

Abstract
We investigated whether phosphorylation of the essential components involved in the 3′ end processing of mRNAs was required for mRNA polyadenylation. The proteins in HeLa nuclear extract were dephosphorylated with alkaline phosphatase, which is known to remove the phosphate moieties from serine and tyrosine. The dephosphorylated extract was used for analyzing cleavagedependent polyadenylation of SV40 late pre-mRNA. The phosphatase treatment of the extract completely blocked the polyadenylation reaction, whereas dephosphorylation of the extract did not inhibit the cleavage reaction. Since the cleavage depends upon functional integrity of the specificity factor, it is unlikely that the phosphorylated state of the latter factor is required for the 3′ end processing. Sodium vanadate, a potent inhibitor of alkaline phosphatase, markedly reduced the inhibitory effect of the phosphatase on the polyadenylation reaction. Dephosphorylation of the extract also prevented formation of the polyadenylation-specific complex with pre-mRNA, whereas the cleavage-specific complexes were formed under this condition. The Mn-dependent polyadenylation, which is largely poly(A) extension reaction, was relatively resistant to the phosphatase treatment. These data indicate that phosphorylation of a key factor is essential for the 3' end processing of pre-mRNA, and suggest that the factor may be poly(A) polymerase.
Polyadenylation is one of the important processing reactions which occur in the generation of a mature mRNA from its precursor. This reaction consists of two tightly coupled steps, namely the cleavage of the precursor RNA molecule, followed by the addition of a poly(A) tail at the cleavage site (for reviews see Manley, 1988; Jacob et al., 1989; Wickens, 1990; Proudfoot, 1991). Several cis-acting elements are critical for polyadenylation. These include the highly conserved hexamer sequence AAUAAA found 10–30 nucleotides upstream of the site of poly(A) addition (Fitzgerald and Shenk, 1981; Manley et al., 1985; Wickens and Stephenson, 1984) and the less conserved elements referred to as the GU- or U-rich sequence downstream of the poly(A) site (Gill and Proudfoot, 1984; Zarkower and Wickens, 1988), which functions only in concert with the upstream hexamer signal. The development of in vitro systems which accurately mimic the in vivo polyadenylation reaction (Moore and Sharp, 1985) has greatly advanced our knowledge of this unique 3′ end processing reaction. Fractionation of the HeLa extracts used in the in vitro system has allowed identification and characterization of the various components involved in the polyadenylation reaction (Takagaki et al., 1989; Gilmartin and Nevins, 1990; Christofori and Keller, 1989). The basic components of the polyadenylation reaction include poly(A) polyermase (PAP), which functions by adding a poly(A) tail to the cleaved substrate; the specificity factor (SF or PF2 or CPF), which confers dependence of the upstream hexamer sequence; and two cleavage factors (CF1 and CF11). Another component, designated CstF, contributes to the function of the cleavage factor by enhancing its activity.
Antibodies raised against purified poly(A) polymerase from a rat hepatoma (Stetler and Jacob, 1984) have been shown to inhibit both cleavage and polyadenylation (Terns and Jacob, 1989), which was consistent with concurrent studies showing requirement of poly(A) polymerase for the cleavage reaction (Christofori and Keller, 1988). With the identification and purification of each of these components, the exact role of each factor can now be examined. An unexplored area in the 3′ end processing of pre-mRNA is the potential regulation of the cleavage/polyadenylation reaction. Phosphorylation of one or more of the factors involved in the mRNA processing reaction could be a potential regulatory mechanism. Phosphorylation appears to modulate the activities of several transcription factors, including the cAMP response element binding protein or CREB (Gonzalez and Montminy, 1989; Ofir et al., 1991); the yeast transcriptional activator ADR1 (Cherry et al., 1989); the two octamer motif binding proteins, Oct-1 and Oct-2 (Tanaka and Herr, 1990); adenovirus-induced E4F transcription factor (Raychaudhuri et al., 1989); heat shock transcription factor (Sorger and Pelham, 1988); GC box-binding factor SP1 (Jackson et al., 1990); and the hepatitis B virus-encoded transcriptional trans-activator hbx (Wu et al., 1990). Phosphorylation/dephosphorylation can also regulate transcription of the beta-Glucoside operon in E. coli (Amster-Choder et al., 1989), as well as the function of the retinoblastoma gene product (RB protein) during the cell cycle (Chen et al., 1989). Poly(A) polymerase purified from the Morris hepatoma 3924A has been previously demonstrated to be a phosphoprotein (Rose and Jacob, 1979). Phosphorylation of this enzyme in vitro by purified exogenous kinase activated the enzyme (Rose and Jacob, 1979, 1980). The availability of the in vitro system for studying cleavage and polyadenylation prompted us to explore the potential role of phosphorylation of the components in the mRNA 3′ end processing machinery on the cleavage/polyadenylation of specific pre-mRNAs. The present study is an attempt to address this issue.
Materials and methods
Preparation of pre-mRNA substrate
Substrate mRNA was generated from a plasmid containing the SV40 late poly(A) site (generously provided by Dr. M. Wickens). Plasmid pSPSV −58/+70 contains the wild-type hexamer AAUAAA. This plasmid was linearized with Dra I and transcribed in vitro under the direction of SP6 promoter, as described by Melton et al., 1984. This generated a wild-type RNA, 125 nucleotides in length. The wild-type substrate which contains the upstream and downstream recognition sequences yielded a 70 nucleotides-long upstream cleavage product in a specific in vitro reaction.
