

Abstract
Previous studies designed to map the transcriptional regulatory sequences of the human immunodeficiency virus (HIV) long terminal repeat (LTR) have shown disparate results depending on the method of analysis. Experiments have shown that deletions 5′ to −104 (relative to the transcription start site, +1) are not required for transcription in vitro, while other experiments have shown that various mutations in this 5′ region of the HIV-1 LTR affect both reporter gene activity in transient expression systems and viral growth. To correlate in vitro and in vivo findings, we performed in vitro transcription competition studies to define minimal sequences necessary for competitive factor binding or competitive transcription complex formation. Using normal HeLa cell nuclear extracts, we found that transcription of a reporter gene ru n by the U3-R region was efficiently competed only by intact LTR DNA fragments representing virtually the entire U3-R region (−453 to +80). Smaller subfragments of the LTR were less effective competitors; these included fragments from −453 to −159, which had a modest competitive ability at higher competitor concentrations, −159 to +80, and −402 to −34, which were both relatively poor competitors. These findings indicate that although the U3-R region truncated to −104 is able to promote in vitro transcription, a more stable transcription complex appears to form on the entire U3-R region. Hence sequences between −453 and −104 appear to be significant in transcription complex formation. In vivo transfection competition studies confirmed these findings. Specific sequences between −453 and −104 which may affect expression or transcription complex formation were mapped using a set of linker-scanning mutants spanning the LTR. Transient transfection analysis of the transcriptional activity of the mutants not only confirmed the location of the well known promoter elements in the 3′ end of the LTR, but also identified three broad regions in the 5′ end (−453 to −328, −273 to −220, and −180 to −130) that reduced LTR-driven expression 20 to 50% when mutated. Thus multiple elements in the 5′ end of the LTR appear to cooperate with elements in the 3′ half to mediate the formation of the wild-type transcription complex.
The mechanisms used by HIV to regulate transcription from its long terminal repeat (LTR) govern the progress of the productive infection and appear to control the establishment and duration of the latent state (for review see Varmus, 1988; Jones, 1989; Pavlakis and Felber, 1990). Using a variety of experimental approaches, a number of groups have defined important cis-acting regulatory elements. These include the TAR region, the TATA box, the Spl binding sequences, the NF-kappa direct repeat sequences, a negative regulatory element (NRE), an AP1 binding consensus sequence and sequences homologous to elements found in the promoters of the IL-2 and IL-2 receptor genes (Arya et al., 1985; Jakobovits et al., 1988; Jones et al., 1988; Muessing et al., 1987; Okamoto and Wong-Staal, 1986; Patarca et al., 1987; Peterlin et al., 1986; Rice and Matthews, 1988; Rosen et al., 1985; Sodroski et al., 1985a; Sodroski et al., 1985b; Jakobovits et al., 1988; Rosen et al., 1985; Jones et al., 1986; Bohnlein et al., 1988; Franza et al., 1987; Kaufman et al., 1987; Nabel and Baltimore, 1987; Tong-Starksen et al., 1987; Garcia et al., 1987; Rosen et al., 1985; Siekevitz et al., 1987; Franza et al., 1988). Such a variety of elements, which respond to both viral and cellular factors, suggest a complex pattern of transcriptional control at several discernible levels. Some of the elements are needed for basal transcription, while others are believed to be involved in the regulation of latency or in high level expression during productive infection (Dinter et al., 1987; Rosen et al., 1985; Siekevitz et al., 1987; Kaufman et al., 1987; Nabel and Baltimore, 1987; Nabel et al., 1988; Tong-Starksen et al., 1987; Arya et al., 1985; Dayton et al., 1986; Jakobovits et al., 1988; Muessing et al., 1987; Okamoto and Wong-Staal, 1986; Peterlin et al., 1986; Rice and Matthews, 1988; Siekevitz et al., 1987; Sodroski et al., 1985a; Sodroski et al., 1985b; Orchard et al., 1990; Franza et al., 1988; Smith and Greene, 1989; Lu et al., 1989, 1990). The number of possible interactions between factors, and between factors and DNA, provide potential for the formation of a variety of different transcriptional complexes on the LTR. Formation and alteration of specific complexes may mediate the various phases of the viral infection.
