraction kit, and cDNA was prepared using a High-Capacity cDNA Archive kit (Applied Biosystems, USA) and analyzed on a 7300 real-time PCR system (Applied Biosystems, USA). Sequences of the primers used for HEV-3 quantitation are given in Table S2. For normalization, the mean numbers of HEV RNA copies in cells harvested 4 h posttransfection were taken as baseline/input RNA copy numbers and subtracted from the respective values at different time points.
Gene expression analysis.
Individual SYBR green-based quantitative reverse transcription-PCR assays were performed for selected antiviral genes as described earlier (20). Briefly, total cellular RNA was extracted using a RiboPure RNA extraction kit, RNA integrity was checked on denaturing agarose gel, and quantification was done on a spectrophotometer (ND-1000; NanoDrop Technologies). cDNA was prepared using a High-Capacity cDNA Archive kit (Applied Biosystems, USA) and analyzed on a 7300 real-time PCR system (Applied Biosystems, USA). β-Actin was used to normalize input RNA. RNA from mock-transfected cells was used as a calibrator, and analysis of relative levels of gene expression was done using SDS2.2 software (Applied Biosystems, USA).
Ago2-RNA coimmunoprecipitation.
Immunoprecipitation of the HEV RNA–miR-122 complex along with the RNA-induced silencing complex was carried out using anti-Ago2 antibodies (11A9; Sigma, USA) (39). Briefly, S10-3 cells were transfected with miR-122 mimics (hsa–miR-122-5p) followed by transfection with HEV-Rluc, mut4, mut6, or mut7 RNA after 24 h. HEV replication was monitored by dual-luciferase reporter assays. After 96 h, cell lysates were prepared using lysis buffer (150 mM KCl, 25 mM Tris-HCl [pH 7.4], 5 mM EDTA, 1% Triton X-100, 5 mM dithiothreitol, protease inhibitor cocktail), clarified, and incubated with anti-Ago2 antibodies or isotype control IgG2a at 4°C overnight. Lysates were incubated further with protein G-agarose beads (Invitrogen, USA) for 2 h, and beads were collected and washed 4 times. Resuspended beads were split into two equal parts. One part of each bead was processed for RNA extraction using a viral RNA kit (Qiagen, Hilden, Germany), and HEV genomic copies were quantitated by real-time PCR as described previously (22). The other part was processed for miRNA isolation (using a mirVana miRNA isolation kit) followed by miR-122 quantitation.
miR-122 pulldown assay.
miR-122 mimics and a nonspecific miRNA control were biotinylated using a Pierce RNA 3′ End Biotinylation kit (Thermo Fisher Scientific, USA). The labeling efficiency was checked by a dot blot assay using avidin horseradish peroxidase (HRP) conjugate. S10-3 cells were cotransfected with HEV-Rluc-RNA plus biotin-labeled miRNAs (either control miRNA or miR-122 mimics) as described above. HEV replication was confirmed using dual-luciferase reporter assays. Cells were lysed after 48 h, and lysates were clarified and incubated with streptavidin agarose beads (Pierce Biotechnology, Thermo Fisher Scientific, USA) for 2 h. Beads were collected and washed 3 times, and HEV RNA was extracted and quantitated by real-time PCR as described above.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Emerson (NIH, USA) and X. J. Meng (Virginia Tech, Blacksburg, VA, USA) for providing us the S10-3 cell line and HEV luciferase subgenomic replicon, respectively.
We gratefully acknowledge financial support provided by the Indian Council of Medical Research (ICMR) and National Institute of Virology (NIV), India. B.H. acknowledges UGC for providing the Senior Research Fellowship.
K.L., V.A., and B.H. conceived the study; K.L. and B.H. planned the experiments and wrote the manuscript. B.H. and P.L.B. conducted the experiments and analyzed the results. All of us reviewed the manuscript.
We declare that we have no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01999-17.
