

Abstract
Vertebrate premessenger RNAs are usually spliced and polyadenylated. In vivo analysis of the relative kinetics of the two reactions is difficult. We have used in vitro processing systems to investigate the order of splicing and polyadenylation of chimeric precursor RNAs containing a single intron and a poly(A) site. Polyadenylated, but not spliced, intermediate RNA appeared first and reached a low steady-state level early during incubation, properties consistent with its being a reaction intermediate in the production of doubly-processed spliced and polyadenylated product RNA. The kinetics of polyadenylation suggested that polyadenylated RNA was the only intermediate in the production of doubly-processed RNA. Spliced, but not polyadenylated, RNA also appeared. This species, however, continued to accumulate during reaction, and could not be chased into product spliced and polyadenylated RNA. These data support a preferred order of reaction for 3′ terminal introns and exons in which polyadenylation precedes splicing.
Although the majority of vertebrate pre-mRNAs undergo both splicing and polyadenylation, the relationship between the two processing steps has been unclear. The two reactions are easily uncoupled in vitro, and purification studies indicate that the activities that catalyze them are distinct (Christofori and Keller, 1988; Gilmartin et al., 1988; Kramer, 1988; Takagaki et al., 1988). In contrast, several reports have indicated that mutation of splicing signals alters polyadenylation both in vitro and in vivo (Villarreal and White, 1983; Niwa et al., 1990). Furthermore, polyadenylation sites only work within context. They are efficiently ignored when placed within vertebrate introns (Adami and Nevins, 1988; Brady and Wold, 1988; Levitt et al., 1989), and cause interrupted histone genes to polyadenylate at cryptic sites (Pandey and Marzluff, 1987; Pandey et al., 1990). These latter observations suggest that the two processing steps are related.
In this communication we report the kinetics of polyadenylation and splicing of an artificial gene consisting of two exons and a poly(A) site which efficiently splices and polyadenylates in vitro. Polyadenylation was observed first, and polyadenylated RNA was spliced efficiently. Some RNA was also spliced first; this RNA, however, appeared recalcitrant to polyadenylation, suggesting that the only successful pathway to the production of doubly-processed RNA required initial polyadenylation.
Materials and methods
Splicing reactions
MXSVL precursor RNA has been previously described and its reaction products characterized (Niwa et al., 1990). Unless indicated otherwise, processing reactions contained 1 mM creatine phosphate; 0.25 mM ATP; 1.0 mM 3′-dATP; 1.5 mM MgCl2; 0.37 mM dithiothreitol; 0.9% polyethylene glycol; 44 mM KC1; 8.8 mM TrisCl, pH 7.9; 8.8% glycerol; 0.2 mM EDTA; and 44% nuclear extract. RNAs were extracted after reaction and analyzed as previously described (Robberson et al., 1990).
Quantitation of processing reactions
Polyacrylamide gels of RNA products from processing reactions were quantitated by scanning in a Betagen Betascope 603 Blot Analyzer. Amount of product produced was calculated from a knowledge of the specific activity of the 32P-UTP labeled precursor RNA, U content of each RNA species, and the determined efficiency of counting in the instrument (15%). When wild-type and mutant substrates were compared, substrates were carefully prepared to the same specific activity and reacted using the same reaction/extract mix. Polyadenylation and splicing rates were identical over at least a 20-fold concentration of precursor RNA, indicating that reactions were performed under non-saturating conditions. Precursor RNAs both react and are degraded in processing extracts. Analysis of total counts for each substrate during reaction indicated that each of the precursors used in this study had a similar half-life in extract. Rates were calculated from early time points in the reaction before product decay contributed significantly to disappearance of total counts.