Despite the many potential regulatory elements and binding sites identified in the 5′ half of the LTR several studies have indicated that sequences upstream of −104 in the HIV-LTR (see Fig. 1; the transcriptional start site is +1) are not necessary for transcription in vitro (Dinter et al., 1987; Patarca et al., 1987; our unpublished observations). However, in vivo transfection analysis of similar mutants has, in some cases, indicated that transcriptional functions may be mediated by these upstream sequences; for example, a three- to four-fold increase in transcriptional activity was noted when these sequences were deleted (Rosen et al., 1985). This disparity suggests that effects of sequences upstream of −104 may not be detected by standard in vitro transcription analyses. Thus, the complexity of the LTR may be greater than that defined by the in vitro transcription activity of deletion mutants.
Figure 1.
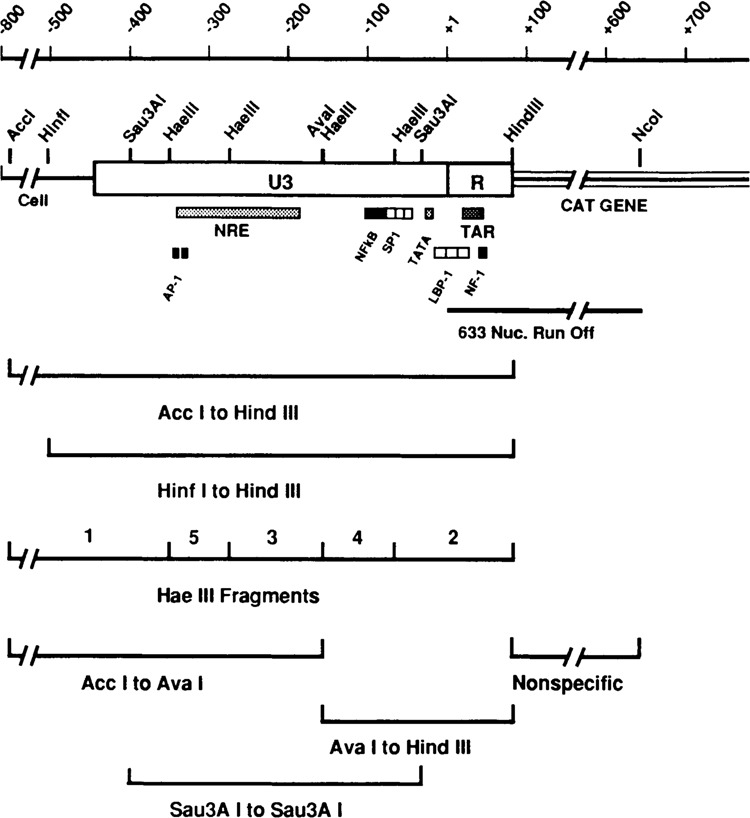
Map of the HIV LTR region of p5′ LTR-CAT and location of the fragments used for competitors of in vitro transcription. The diagram shows the U3 and R regions with the adjacent bacterial chloramphenicol acetyl transferase gene and immediate flanking sequences on the 5′ side. The Acc I and Hind III sites, used for the excision of the major competitor fragment, are indicated. In addition, other important restriction sites are shown by letters: A, Ava I; B, BstN I; H, Hae III; I, Hinf I; P, Pvu II; S, Sau3A I. The location of many of the identified cisacting regulatory sequences are shown below the LTR diagram. The exact nucleotide positions of the restriction sites and the cisacting elements can be found in Materials and Methods. The bottom part of the figure shows the location of the various competitor fragments used in the experiments; all of these fragments were derived from the Acc I-Hind III fragment (see Materials and Methods).
To examine this possibility we used in vitro transcription competition analyses instead of deletion analysis to define the regions of the LTR necessary for basal transcriptional activity or basal transcription complex formation. Previous studies have shown the in vitro transcription competition analyses can furnish insight into promoter structure and transcription complex formation which may not be detected by deletion analysis (McStay and Reeder, 1986). In addition, competition analysis has demonstrated the existence of cis-acting elements in promoters from viruses such as cytomegalovirus, SV40, and pseudorabies virus (Ghazal et al., 1988; Ghazal et al., 1987; Sassone-Corsi et al., 1985).