Assay for cleavage and polyadenylation of pre-mRNA
The 3′ end processing reaction was carried out as described (Terns and Jacob, 1989). Reactions were carried out in final volumes of 12.5 or 25 μl and contained 40% HeLa nuclear extract (32P)-labeled RNA substrate, 1mM ATP, 20mM creatine phosphate, 0.04μg/μl tRNA, 0.2mM DTT, 2.5% polyvinyl alcohol, and 0.5mM MgCl2 or 0.5mM MnCl2. Samples were incubated at 30 °C for 1 hour. Reaction products were isolated with phenol:chloroform:isoamyl alcohol (25:24:1) precipitated with ethanol and separated by electrophoresis on an 8% polyacrylamide-8.3M Urea gel followed by autoradiography. [32P]-labeled Hind III fragments of ΦX 174 DNA were used as markers. Accurate cleavage of the substrate RNA was assayed by replacing ATP with 0.5mM cordycepin triphosphate (Moore and Sharp, 1985). For dephosphorylation experiments, calf intestinal alkaline phosphatase of the highest purity (Boehringer Mannheim), which had been dialyzed against 10mM Hepes pH 7.6, was used. The alkaline phosphatase was added to these samples and incubated for 30 minutes at 30°C. Reaction components were then added to these samples and allowed to proceed as described. In studies requiring inhibition of alkaline phosphatase, 1mM sodium vanadate was pre-incubated with the phosphatase for 30 minutes at 30°C prior to its addition to the samples.
Analysis of complex formation with pre-mRNA before and after alkaline phosphatase treatment
HeLa nuclear extract was dephosphorylated with 0.04 U/μg of phosphatase, as stated above. Control extract was pre-incubated with 10mM Hepes pH 7.6 for 30 minutes at 30°C. In vitro processing reactions (25 μl) were set up under polyadenylation conditions and terminated at various time points (0, 5, 30, and 90 minutes) to study the complex formation at different stages of the reaction. Half of the reaction in each case (control and phosphatase-treated) was deproteinized with Proteinase K stop buffer, phenol extracted, ethanol precipitated, and analyzed on an 8% polyacrylamide 8.3 M Urea gel to determine the polyadenylated state of the RNA over the time course. The remaining half of the reaction was terminated with the addition of heparin (final concentration of 5 mg/ml) and incubated at 30°C for an additional 10 minutes. The samples were then chilled on ice and mixed with 2.5 μl of 50% glycerol 0.05% xylene cyanol. Samples were analyzed on a 4% non-denaturing polyacrylamide gel, as previously described (Terns and Jacob, 1989).
Results
Dephosphorylation of components in HeLa nuclear extract with alkaline phosphatase specifically inhibits polyadenylation
HeLa extract was treated with calf intestine alkaline phosphatase, which removes phosphate groups from serine (Fernley, 1971) and tyrosine (Swarup et al., 1981) residues. The alkaline phosphatase was RNase free and showed a single band upon electrophoresis and silver staining (data not shown). Increasing amounts of phosphatase were added to the extract, ranging from 0.01U/μg to 0.08U/μg of protein. The dephosphorylated samples were assayed using a wild-type SV40 late pre-mRNA substrate (see Fig. 1). Polyadenylation of the substrate in the presence of Mg2+, which directs cleavage-dependent polyadenylation, was inhibited by the phosphatase at a concentration as low as 0.02U/μg (compare lane 2 with lane 4). Complete inhibition of polyadenylation was achieved at a concentration of 0.04U/μg alkaline phosphatase. Control reactions included samples treated with boiled alkaline phosphatase and incubated in 10mM Hepes pH 7.6 at a volume equivalent to that used for the highest concentration of the phosphatase. Lack of inhibition of polyadenylation in these control samples demonstrated the specificity of the inhibition following treatment with the phosphatase (data not shown). The inhibition of poly(A) addition to mRNA was proportional to the accumulation of cleaved product as the alkaline phosphatase concentration increased (see lanes 3 to 8).
Figure 1.
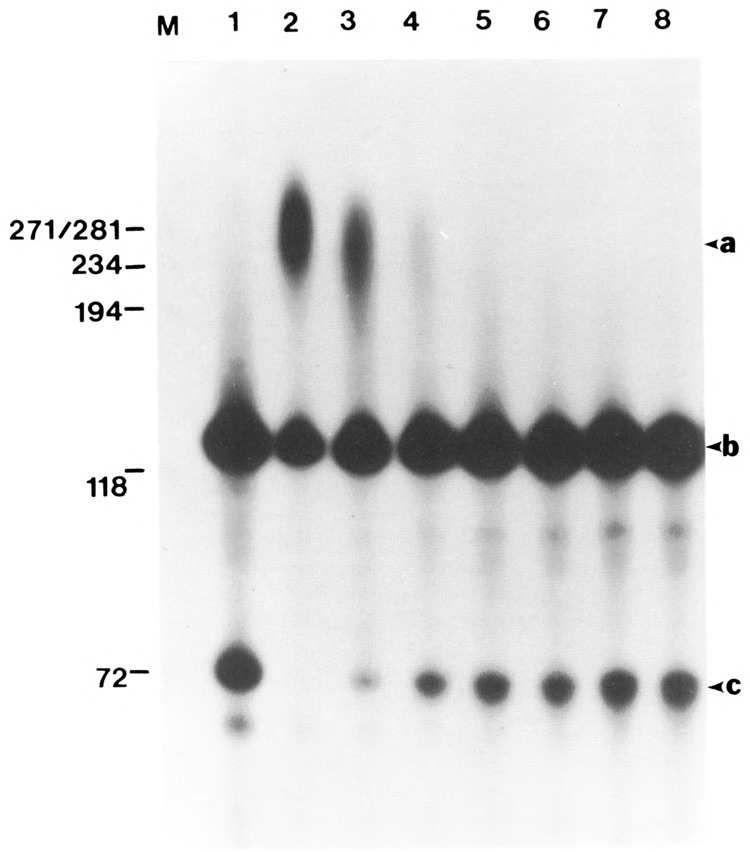
Inhibition of polyadenylation by dephosphorylation of components in the HeLa nuclear extract. HeLa nuclear extract was incubated with increasing amounts of calf intestinal alkaline phosphate (0.01 U/μg to 0.08 U/μg) for 30 minutes at 30°C. The samples were then assayed for 3′ end processing using a full-length SV40 mRNA substrate under Mg2+ conditions. One sample (lane 1) consisted of untreated HeLa extract in the presence of 3′ dATP and Mg2+ to detect the upstream cleavage product. Arrows a, b, and c correspond to the polyadenylated RNA, −58/+55 SV40 late pre-mRNA (containing the upstream and downstream recognition sequences and the cleavage site), and the 5′ cleavage product, respectively. Lanes 2 to 8 correspond to samples incubated with 0, 0.015, 0.02, 0.03, 0.04, 0.06, and 0.08 U/μg of alkaline phosphatase, respectively.