Two dimensional electrophoresis
Analysis for complexes was performed on native RNP gels (Zillmann et al., 1988). Reaction samples were mixed with a stop solution of final concentration 2.4 mg/ml heparin; 5% glycerol, .02% bromphenol blue. Samples were directly loaded onto a horizontal gel consisting of 3.0% Nusieve agarose in 25 mM Tris, 25 mM glycine, pH 8.8. Gels were run at 10 volts/cm for 4 hrs at 4°C. To analyze RNA content of individual RNP gel bands, the gel was sliced into 2.5 mm slices. Each slice was melted at 65°C and extracted once with phenol. The aqueous phase was ethanol precipitated with carrier glycogen, and analyzed for RNA content on denaturing acrylamide gels containing 8M urea.
Results
To examine the interaction of polyadenylation and splicing factors during pre-messenger RNA processing, we constructed precursor RNAs that contained both splicing and polyadenylation signals (Fig. 1). The chimeric DNA used for most of the experiments in this study (MXSVL) consists of 217 nucleotides from the major late transcription unit of adenovirus containing two exons and a single intron (MX) fused within exon 2 to a standard polyadenylation cassette of 196 nucleotides from the late transcription unit of SV40 (SVL). Precursor RNAs made from only the splicing or polyadenylation portion of the chimeric gene splice or polyadenylate well, respectively, in in vitro HeLa cell extracts (Sperry and Berget, 1986; Zillmann et al., 1988). They were chosen for this study because of both their high in vitro activity and the available wealth of information about their behavior in vitro. The resulting chimeric RNA polyadenylates and splices efficiently (Niwa et al., 1990).
Figure 1.

Possible pathways of splicing and polyadenylation of chimeric precursor RNAs. Dark boxes and hatched boxes correspond to sequences from the human adenovirus major late transcription unit and SV40 late transcription unit, respectively.
In vitro splicing and polyadenylation are routinely performed using the same nuclear extract but under slightly different conditions. Splicing assays contain higher magnesium (1.5–2.5 mM versus 0–1.0 mM), and salt. We began our investigation using a set of conditions representing a compromise between the two maxima (1.5 mM MgCl2; 44 mM KC1). In addition, to facilitate observation of cleavage products without interfering poly(A)-addition, ATP was replaced with 3′-dATP (cordycepin). In the presence of cordycepin, polyadenylation cleavage occurs, and poly(A)-addition is terminated after the addition of a single A residue (Jacob and Rose, 1983; Sheets et al., 1987). We will refer to molecules that have undergone single A-addition as being polyadenylated. Control reactions indicated that replacement of ATP with 3′-dATP had no effect on splicing efficiency in templates lacking poly(A) sites (data not shown).
Polyadenylation precedes splicing
Theoretically, a chimeric RNA could polyadenylate or splice in the first step to produce either a polyadenylated, but not spliced (A+ S−) intermediate RNA or a spliced, but not polyadenylated (S+ A−) intermediate RNA, respectively (Fig. 1). In a second step, either intermediate could be converted to spliced and polyadenylated (S+ A+) product RNA. We designate these RNA species as A+ S−, S+A−, and S+ A+ in this communication. The chimeric MXSVL substrate both spliced and polyadenylated (Fig. 2A). A reaction product band of the correct molecular weight to be spliced and polyadenylated RNA (S+ A+) was observed after 20 minutes of incubation. Mapping of the 3′ terminus using complementary SV40 probes indicated that correct cleavage had occurred at the poly(A)-addition site (Niwa et al., 1990). Production of S+ A+ RNA indicated that both reactions could occur in a single RNA molecule. Two RNA species, corresponding in molecular weight to A+ S− and S+ A− intermediates, also appeared during the reaction. A+ S− RNA was detected first, as early as 8 minutes of incubation. S+A− RNA appeared later, at approximately the same time as the doubly processed S+ A+ RNA.
Figure 2.