The in vitro and in vivo transcription competition results presented below suggest that stable transcription complex formation on the known promoter elements of the 3′ half of U3-R (−158 to +80) is significantly aided by the sequences of the 5′ half of the LTR (−453 to −159). This indication of significance of the 5′ half of U3-R in transcription activity was confirmed by determining the effect on expression of a set of linker scanning mutants which extend across the U3-R region. Between −453 and −130, three regions were found in which mutations significantly lowered transcriptional activity of the LTR in HeLa cells.
Materials and methods
Plasmid construction
The 5371 base pair plasmid, p5′LTR-CAT, was the gift of L. Bachelor, E.I. DuPont de Nemours & Co. It was constructed by inserting the Hpa II-Hind III fragment of the HIV infectious pro-viral clone pHXB2-gpt (Ratner et al., 1987; Shaw et al., 1984) into pSVO-CAT (Gorman et al., 1982) which had been cleaved with Pvu II and Hind III. The chloramphenicol acetyl transferase (CAT) gene coding sequences are immediately downstream from the R region of the 5′ HIV LTR (plus adjacent flanking sequences upstream of the LTR; see Figure 1). In transfected lymphocytic cells CAT gene expression from this plasmid is significantly increased in the presence of the tat protein and in response to phorbol ester and lectin stimulation (data not shown). The restriction sites used in our experiments were numbered relative to the transcription start site (+1): Acc I −796, Ava I −158, Hae III −352, −276, −159, −68; Hind III +80, Nco I +633, Sau3A I −402, −34.
Similar plasmid constructions were used for the transfection competition studies. The reporter plasmid for the transfection experiments, pXLTR-CAT, was derived from p5′LTR-CAT. In it the sequences flanking the LTR on the 5′ side, between Acc I and Hinf I (see Figure 1), are replaced by an Xba I linker. The Xba I to Hind III fragment of pXLTR-CAT, representing the intact LTR, was cloned into pGEM3Zf+, providing the intact LTR competitor plasmid, pGXHLTR. The 5′ half competitor plasmid, p5XX8, was made from a derivative of p5′LTR-CAT which had the Hinf I site replaced with an Xba I linker and the Ava I site replaced with an Xho I linker. Hence the 5′ half of the LTR could be removed as an Xba I to Xho I fragment (equivalent to the Hinf I to Ava I fragment of a WT LTR, Figure 1); this was cloned into pGEM7Zf−. The 3′ half competitor plasmid, p3X8H, was similarly constructed: the Xho I to Hind III fragment (equivalent to the Ava I to Hind III fragment of the WT LTR, Figure 1) was cloned into pGEM7Zf−.
The location of the described cis-acting regulatory sequences were: “Site A”: −379 to −367, “SiteB”: −350 to −327, API-like: −335 to −330, −341 to −348; NRE: −342 to −187; IL2 and IL2 receptor promoter homologies: −274 to −256, −253 to −213, −170 to −160, −221 to −203; “Negative Regulatory Factor”: −173 to −159, NF-kappa B: −105 to −93, −91 to −81; Spl: − 78 to − 69, − 67 to − 58, − 56 to − 47; TATA: −27 to −23; Initiator −6 to +5, LBP −1: −17 to + 27, UBP−1: −18 to +23;TAR: +19 to +42 (Arya et al., 1985; Bohnlein et al., 1988; Franza et al., 1987, 1988; Garcia et al., 1987; Jakobovits et al., 1988; Jones et al., 1986, 1988; Harrich et al., 1989; Kaufman et al., 1987; Lu et al., 1990; Muessing et al., 1987; Nabel and Baltimore, 1987; Okamoto and Wong-Staal, 1986; Okamoto, et al., 1986; Orchard et al., 1990; Peterlin et al., 1986; Rosen et al., 1985; Shaw et al., 1988; Siekevitz et al., 1987; Sodroski et al., 1985a; Smith and Green, 1989; Sodroski et al., 1985b; Tong-Starksen et al., 1987; Wu et al., 1988a, 1988b).