REFERENCES
- 1.Arankalle VA, Chobe LP, Jha J, Chadha MS, Banerjee K, Favorov MO, Kalinina T, Fields H. 1993. Aetiology of acute sporadic non-A, non-B viral hepatitis in India. J Med Virol 40:121–125. doi: 10.1002/jmv.1890400208. [DOI] [PubMed] [Google Scholar]
- 2.Dalton HR, Bendall R, Ijaz S, Banks M. 2008. Hepatitis E: an emerging infection in developed countries. Lancet Infect Dis 8:698–709. doi: 10.1016/S1473-3099(08)70255-X. [DOI] [PubMed] [Google Scholar]
- 3.Krain LJ, Nelson KE, Labrique B, Labrique AB. 2014. Host immune status and response to hepatitis E virus infection. Clin Microbiol Rev 27:139–165. doi: 10.1128/CMR.00062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamar N, Dalton HR, Abravanel F, Izopet J. 2014. Hepatitis E virus infection. Clin Microbiol Rev 27:116–138. doi: 10.1128/CMR.00057-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khuroo MS, Teli MR, Skidmore S, Sofi MA, Khuroo MI. 1981. Incidence and severity of viral hepatitis in pregnancy. Am J Med 70:252–255. doi: 10.1016/0002-9343(81)90758-0. [DOI] [PubMed] [Google Scholar]
- 6.Kamar N, Selves J, Mansuy J-M, Ouezzani L, Péron J-M, Guitard J, Cointault O, Esposito L, Abravanel F, Danjoux M, Durand D, Vinel J-P, Izopet J, Rostaing L. 2008. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med 358:811–817. doi: 10.1056/NEJMoa0706992. [DOI] [PubMed] [Google Scholar]
- 7.Smith DB, Simmonds P, Jameel S, Emerson SU, Harrison TJ, Meng X-J, Okamoto H, Van der Poel WHM, Purdy MA, Purdy MA. 2014. Consensus proposals for classification of the family Hepeviridae. J Gen Virol 95:2223–2232. doi: 10.1099/vir.0.068429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koonin EV, Gorbalenya AE, Purdy MA, Rozanov MN, Reyes GR, Bradley DW. 1992. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: delineation of an additional group of positive-strand RNA plant and animal viruses. Proc Natl Acad Sci U S A 89:8259–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anang S, Subramani C, Madhvi A, Bakshi K, Srivastava A, Shalimar, Nayak B, Ranjith Kumar CT, Surjit M. 2016. Endoplasmic reticulum stress induced synthesis of a novel viral factor mediates efficient replication of genotype-1 hepatitis E virus. PLoS Pathog 12:e1005521. doi: 10.1371/journal.ppat.1005521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girard M, Jacquemin E, Munnich A, Lyonnet S, Henrion-Caude A. 2008. miR-122, a paradigm for the role of microRNAs in the liver. J Hepatol 48:648–656. doi: 10.1016/j.jhep.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 11.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi J-TA, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR. 2007. A viral microRNA functions as an orthologue of cellular miR-155. Nature 450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 13.Ouda R, Onomoto K, Takahasi K, Edwards MR, Kato H, Yoneyama M, Fujita T. 2011. Retinoic acid-inducible gene I-inducible miR-23b inhibits infections by minor group rhinoviruses through down-regulation of the very low density lipoprotein receptor. J Biol Chem 286:26210–26219. doi: 10.1074/jbc.M111.229856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. 2007. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimakami T, Yamane D, Welsch C, Hensley L, Jangra RK, Lemon SM. 2012. Base pairing between hepatitis C virus RNA and microRNA 122 3′ of its seed sequence is essential for genome stabilization and production of infectious virus. J Virol 86:7372–7383. doi: 10.1128/JVI.00513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Shen A, Rider PJ, Yu Y, Wu K, Mu Y, Hao Q, Liu Y, Gong H, Zhu Y, Liu F, Wu J. 2011. A liver-specific microRNA binds to a highly conserved RNA sequence of hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J 25:4511–4521. doi: 10.1096/fj.11-187781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S, Qiu L, Yan X, Jin W, Wang Y, Chen L, Wu E, Ye X, Gao GF, Wang F, Chen Y, Duan Z, Meng S. 2012. Loss of MiR-122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G1 modulated P53 activity. Hepatology 55:730–741. doi: 10.1002/hep.24809. [DOI] [PubMed] [Google Scholar]