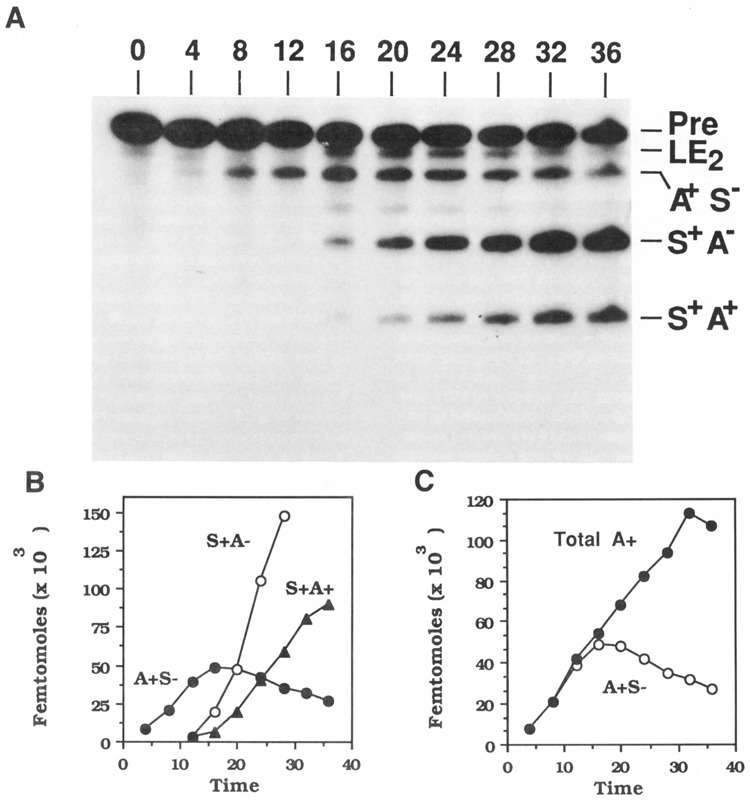
Time course of splicing of MXSVL RNA. MXSVL precursor RNA was incubated in in vitro processing reactions for the indicated times, prepared, and displayed on a 5% denaturing acrylamide gel. A. Short time course. Splicing and polyadenylation product bands are identified as on Figure 1B at various times of reaction (minutes). B. Quantitation of intermediate and product bands. The gel in A was scanned in a Betagen 603 gel analyzer, and radioactivity was converted to fmoles product and plotted versus time of reaction. Product species are indicated: A+S−, filled circles; S + A−, open circles; S + A +, triangles. LE2 is lariat-exon 2. Released lariat and exon 1 are off the bottom of the gel. C. Quantitation of the total amount of polyadenylation. The total fmoles of polyadenylated RNA was calculated as the sum of the A + S− and S + A+ RNA (filled circles) and plotted versus time of reaction and with the amount of A + S− RNA (open circles).
The appearance of both S+ A− and A+ S− RNAs suggested that either could be an intermediate in the production of fully processed S+ A+ product RNA. A+S− RNA consistently appeared before S+ A− RNA. Analysis of the kinetics of reaction indicated that the A+ S− RNA behaved as an intermediate. Amounts of A+S− RNA rose quickly and then leveled off, implying that it reached steady-state. At later times, as S+A+ RNA began to increase, the levels of A+S− RNA began to decrease, suggesting that the population of A+S− RNA was being depleted by conversion to other species.
To quantitate the kinetics of reaction, gels were scanned in a Betagen Betascope 603 Blot Analyzer. A plot of product versus time is shown in Figure 2B. The amount of S+ A+ RNA began to increase just as the amounts of A+ S− RNA leveled off, supporting a precursor-product relationship between the two RNAs. When the total amount of polyadenylated RNA (the sum of the A+ S− and S+ A+ RNAs) was plotted versus time (Fig. 2C), it became apparent that the rate of polyadenylation remained linear during the course of the reaction, and that the rate of appearance of S+ A+ RNA compensated for the decrease in production of A+ S− RNA at later times. These relative kinetics of appearance of A+ S− and S+ A+ RNAs again supports a precursor-product relationship between the two species of RNA.
The spliced, but not polyadenylated (S+ A−) intermediate, on the other hand, continued to increase in amount during incubation, suggesting that spliced RNAs were not precursors to spliced and polyadenylated S+ A+ RNA. The observation of a single linear rate for polyadenylation (Fig. 2C) also suggests that there was only one precursor pool for polyadenylation. Therefore, it seems likely that the bulk of the S+ A− RNA was not competent for polyadenylation.