Linker scanning mutants
The linker scanning mutants, which consecutively replaced 18 bp of wild type sequence with an Nde I-Xho I-Sal I polylinker CATATGCTC-GAGGTCGAC across the U3 and R regions, were based on pXLTR-CAT using a PCR-directed technique (Zaret et al., 1990). Briefly, two external primers, common to all mutants, were synthesized; one was complementary to sequences 5′ of the Xba I site of pXLTR-CAT (in plasmid sequences) and the other complementary to sequences on the opposite strand located 3′ of the Hind III site (in the CAT coding sequence). For each mutant, a specific set of internal primers were synthesized. The primer for the 5′ half product included CGC, 12 bases making up Nde I and Xho I sites and 17 bases of wild type coding sequence. The primer for the 3′ half product included CGC, 12 bases making up an Xho I site and a Sal I site and 17 bases complementary to wild type sequence. Using the matched external and internal primers, the two half-products were synthesized via the PCR reaction. The 5′ half product was cleaved with Xba I and Xho I; the 3′ half product was cleaved with Xho I and Hind III. Each was gel purified. The two fragments were ligated, cut with Xba I and Hind III, and the products were separated on poly-acrylamide gels. The appropriate dimer containing both 5′ and 3′ halves was gel isolated and ligated into pXLTR CAT in place of the wild type LTR sequences between the Hind III and Xba I sites. The mutations were verified by sequencing.
In vitro transcription and competition
Three liters of spinner-adapted HeLa cells were grown to log phase (106 cells/ml) in Joklik’s medium supplemented with 10% horse serum (Gibco), pen-strep and 10 mM Hepes pH 7.4. The cells were extracted using the procedure of Shapiro et al. (1988). After extraction the final protein concentrations were 12–22 μg/μl in nuclear dialysis buffer (Shapiro et al., 1988).
In vitro transcription reaction volumes were 20 gl containing approximately 50 gg of protein and 400 ng of template DNA (Nco I linearized p5′LTR-CAT). The in vitro system was optimized for extract and template concentrations (not shown). For competition experiments, the competitor fragment was ethanol precipitated, dissolved in TE (10 mM Tris-HCl, pH 7.5, 1 mM EDTA), and added to the reaction mix along with the template fragment and prior to the addition of the extract. Each reaction also contained 1% polyvinyl alcohol, 30 mM KC1, 3.8 μM MgCl2, 80 μM ATP, 80 μM CTP, 80 μM GTP, 5 μM UTP, 4 mM creatine phosphate, 2 μCi 32P-UTP (Amersham, specific activity 3000 Ci/mmol). Reactions were incubated for 40 min. at 30°C. At the end of the incubation 20 μl stop buffer (Shapiro et al., 1988) was added, and the reaction mixes were extracted once with phenol/chloroform/isoamyl alcohol (50:49:1) and ethanol precipitated. Precipitates were dissolved in formamide and analyzed by electrophoresis on 4% polyacrylamide gels containing 8M Urea and 0.5 X TBE (45 mM Tris-borate, 45 mM boric acid, 1 mM EDTA, pH 8.5). Gels were run at 26 V/cm for 3 h; under these conditions RNA or markers less than 370 nucleotides ran off the gel. Gels were dried and autoradiographed. Molecular weight markers were end-labeled 123 base pair DNA ladders (BRL).
Preparation of competitor fragments
The Acc I to Hind III fragment was cleaved from p5′LTR-CAT and purified on a 1.5% preparative agarose gel. The fragment was eluted using the DEAE-nitrocellulose procedure (Schleicher and Schuell). The Acc I to Hind III fragment was prepared in large amounts, since it served as the starting material for the preparation of all other competitor fragments. To make sub-fragments, purified Acc I-Hind III fragment was cleaved with a specific enzyme, and smaller fragments were isolated on polyacrylamide gels. The fragments were electroeluted, purified, and stored in TE. The nonspecific competitor fragment was the 553 base pair Hind III to Nco I fragment from the CAT gene coding region of p5′LTR-CAT with Hind III and Nco I. Competitor fragments were quantitated spectrophotometrically.