Dephosphorylation does not affect the cleavage reaction
Next, the effect of alkaline phosphatase on the cleavage reaction was examined (Fig. 2). The ATP analogue, cordycepin triphosphate (3′ dATP), which terminates poly(A) addition, facilitated detection of the cleaved product (lane 1). HeLa nuclear extract was treated with alkaline phosphatase at concentrations used above and assayed for cleavage of the pre-mRNA substrate. Unlike the polyadenylation reaction, the cleavage reaction was not affected following dephosphorylation (compare lanes 8 to 11 with lanes 1 to 7). The lack of inhibition of the cleavage reaction indicates that the various cleavage factors were unaffected by dephosphorylation. Since the cleavage factors were unaffected, the observed accumulation of cleavage product in the absence of cordycepin triphosphate (Fig. 1) can be attributed solely to inhibition of poly(A) addition, so that the reaction stalls at the cleavage step.
Figure 2.
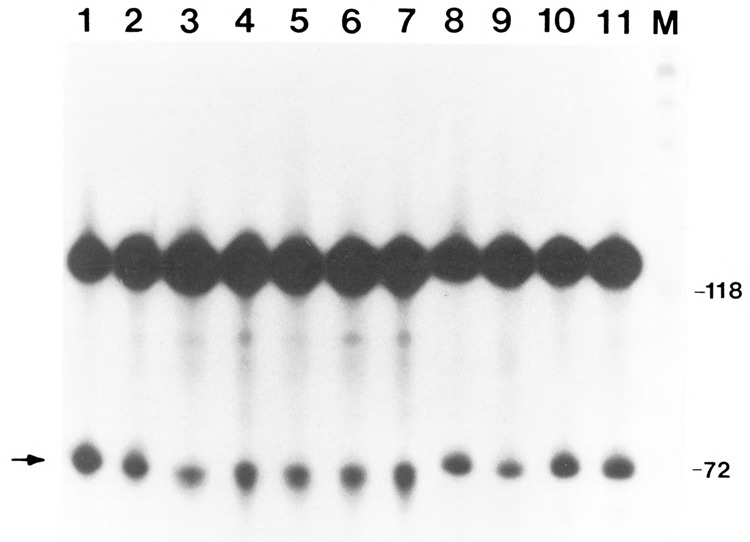
Effect of alkaline phosphatase on the cleavage of SV40 L mRNA. HeLa nuclear extract was incubated with calf intestinal alkaline phosphatase, as in Figure 1. The samples were then assayed for cleavage in the presence of 3′ dATP and Mg2+ and varying amounts of alkaline phosphatase. Lanes 1–7 correspond to 0.01, 0.015, 0.02, 0.03, 0.04, 0.06, and 0.08 U/μg of alkaline phosphatase, respectively. Control samples consisted of incubation of the samples under the following conditions: lane 8, incubation for 30 minutes at 30°; lane 9, l0mM Hepes pH 7.6 in a volume to match that of the 0.08 U/μg; lane 10, 0.08 U/μg of alkaline phosphatase that was previously boiled for 5′; lane 11, a sample processed under normal conditions Without alkaline phosphatase.
Inactivation of alkaline phosphatase activity with sodium vanadate reduces inhibitory effect of the phosphatase on the polyadenylation reaction
We then investigated whether inactivation of alkaline phosphatase can prevent its inhibitory effect on polyadenylation. Several inhibitors of alkaline phosphatase have been reported, including sodium phosphate and sodium fluoride (Ludlow et al., 1989). Perhaps the most commonly used inhibitor is sodium vanadate (Klarlund, 1985; Cantley et al., 1977; Lopez et al., 1976; Morrison et al., 1989). To determine further the specificity of the inhibitory effect of alkaline phosphatase on the polyadenylation reaction, HeLa nuclear extract was incubated with alkaline phosphatase (ranging from 0.01U/μg to 0.024U/μg) that had been pretreated with 1mM sodium vanadate. Pretreatment of phosphatase with vanadate prevented its inhibitory effects even at the highest concentration of alkaline phosphatase (Fig. 3, lanes 3 and 4). In contrast, incubation with untreated alkaline phosphatase under the same reaction conditions did not permit the polyadenylation to proceed (see lanes 5–7), resulting in accumulation of the cleaved product. Inhibition of the phosphatase and the resultant increase in polyadenylation were pronounced even at the lower concentration of alkaline phosphatase (compare lane 2 with lane 5). The cleavage product did not accumulate when the lowest concentration of alkaline phosphatase was used with the inhibitor (compare lane 2 with 5). The absence of detectable cleavage product demonstrates that the phosphatase effect was completely overcome at this concentration of the phosphatase inhibitor, allowing polyadenylation to continue unabated.