In an attempt to establish a precursor-product relationship between the two intermediate RNAs and the final S+ A+ RNA, several experimental approaches were tried. In none of these did S+ A− RNA behave as an intermediate. We first examined the behavior of S+ A− RNA during long incubations (Fig. 3A). Incubation for over one hour caused complete conversion of input precursor RNA to processed RNA. Polyadenylated intermediate, A+ S− RNA, was also completely converted to other species. S+ A− RNA remained at late times, however. There was no net conversion of S+ A− RNA to S+ A+ RNA. Instead, both RNAs were slowly degraded as incubation continued. The amount of S + A− RNA remained constant even if fresh extract was added after 60 minutes (data not shown), indicating that incomplete conversion was not a result of loss of polyadenylation activity of the extract after extended incubation.
Figure 3.

(opposite). Properties of S + A− RNA. A. Extended time course. MXSVL was incubated for the indicated times (hours). RNA species are as in Figure 1B. C. Reintroduction of S + A− RNA into extract. A standard processing reaction of MXSVL was performed for 30 minutes. S + A− RNA intermediate was located by auto radiography of an RNA gel. RNA was extracted from the gel and reintroduced into a standard processing reaction (S + A− lanes). The resulting product polyadenylated RNA is indicated with an A +. As a control, SVL precursor RNA containing no splicing signals and all of the SV40 polyadenylation signals present in MXSVL was isolated from a gel of a 30-minute time point and reintroduced into extract to monitor its level of polyadenylation and compare it to that of the reintroduced S + A− RNA. Product polyadenylated RNA is indicated with an A+ (SVL lanes). A standard processing reaction of MXSVL (MXSVL lanes) is added for band identification. Structures of the utilized precursor RNAs are shown in B.
The above observation suggested that S+ A− RNA was a dead-end product. To ascertain if inhibition was a property of the RNA, S+ A− intermediate RNA was isolated from a gel and added back to fresh extract to see if it could be polyadenylated under these conditions (Fig. 3B and C). S+ A− RNA contains no intron-located splicing signals, but it does contain all normal polyadenylation signals. Therefore, we compared the polyadenylation activity of the gel-isolated S + A− RNA to that of a standard SVL polyadenylation precursor RNA containing essentially the same SV40 sequences as MXSVL. This RNA was also isolated from a gel of a 30-minute standard reaction prior to reintroduction into extract. The reintroduced S+ A− RNA was polyadenylated with kinetics similar to that of the SVL RNA. We conclude that the lack of conversion of S + A − RNA to product polyadenylated S + A+ RNA was not an inherent property of the RNA, but rather some property of the processing reaction occurring on chimeric templates.
Splicing requires magnesium; polyadenylation does not (Moore and Sharp, 1985). To ask if S + A− RNA could be chased into S + A+ RNA if splicing was prevented, reactions were performed in which EDTA was added after 30 minutes. Under these conditions, no further splicing occurred after the addition of the EDTA. Although precursor RNA continued to polyadenylate after the addition, no net conversion of S + A− RNA to S + A+ RNA occurred (data not shown), again suggesting that the S + A− RNA was recalcitrant to polyadenylation.
Magnesium concentration influences the pathway of processing of chimeric RNAs
The relative kinetics of appearance of the two spliced species, S + A− RNA and S + A+ RNA, was dependent upon the magnesium concentration in the assay (Fig. 4A). At low magnesium (0–0.5 mM), the major spliced species was A+S + RNA. Less S + A− RNA was created under these conditions. At intermediate magnesium concentrations (1.0 mM), both spliced RNAs appeared at roughly equal amounts. At higher magnesium, splicing produced mostly the S + A− RNA species. Total polyadenylation activity was greater at low magnesium. Thus, under conditions favoring polyadenylation, more splicing occurred via the branch of the pathway in which polyadenylation was the initial reaction of precursor RNA.