Transfection competition experiments
The established human T cell line, Jurkat, was used for transfection experiments. Cells were transfected by the DEAE-dextran procedure. Briefly, cells were grown in suspension culture in RPMI1640 medium supplemented with 10% FCS at 37°C in 5% CO2. Cells in log phase (approximately 106 cells/ml) were removed from culture and washed with tris-buffered saline (TBS; 25 mM Tris-HCl, pH 7.4, 137 mM NaCl, 5 mM KC1, 1 mM CaCl2, 1 mM MgCl2, 0.7 mM Na2HPO4). Aliquots of 107 cells were incubated at room temperature for 30 min in 600 μL of transfection mixture: TBS containing 500 μg/ml DEAE-dextran, 0.5 μg pXLTR-CAT and varied amounts of competitor plasmid ranging from 2 to 16 μg (corresponding to molar excesses between 5− to 40-fold). Cells were subsequently washed with TBS, diluted in fresh medium, and grown as described above. In some experiments Jurkat cells were stimulated 24 hours after transfection by the addition of phytohaemagglutinin (PHA) and phorbol 12-myristate acetate (PMA) at concentrations of 2 μg/ml and 10 μg/ml, respectively. All cells were harvested 48 hrs after transfection, washed with TBS, resuspended in 100 μl 250 mM Tris-HCl, pH 7.5 and prepared for CAT assay (Gorman et al., 1982). CAT assay results were quantitated by scanning TLC plates using a Molecular Dynamics Phosphoimager. Experiments were repeated at least six times to verify the consistency of the results. Transfection efficiency variations were determined by quantitating the amount of reporter plasmid, pXLTR-CAT, in the transfected cells at the time of harvest, as previously described (Alwine, 1985).
Transfection analysis of linker scanning mutants
Triplicate one μg quantities of the wild type plasmid and one μg of each linker-scanning mutant were transfected into 60 mm dishes of HeLa cells with DOTMA, as described (Felgner et al., 1987). The results presented represent 3 to 6 separate experiments. CAT activity was assayed 48 h after transfection by the TLC method (Gorman et al., 1982). Unacetylated and acetylated chloramphenicol was quantitated using a Molecular Dynamics Phosphoimager. Results are expressed as CAT activity of each mutant plasmid divided by wild type CAT activity.
Results
Experimental system and design for the in vitro studies
The in vitro transcription system used was normal HeLa cell nuclear extracts. The transcription template contained the U3 and R regions of the LTR (Fig. 1; see also Materials and Methods), producing a 633 nucleotide run-off transcript when cut at the Ncol site in CAT. Transcriptional activity from this template was competed using a variety of LTR competitor fragments (Fig. 1). At relatively low molar ratios of competitor to template, an efficient competitor interacts with transcription factors, preventing them from forming active transcription complexes on the template. Thus efficient competition by specific LTR fragments indicates minimal amounts of the LTR necessary (1) to define interactions by factors present in limiting amounts or (2) to define the smallest region necessary for the formation of a transcription complex capable of competing with the template LTR.
Competition by an intact LTR
The 875 nucleotide Acc I to Hind III fragment (−796 to + 80, relative to the transcription start site +1; Fig. 1) was used as an LTR self-competitor, since it represents the entire LTR plus some 5′-flanking sequences. As shown in Figure 2A, the Acc I to Hind III fragment was a highly efficient competitor. Production of the 633 nucleotide run-off transcript from the transcription template was completely inhibited at less than a 7.5-fold molar excess of competitor to template (99% competition at 7.5 fold molar excess as measured by laser densitometry). The Acc I to Hind III competitor fragment is itself capable of producing an 80 nucleotide run-off transcript; however, under the electrophoretic conditions used, transcripts in this size range ran off the gels. A similar LTR competitor, the 593 base pair Hinf I to Hind III fragment (−514 to +80; Fig. 1), competed as well as the Acc I to Hind III fragment (Fig. 3). This fragment removes the majority of the sequences flanking the LTR on the 5′ side. A nonspecific competitor (553 base pairs of CAT gene coding sequence, Fig. 1) had no effect on specific transcription at 7.5-fold molar excess (Fig. 2A), or at much higher molar excesses (e.g., 30-fold excess, Fig. 3). Similar results were obtained using several different extract and competitor DNA fragment preparations.
Figure 2.