- 18.Jopling CL, Schütz S, Sarnow P. 2008. Position-dependent function for a tandem MicroRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4:77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devhare PB, Chatterjee SN, Arankalle VA, Lole KS. 2013. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS One 8:e63793. doi: 10.1371/journal.pone.0063793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devhare PB, Desai S, Lole KS. 2016. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci Rep 6:26827. doi: 10.1038/srep26827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamad IAY, Fei Y, Kalea AZ, Yin D, Smith AJP, Palmen J, Humphries SE, Talmud PJ, Walker AP. 2015. Demonstration of the presence of the “deleted” MIR122 gene in HepG2 cells. PLoS One 10:e0122471. doi: 10.1371/journal.pone.0122471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee SN, Devhare PB, Pingle SY, Paingankar MS, Arankalle VA, Lole KS. 2016. Hepatitis E virus (HEV)-1 harbouring HEV-4 non-structural protein (ORF1) replicates in transfected porcine kidney cells. J Gen Virol 97:1829–1840. doi: 10.1099/jgv.0.000478. [DOI] [PubMed] [Google Scholar]
- 23.Cao D, Huang Y-W, Meng X-J. 2010. The nucleotides on the stem-loop RNA structure in the junction region of the hepatitis E virus genome are critical for virus replication. J Virol 84:13040–13044. doi: 10.1128/JVI.01475-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla P, Nguyen HT, Faulk K, Mather K, Torian U, Engle RE, Emerson SU. 2012. Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. J Virol 86:5697–5707. doi: 10.1128/JVI.00146-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forster SC, Tate MD, Hertzog PJ. 2015. MicroRNA as type I interferon-regulated transcripts and modulators of the innate immune response. Front Immunol 6:334. doi: 10.3389/fimmu.2015.00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li A, Qian J, He J, Zhang Q, Zhai A, Song W, Li Y, Ling H, Zhong Z, Zhang F. 2013. Modulation of miR-122 expression affects the interferon response in human hepatoma cells. Mol Med Rep 7:585–590. doi: 10.3892/mmr.2012.1233. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. 2011. Biological functions of microRNAs: a review. J Physiol Biochem 67:129–139. doi: 10.1007/s13105-010-0050-6. [DOI] [PubMed] [Google Scholar]
- 28.Hao J, Jin W, Li X, Wang S, Fan H, Li C, Chen L, Gao B, Zhang X. 2013. Inhibition of alpha interferon (IFN-α)-induced microRNA-122 negatively affects the anti-hepatitis B virus efficiency of IFN-α. J Virol 87:137–147. doi: 10.1128/JVI.01710-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jangra RK, Yi M, Lemon SM. 2010. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol 84:6615–6625. doi: 10.1128/JVI.00417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conrad KD, Giering F, Erfurth C, Neumann A, Fehr C, Meister G, Niepmann M. 2013. microRNA-122 dependent binding of Ago2 protein to hepatitis C virus RNA is associated with enhanced RNA stability and translation stimulation. PLoS One 8:e56272. doi: 10.1371/journal.pone.0056272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasudevan S, Tong Y, Steitz JA. 2007. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 32.Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR. 2013. Treatment of HCV infection by targeting microRNA. N Engl J Med 368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 34.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cha M-Y, Ryu D-K, Jung H-S, Chang H-E, Ryu W-S. 2009. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J Gen Virol 90:978–986. doi: 10.1099/vir.0.009928-0. [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Qi Y, Ruan Q, Ma Y, He R, Ji Y, Sun Z. 2011. A rapid method to screen putative mRNA targets of any known microRNA. Virol J 8:8. doi: 10.1186/1743-422X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M. 2008. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A 105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lole KS, Arankalle VA. 2006. Quantitation of hepatitis B virus DNA by real-time PCR using internal amplification control and dual TaqMan MGB probes. J Virol Methods 135:83–90. doi: 10.1016/j.jviromet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Mukherjee A, Shrivastava S, Bhanja Chowdhury J, Ray R, Ray RB. 2014. Transcriptional suppression of miR-181c by hepatitis C virus enhances homeobox A1 expression. J Virol 88:7929–7940. doi: 10.1128/JVI.00787-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.