Figure 4.



Effect of magnesium concentration on processing of MXSVL RNA. A. Time course of reaction. MXSVL RNA was incubated under the listed final magnesium concentrations for the indicated times (minutes). RNA species are indicated. B. Quantitation of splicing at different magnesium concentrations. The amount of spliced RNA that was polyadenylated (S +A+ RNA, open circles) or was not polyadenylated (S + A− RNA, filled circles) was calculated for the 30-minute time points from the gel in A and plotted as a function of magnesium concentrations. The ratio of the two values was also plotted (triangles). C. Effect of magnesium concentration on the processing of metallothionein precursor RNA. The structure of the precursor is diagrammed below the panel. Black boxes indicate natural exon sequences from the murine metallothionein II gene. The entire 143 nucleotide second intron is included in the transcript. This precursor is less active for processing than MXSVL. The reaction in C included increased concentrations of polyethylene glycol (2.5% final) to aid in visualizing activity.
The amount of product RNA formed in the gel in Figure 4 was quantitated by scanning the gel. The amounts of spliced S + A− RNA (Fig. 4B, closed circles) and spliced and polyadenylated S + A+ RNA (Fig. 4B, open circles) formed at 30 minutes were calculated and plotted versus magnesium concentration. At low magnesium concentrations, more S + A+ RNA was observed than S + A− RNA. Thus, more RNA was being spliced by that branch of the pathway involving initial polyadenylation than by that branch of the pathway involving direct splicing of unreacted precursor RNA. This splicing preference was not caused by complete conversion of precursor RNA to polyadenylated RNA at low magnesium. At 30 minutes, when splicing was occurring at maximal rate, considerable unprocessed RNA remained at all magnesium concentrations. Thus, there was a rate enhancement for splicing to produce S + A + RNA at low magnesium. The ratio of S + A + /S + A− RNA is also plotted in Figure 4B (triangles). Five-fold more precursor RNA was spliced by that branch of the pathway involving initial polyadenylation, indicating a five-fold difference in the rate of the two splicing reactions at low magnesium.
MXSVL consists of a cap proximal intron fused to the last several hundred nucleotides of the SV40 late transcription unit. Thus, although the polyadenylation portion of the last exon was derived from a 3′ terminal exon, the intron was not a 3′ terminal intron. We also examined splicing and polyadenylation from a natural last intron and exon in vitro. We used the three exon mouse metallothionein II gene fused to an SP6 promoter within exon 2 (Fig. 4C). The precursor RNA made from this construct both spliced and polyadenylated in vitro, although at a lower rate than the MXSVL precursor RNA. It produced the same intermediate RNAs in the same ratios as did MXSVL RNA. Furthermore, it had an identical response to magnesium concentration as MXSVL, in which splicing via polyadenylation occurred at a higher rate than splicing of unreacted precursor RNA at low magnesium. Thus the preference for initial polyadenylation was observed both with precursor RNAs from natural genes and from artificial chimeras.
S+A− RNA accumulates in unique RNP complexes
The above results suggested that S + A− RNA was not an intermediate in the production of S +A+ RNA, suggesting that when splicing occurred first, polyadenylation was somehow inhibited. Because S + A− RNA isolated from a reaction and reintroduced into extract polyadenylated efficiently, the lack of polyadenylation must have been the result of the reaction rather than an inherent property of the RNA. It also suggested that S + A− RNA might have improperly associated with factors in the extract. To examine complexes formed during reaction of chimeric RNA, reactions of MXSVL were analyzed by native gel electrophoresis (Fig. 5A). Multiple complexes quickly formed on MXSVL precursor RNA which presumably represent association of both splicing and polyadenylation factors with this precursor RNA. In addition to the early complexes, two later complexes (denoted D and E in Fig. 5A) formed after 30 minutes of reaction and appeared with the kinetics of splicing. These two complexes exhibited faster mobility in the RNP gel than the specific complexes formed on unreacted precursor RNA (denoted A–C in Fig. 5A), suggesting that they were missing several factors compared to complexes appearing earlier.