Figure 3.
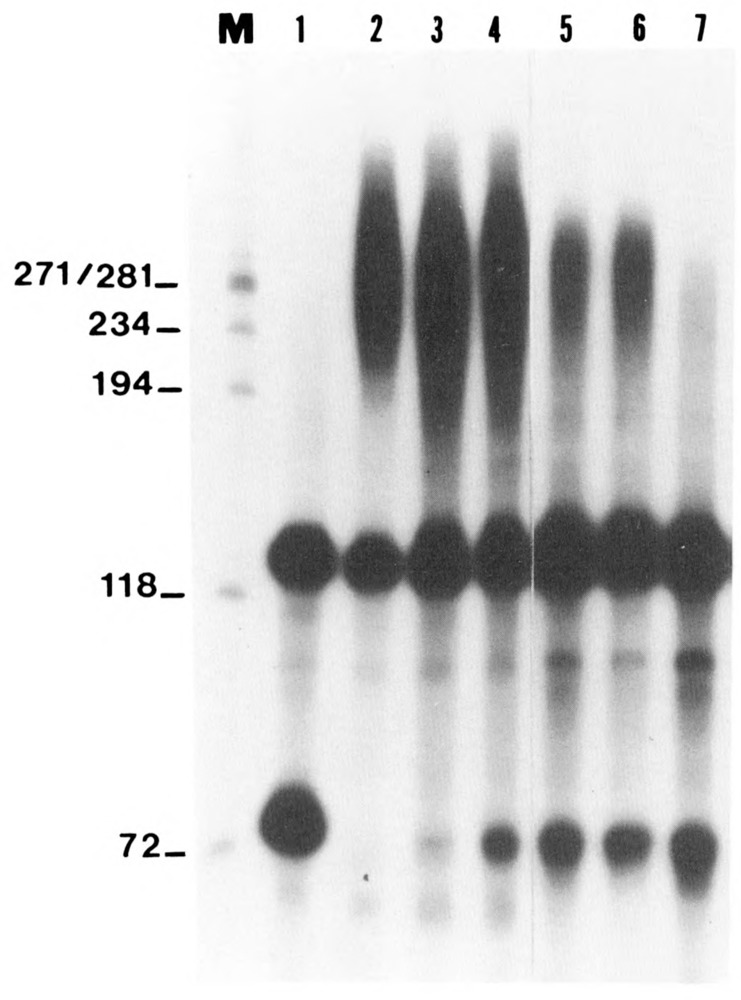
Effect of sodium vanadate on the inhibitory effect of alkaline phosphatase. Sodium vanadate, a potent inhibitor of alkaline phosphatase, was preincubated with alkaline phosphatase at concentrations previously shown to be inhibitory for polyadenylation. HeLa nuclear extract was then reacted with the pretreated phosphatase and further processed as described in the legend to Figure 1. Lanes 5–7 correspond to samples incubated with 0.01,0.015, and 0.02 U/μg of alkaline phosphatase, respectively, and lanes 2–4 represent samples incubated with corresponding concentrations of pretreated phosphatase. Inhibition of alkaline phosphatase by vanadate negated the inhibitory effects of dephosphorylation on polyadenylation compared to the sample incubated with untreated phosphatase (compare lane 4 with 7). Note that at the lower concentration of the pretreated phosphatase, the 5′ cleavage product was not detected, in contrast to the sample incubated with untreated phosphatase (compare lane 2 with lane 5).
Mn2+-activated hexamer-dependent polyadenylation is relatively resistant to phosphatase treatment
Previous studies have shown that the polyadenylation reaction in HeLa nuclear extract can be activated by Mn2+ and that the increased activity in the presence of this divalent metal ion is largely due to longer poly(A) tail added to mRNA (see Terns and Jacob, 1989). In general, the Mn2+-dependent addition of poly(A) to pre-mRNA is cleavage-independent, as this non-specific activity can be separated from cleavage activity by chromatographic fractionation (see Manley, 1988). To determine whether the Mn2+-activated polyadenylation of SV40L pre-mRNA is affected by dephosphorylation, alkaline phosphatase was added to the samples at varying concentrations (ranging from 0.01U to 0.06U/μg) and the reaction carried out as before (Fig. 4).
Figure 4.