Figure 5.

Complexes formed by MXSVL. A. Reactions of MXSVL RNA in the presence of cordycepin and 2.0 mM MgCl2 were analyzed by RNP gel electrophoresis (Materials and Methods) at the indicated times (min). Note that the early time points are out of order to facilitate complex comparison. Individual bands are denoted by letters. Noncomplexed RNA migrated off the bottom of the gel. The 50-minute time point lane was sliced, as indicated by the slice numbers to the right of the gel. B. RNA from each slice was isolated and displayed on a denaturing acrylamide gel. Lane numbers correspond to slice numbers in A. A total reaction from a 50-minute time point was loaded in the lane marked T; markers are in lane M. Individual RNA species are indicated. Because of the late time and high magnesium, S + A− RNA was the prominent product. A small amount of S + A+ doubly processed RNA was made (see lane T) and was broadly dispersed through the gel from slices 18 downward, presumably reflecting its release from complexes upon completion of both reactions.
To analyze which RNA species were in which bands, the lane from the 50-minute reaction in Figure 4A was sliced into 22 pieces as diagrammed. RNA was extracted from each slice and displayed on a denaturing acrylamide gel (Fig. 5B). S + A− RNA was located in two regions of the first dimensional RNA gel that corresponded to slices 13–14 and 19–21. These were the two regions of the RNP gel containing the complexes D and E. Therefore, it is likely that these two complexes represent assemblies containing S+A− RNA and nuclear factors. Their relatively faster migration compared to earlier complexes suggests that they are missing certain factors present on unreacted precursor RNA. Presumably it is the nature of these factors and their association that prevented polyadenylation of S + A− RNA.
Discussion
We have examined the kinetics of in vitro splicing and polyadenylation of simple precursor RNAs containing two exons, terminated by a poly(A) site. A product RNA that had been both spliced and polyadenylated was created by 20 minutes of reaction. Theoretically this RNA could have been created by a pathway that polyadenylated first or by an alternative pathway that spliced first. Although both reactions occurred, product RNA appeared to be created only by the pathway in which polyadenylation preceded splicing. Polyadenylated RNA appeared very quickly and reached steady-state by 20 minutes, at which time product polyadenylated and spliced RNA began to accumulate. Spliced but not polyadenylated RNA, in contrast, continued to accumulate and could not be chased into final product by increased time, fresh extract, or conditions that permitted polyadenylation but inhibited splicing. Therefore, only polyadenylated RNA appeared to be a precursor for final product. These results support older in vivo data suggesting that polyadenylation precedes splicing of adenoviral RNAs (Nevins and Darnell, 1978).
Spliced but not polyadenylated intermediate RNA accumulated in two complexes as revealed by native gel electrophoresis. Thus, following polyadenylation, the processed RNAs were not released as free RNAs. It is presumably the nature of these complexes which prevents polyadenylation of the spliced intermediate RNA. We and others have observed that splicing factors disassociate from spliced product RNA (Frendewey and Keller, 1985; Konarska and Sharp, 1986; Zillmann et al., 1988). We would anticipate that the complexes containing S + A− RNA, therefore, would contain only polyadenylation factors. The two observed S + A− complexes had gel mobilities similar to those of complexes assembled using SV40 late poly(A) site without splicing signals (Zarkower and Wickens, 1987). We do not yet know, however, if the standard polyadenylation factors are present on these complexes, or, if they are, why the complex is not able to polyadenylate.
We have previously reported that the presence of an intron accelerated the initial rate of polyadenylation of chimeric RNAs compared to precursors containing just a poly(A) site, or precursors containing an upstream intron with a mutated 3′ splice site; and have suggested that splicing and polyadenylation factors interact so as to recognize terminal exons (Niwa et al., 1990). The data in this communication supports that suggestion. Here we suggest that nuclear factors interact with terminal exons (and introns) so as to set them up for polyadenylation and subsequent splicing. Our data also raise the possibility that the splicing factors that recognize and process 3′ terminal introns are somewhat different from the factors that recognize internal introns.