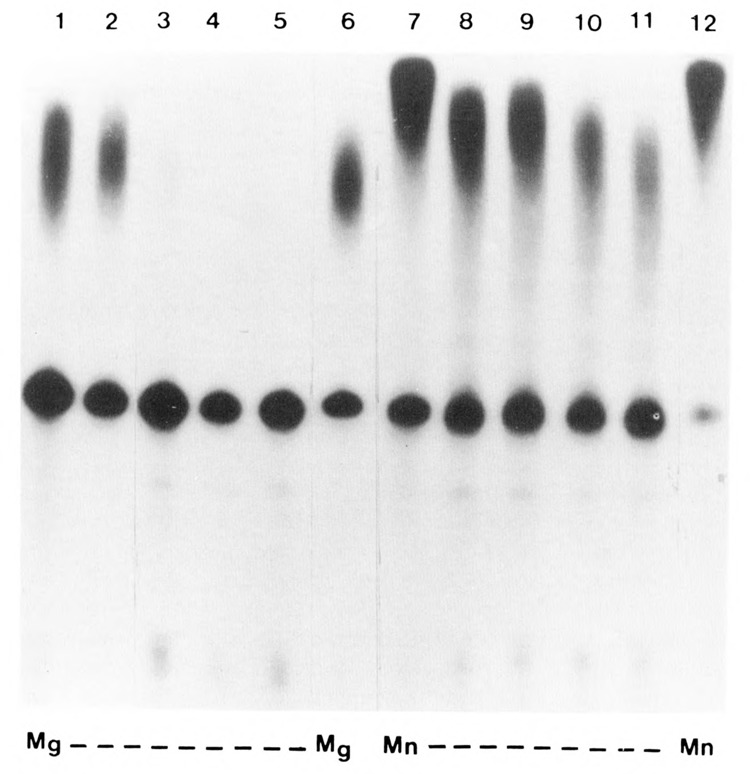
Effect of alkaline phosphatase on the Mn2+ activated cleavage-independent polyadenylation. HeLa nuclear extract was incubated with varying amounts of alkaline phosphatase, as previously described. The samples were then processed for polyadenylation of SV40 L pre-mRNA in the presence of Mg2+ (lanes 1–6) or Mn2+ (lanes 7–12). Lanes 2 to 5 contained 0.01,0.02,0.04, and 0.06 units/μg of alkaline phosphatase, respectively. Lanes 8 to 11 contained similar units of the phosphatase. Lanes 1 and 7 represent untreated samples, whereas lanes 6 and 12 represent samples incubated with Hepes buffer in a volume equal to that of the highest concentration of the phosphatase used.
Control samples were incubated under identical conditions, namely that Mg2+ was replaced by Mn2+ as the divalent ion. As anticipated, Mg2+-dependent polyadenylation reaction was inhibited completely by 0.02U/μg of alkaline phosphatase (Fig. 4, lanes 3 to 5), whereas the Mn2+-activated reaction was inhibited only by about 20% even at the highest concentration (0.06U/μg) of phosphatase used in this experiment. It is evident from this study that phosphorylation of the factor, presumably of poly(A) polymerase, is not essential for the Mn2+-dependent reaction. Since Mn2+-dependent polyadenylation is largely a poly(A) extension reaction, the AAUAAA-dependent coupled cleavage-polyadenylation reaction predominantly directed by Mg2+ appears to be more sensitive to the phosphorylated state of the factor(s).
Inhibition of polyadenylation reaction by alkaline phosphatase prevents formation of polyadenylation-specific complex
Previous studies from several laboratories have shown that incubation of pre-mRNA substrate with HeLa nuclear extract can result in the formation of cleavage-specific and polyadenylation-specific complexes. In particular, Zarkower and Wickens (1987) have shown that pre-cleavage and post-cleavage complexes, designated B and B′ respectively, are formed initially. These complexes contain pre-mRNA and 5′ cleaved product, respectively. Even when −55/+70 pre-mRNA substrate was incubated with treated or untreated HeLa extract and immediately terminated with heparin (Fig. 5, lanes 2 and 6), the two cleavage complexes were formed, along with the pre-active complex (PA). When incubation was continued for 30 minutes, a polyadenylation-specific complex (P) was shown to form in the control sample (lane 8), whereas the phosphatase-treated HeLa nuclear extract did not form this polyadenylated complex even after a 90-minute incubation (compare lanes 5 with 9). These data support the earlier contention that dephosphorylation specifically inhibits the polyadenylation reaction. This was further confirmed by identifying the polyadenylated RNA following electrophoresis of the same samples under denaturing conditions. Only the samples which yielded the (P) complex contained the polyadenylated RNA (Fig. 5B, lanes 7 and 8).
Figure 5.
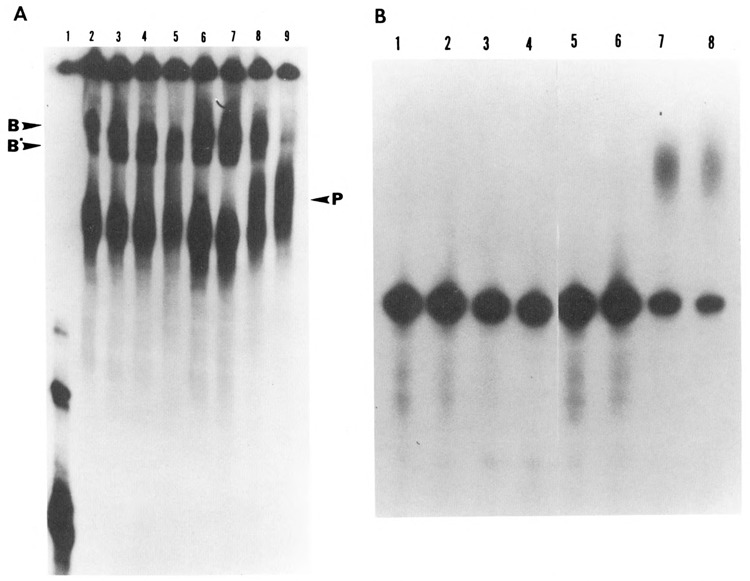
Effect of dephosphorylation on the formation of RNP complexes under polyadenylation conditions. HeLa nuclear extract was dephosphorylated with alkaline phosphatase (0.04 U/μg). Control samples consisted of extract incubated with 10 mM Hepes pH 7.6 in a volume equal to that of treated samples. Polyadenylation reactions were carried out for varying time intervals ranging from 0 to 90 minutes using [32P]-labeled SV40 pre-mRNA substrate. Half of the reaction (12.5 μl) was terminated by the addition of heparin to a final concentration of 5 mg/ml and incubated for an additional 10 minutes at 30°C. These samples were analyzed on a 4% non-denaturing polyacrylamide gel at 4°C (A). The remaining samples were deproteinized and analyzed on an 8% acrylamide, 8.3 M Urea gel (B). A. Lanes 2 to 5 correspond to the dephosphorylated samples incubated for 0, 5, 30, and 90 minutes respectively, while lanes 6 to 9 correspond to the untreated sample incubated for the same time periods. Electrophoresis on the native gel showed that in the primary stages of the RNP complex formation, where cleavage takes place, the dephosphorylated and non-dephosphorylated samples formed comparable complexes with pre-mRNA (compare lanes 2 and 3 with lanes 6 and 7). However, as the reaction progressed from cleavage to polyadenylation, the treated samples were unable to form a final polyadenylated RNP complex (compare lanes 4 and 5 with lanes 8 and 9). The arrow on the left corresponds to pre-mRNA used as the substrate. B. Electrophoresis of the remaining half of the samples under denaturing conditions showed the state of the RNA corresponding to the complexes formed in A. The emergence of the polyadenylated RNA correlated with the formation of the polyadenylation-specific complexes (P); compare B, lanes 7 and 8 with A, lanes 8 and 9. The sample corresponding to A, lane 1 was not processed under denaturing conditions, as it represented only the labeled pre-mRNA probe.
When cleavage and polyadenylation reactions were uncoupled with 3′ dATP, only the cleavage-specific complexes B and B′ were formed, and formation of these complexes was again unaffected by phosphatase treatment (Fig. 6, compare lanes 2–4 with lanes 5–7). To emphasize further, when the phosphatase-treated HeLa nuclear extract was used for the 3′ end processing of the SV40 late pre-mRNA substrate, neither polyadenylation complex nor a polyadenylated RNA was formed, whereas formation of the cleavage-specific complexes persisted.
Figure 6.
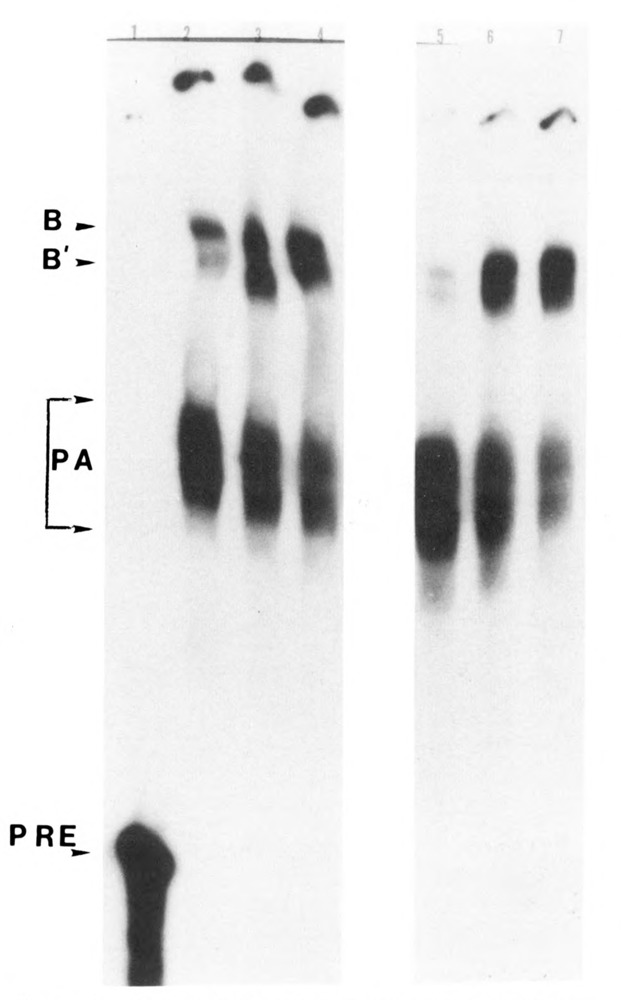
Effect of dephosphorylation on the formation of RNP complexes under cleavage conditions. HeLa nuclear extract was dephosphorylated with 0.04 U/μg of alkaline phosphatase. Control samples were incubated with 10 mM Hepes pH 7.6 in a volume equal to that of the treated samples. Cleavage reactions were carried out for varying time intervals ranging from 0 to 30 minutes. The reactions were terminated by the addition of heparin to a final concentration of 5 mg/ml and incubated for an additional 10 minutes at 30°C. These samples were analyzed on a 4% non-denaturing poly-acrylamide gel. Lanes 2 to 4 correspond to the dephosphorylated samples incubated for 0, 5, and 30 minutes, respectively, whereas lanes 5 to 7 correspond to the untreated samples incubated for the same time intervals. The native gel showed that the formation of the pre-cleavage complex (B) and the post-cleavage complex (B′) by the dephosphorylated samples was comparable to that formed by the untreated samples (compare lane 2 with lane 5, 3 with 6, and 4 with 7).