The processing reactions in this report occurred using a pretranscribed RNA. In vivo, processing may be co-transcriptional. Polyadenylation has been reported to occur shortly after the polymerase passes the poly(A)-addition site (Nevins and Darnell, 1978). Splicing of capproximal introns also occurs before transcription is completed (Berget and Sharp, 1978; Beyer and Osheim, 1988). We do not know to what extent the kinetics that we have observed were influenced by the uncoupling of transcription and processing used in these studies. We hope to investigate this question using coupled in vitro reactions.
Acknowledgments
We thank Ed Murphy and the Department of Molecular and Tumor Biology for access to the Betagen Betascope, Dr. H. Gilbert for advice on kinetics, April Kilburn for constructing the metallothionein clones, and Becky Moore and Dixie Brewer for technical assistance. This work was supported by an American Cancer grant NP-695 and a Texas Advanced Technology Program award to S. M. B.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Adami G. and Nevins J. R. (1988), EMBO J 7, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget S. M. and Sharp P. A. (1978), J Mol Biol 129, 547–559. [DOI] [PubMed] [Google Scholar]
- Beyer A. and Ohsheim Y. N. (1988), Genes Dev 2, 754–765. [DOI] [PubMed] [Google Scholar]
- Brady H. A. and Wold W. S. M. (1988), Mol Cell Biol 8, 3291–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofori G. and Keller W. (1988), Cell 54, 875–889. [DOI] [PubMed] [Google Scholar]
- Frendewey D. and Keller W. (1985), Cell 42, 355–367. [DOI] [PubMed] [Google Scholar]
- Gilmartin G. M., McDevitt M. A., and Nevins J. R. (1988), Genes Dev 2, 578–587. [DOI] [PubMed] [Google Scholar]
- Jacob S. T. and Rose K. M. (1983), in Enzymes of Nucleic Acid Synthesis and Modification, vol 2 (Jacob S. T., ed.), CRC Press Inc., Boca Raton, FL, pp. 35–157. [Google Scholar]
- Kramer A. (1988), Genes Dev 2, 1155–1167. [DOI] [PubMed] [Google Scholar]
- Konarska M. M. and Sharp P. A. (1986), Cell 46, 845–855. [DOI] [PubMed] [Google Scholar]
- Levitt N., Briggs D., Gil A., and Proudfoot N. J. (1989), Genes Dev 3, 1019–1025. [DOI] [PubMed] [Google Scholar]
- Moore C. L. and Sharp P. A. (1985), Cell 41, 845–855. [DOI] [PubMed] [Google Scholar]
- Nevins J. R. and Darnell J. E. Jr. (1978), Cell 15, 1477–1487. [DOI] [PubMed] [Google Scholar]
- Niwa M., Rose S. D., and Berget S. M. (1990), Genes Dev 4, 1552–1559. [DOI] [PubMed] [Google Scholar]
- Pandey N. B. and Marzluff W. F. (1987), Mol Cell Biol 7, 4557–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey N. B., Chodchoy N., Liu T.-J., and Marzluff W. E. (1990), Nucl Acids Res 18, 3161–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberson B. L., Cote G., and Berget S. M. (1990), Mol Cell Biol 10, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets M. D., Stephenson P., and Wickens M. P. (1987), Mol Cell Biol 7, 1518–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry A. O. and Berget S. M. (1986), Mol Cell Biol 6, 4734–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y., Ryner L. C., and Manley J. L. (1988), Cell 52, 731–742. [DOI] [PubMed] [Google Scholar]
- Villarreal L. P. and White R. T. (1983), Mol Cell Biol 3, 1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkower D. and Wickens M. (1987), EMBO J 6, 4185–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillmann M., Zapp M. L., and Berget S. M. (1988), Mol Cell Biol 8, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]