Discussion
The present studies have shown that dephosphorylation of HeLa nuclear extract with alkaline phosphatase, which is known to remove the phosphate moieties from serine and tyrosine residues, can completely inhibit the polyadenylation of SV40 pre-mRNA. Under these conditions, the labeled phosphate from the proteins can be removed by phosphatase treatment (data not shown). No detectable RNase or poly(A)-degrading activities were observed at the highest concentration of the alkaline phosphatase used in the present study. Phosphatase treatment may generate ADP from ATP, which in turn may inhibit the polyadenylation reaction. However, persistence of 3′ end processing in the presence of unlabeled ADP rules out such a possibility (unpublished data). The relative resistance of Mn-dependent polyadenylation or the Mg-dependent poly(A) extension reaction to phosphatase treatment (see next paragraph) further negates potential contamination of the phosphatase preparation with a nuclease. Several studies have shown that phosphorylation of certain pol II transcription factors is required for the optimal transcription (see Introduction). More recently, we have shown that phosphorylation of one of the pol I transcription-factor (ElBF) can regulate rDNA transcription (Zhang et al., 1991). To our knowledge, the present report is the first to show that phosphorylation of a trans-factor is essential for mRNA polyadenylation reaction.
The following observations suggest that the factor may indeed be poly(A) polymerase. First, the cleavage reaction was not inhibited by phosphatase treatment. Second, pretreatment of the alkaline phosphatase with sodium vanadate, which is known to block phosphatase activity, prevented the inhibitory action of the phosphatase on the polyadenylation reaction. Third, the Mn2+-dependent, hexamer-independent processing reaction, which is essentially a poly(A) extension reaction, was not significantly affected by phosphatase treatment at concentrations which completely blocked the Mg2+-dependent reaction. We have further confirmed this observation by adding the phosphatase 30 minutes after incubation in the presence of Mg2+, which rendered the poly(A) extension reaction resistant to dephosphorylation (data not shown). Fourth, the formation of polyadenylation-specific complex was selectively inhibited by treatment with alkaline phosphatase, whereas the cleavage-specific complexes were formed even after dephosphorylation. Since the cleavage reaction depends upon the functional specificity factor, and the cleavage reaction remained unaffected by dephosphorylation of HeLa nuclear components, it is unlikely that the phosphorylated state of the specificity factor is crucial for the 3′ end processing. Therefore, it appears that of the three major factors involved in the 3′ end processing of mRNA — namely poly(A) polymerase, cleavage factors (I and II), and specificity factor — only PAP need be phosphorylated for its functional specificity.
The present data are consistent with our observations more than a decade ago (Rose and Jacob, 1979, 1980). These studies showed that poly(A) polymerase is a phosphoprotein, and that phosphorylation can activate the enzyme. Phosphorylated enzyme has relatively higher specific activity and is more stable than poorly phosphorylated or dephosphorylated enzyme. The present studies have extended these earlier observations by using a defined pre-mRNA substrate and a cell-free system which can cleave and polyadenylate the pre-mRNA.
Although we tried to use a reconstituted system containing partially purified PAP and cleavage/specificity factors, it was technically difficult to control sodium vanadate concentrations in these studies without stimulatory effects of the vanadate on the protein kinases associated with the fractions. Further, alkaline phosphatase conjugated to the agarose beads did not work effectively in our hands. Ideally, we should be able to add phosphorylated or dephosphorylated poly(A) polymerase to the in vitro system and show directly that phosphorylation of poly(A) polymerase can control the 3′ end processing reaction in concert with other trans-factors which can apparently function independently of their phosphorylated states. A few technical problems must be circumvented before such an approach can be made. First, the most highly purified poly(A) polymerase from various sources has lost its capacity to reconstitute in a cell-free system. It is quite likely that extensive purification can dephosphorylate the enzyme significantly, which may lead to its inactivation in a functional assay. Indeed, we have shown that purification removes phosphate moieties from poly(A) polymerase (Rose and Jacob, 1979). Although some phosphatase moieties are retained after purification process, the most crucial phosphate residues might be lost during the fractionation steps. Preliminary study in our laboratory has failed to identify the ideal conditions for activating the extensively purified enzyme. It seems highly unlikely that a hitherto unidentified factor associated with poly(A) polymerase is lost during extensive purification. We are currently searching for the protein kinase which would add the phosphate residues to the correct peptide residues under the functional assay conditions. Another potential problem is the possibility that other posttranslational modifications of the enzyme might be indispensable for the enzyme function, together with the phosphorylation. Such modifications of the enzyme molecule could be lost during extensive purification of the enzyme, which could lead to loss of its functional activity. We are exploring these avenues to elucidate the exact mechanisms by which poly(A) polymerase modulates the 3′ end processing reaction. Nevertheless, one potential mode of regulation of this unique cellular reaction does occur at the level of phosphorylation. Since the phosphorylation of cleavage and specificity factors does not appear to be involved in the processing reactions, it is reasonable to suggest that poly(A) polymerase, the only other essential factor, is essential for at least the Mg2+-activated cleavage-dependent polyadenylation of SV40 late pre-mRNA. It is possible that the proper 3′ end processing of some pre-mRNAs may be contingent upon phosphorylation of other factors. Further studies are required to validate this concept. Nevertheless, the need for phosphorylation of at least one factor in the 3′ end processing of pre-mRNA is evident from the present investigation.
Acknowledgments
We thank Dr. Marvin Wickens for providing the plas-mid pSPSV −58/+70 and Katrina Piotrowski for secretarial assistance. This work was supported by grant CA25078 from the National Institutes of Health to S.T.J.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
Jamie A. Hengst-Zhang is currently at the Department of Medicine, Northwestern University, Chicago, IL 60611
Note added in proof
Recent studies in our laboratory have shown that unlike the 3′ end processing of SV40 late pre-mRNA, the cleavage of adenovirus L3 pre-mRNA is inhibited by dephosphorylation of the proteins in the HeLa nuclear extract. This observation is consistent with the known requirement of functional poly(A) polymerase for cleavage of L3 pre-mRNA. The cleavage of SV40 late pre-mRNA in the absence of a functionally active poly(A) polymerase (Y. Takagaki, L. C. Ryner, and J. L. Manley [1988], Cell 52, 731–742) would explain the resistance of the latter reaction to dephosphorylation. These data, taken together, further support the notion that the phosphorylated state of poly(A) polymerase may be crucial for the 3′ end processing of pre-mRNAs.
References
- Amster-Choder O., Houman F., and Wright A. (1989), Cell 58, 847–855. [DOI] [PubMed] [Google Scholar]
- Cantley L. C., Josephson L., Warner R., Yanagisawa M., Lechene C., and Guidotti G. (1977), J Biol Chem 252, 7421–7423. [PubMed] [Google Scholar]
- Chen P. L., Scully P., Shew J. Y., Wang Y. J., and Lee W. H. (1989), Cell 58, 1193–1198. [DOI] [PubMed] [Google Scholar]
- Cherry J. R., Johnson T. R., Dollaid C., Shuster J. R., and Denis C. L. (1989), Cell 56, 409–419. [DOI] [PubMed] [Google Scholar]
- Christofori G. and Keller W. (1989), Mol Cell Biol 9, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernley H. N. (1971), in The Enzymes, vol. 4 (Boyer P. D., ed.), Academic Press, New York, pp. 373–415. [Google Scholar]
- Fitzgerald M. and Shenk T. (1981), Cell 24, 251–260. [DOI] [PubMed] [Google Scholar]
- Gill A. and Proudfoot N. J. (1984), Nature 312, 473–474. [DOI] [PubMed] [Google Scholar]
- Gilmartin G. M. and Nevins J. (1990), Genes Dev 3, 2180–2189. [DOI] [PubMed] [Google Scholar]
- Gonzalez G. A. and Montminy M. R. (1989), Cell 59, 675–680. [DOI] [PubMed] [Google Scholar]
- Jackson S. P., MacDonald J. J., Lees-Miller S., and Tjian R. (1990), Cell 63, 155–165. [DOI] [PubMed] [Google Scholar]
- Jacob S. T., Terns M. P., and Maguire K. A. (1989), Cancer Res 49, 2827–2833. [PubMed] [Google Scholar]
- Klarlund J. S. (1985), Cell 41, 707–717. [DOI] [PubMed] [Google Scholar]
- Lopez V., Stevens T., and Lindquist R. N. (1976), Arch Biochem Biophys 175, 31–38. [DOI] [PubMed] [Google Scholar]
- Ludlow J. W., DeCaprio J. A., Huang C. M., Lee W. H., Paucha E., and Livingston D. M. (1989), Cell 56, 57–65. [DOI] [PubMed] [Google Scholar]
- Manley J. L., Yu H., and Ryner L. C. (1985), Mol Cell Biol 5, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley J. L. (1988), Biochim Biophys Acta 950, 1–12. [DOI] [PubMed] [Google Scholar]
- Moore C. L. and Sharp P. A. (1985), Cell 41, 845–855. [DOI] [PubMed] [Google Scholar]
- Morrison D. K., Kaplan D. R., Escobedo J. A., Rapp U. R., Roberts T. M., and Williams L. T. (1989), Cell 58, 649–657. [DOI] [PubMed] [Google Scholar]
- Ofir R., Dwarki V. J., Rashid D., and Verma I. M. (1991), Gene Expr 1, 55–61. [PMC free article] [PubMed] [Google Scholar]
- Proudfoot N. J. (1988), Cell 64, 671–674. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri P., Bagchi S., and Nevins J. R. (1989), Genes Dev 3, 620–627. [DOI] [PubMed] [Google Scholar]
- Rose K. M. and Jacob S. T. (1979), J Biol Chem 254, 10256–10261. [PubMed] [Google Scholar]
- Rose K. M. and Jacob S. T. (1980), Biochemistry 19, 1472–1476. [DOI] [PubMed] [Google Scholar]
- Sorger P. K. and Pelham H. R. B. (1988), Cell 54, 855–864. [DOI] [PubMed] [Google Scholar]
- Stetler D. A. and Jacob S. T. (1984), J Biol Chem 259, 7239–7244. [PubMed] [Google Scholar]
- Swarup G., Cohen S., and Garbers D. L. (1981), J Biol Chem 256, 8197–8201. [PubMed] [Google Scholar]
- Takagaki Y., Ryner L. C., and Manley J. L. (1989), Genes Dev 3, 1711–1724. [DOI] [PubMed] [Google Scholar]
- Tanaka M. and Herr W. (1990), Cell 60, 375–386. [DOI] [PubMed] [Google Scholar]
- Terns M. P. and Jacob S. T. (1989), Mol Cell Biol 9, 1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens M. and Stephenson P. (1984), Science 226, 1045–1051. [DOI] [PubMed] [Google Scholar]
- Wickens M. (1990), Trends Biochem Sci 15, 277–281. [DOI] [PubMed] [Google Scholar]
- Wu J. Y., Zhou Z. Y., Judd A., Cartwright C. A., and Robinson W. S. (1990), Cell 63, 687–695. [DOI] [PubMed] [Google Scholar]
- Zarkower D. and Wickens M. (1987), EMBO J 6, 4185–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkower D. and Wickens M. (1988), J Biol Chem 263, 5780–5788. [PubMed] [Google Scholar]
- Zhang J., Niu H., and Jacob S. T. (1991), Proc Natl Acad Sci USA, 88, 8293–8296. [DOI] [PMC free article] [PubMed] [Google Scholar]