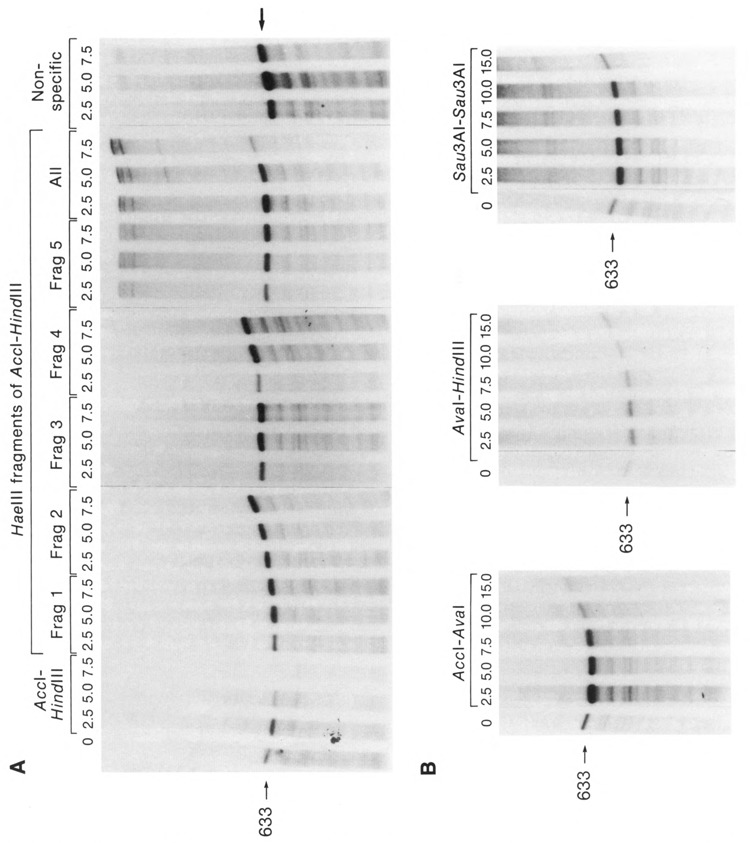
In vitro transcription competition. Various molar ratios (competitor to transcription template) of the indicated LTR or nonspecific fragments were used to compete for transcription from a constant amount of the LTR transcription template (Nco I linearized p5′LTR-CAT). The transcription template produces the 633 nucleotide run-off transcript indicated. A. Competitors used (see also Figure 1): the intact LTR competitor, Acc I to Hind III; the fragments 1 through 5 formed by cleavage of the Acc I to Hind III fragment with Hae III; the nonspecific competitor made up of 593 bp of CAT gene sequences. The lane marked ALL shows the competition produced by the indicated molar excesses of all of the Hae III fragments mixed together in the reaction. B. Competitors which overlapped the Hae III sites; the Acc I to Ava I fragment which approximately represents the sequences of the 5′ half of the LTR; the Ava I to Hind III fragment which approximately represents the sequences of the 3′ half of the LTR; and the Sau3A I to Sau3A I fragment (all shown in Figure 1).
Figure 3.
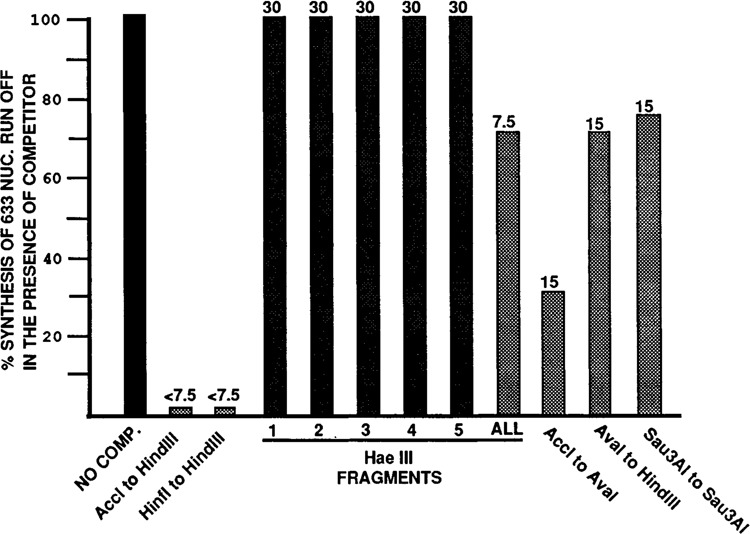
Summary of competition data. The bar graphs indicate the amount of specific 633 run-off transcript produced in the presence of the competitor indicated. All percentages are determined relative to the amount of transcript produced in the absence of competitor (No Comp.). As explained in the text, some fragments acted as nonspecific carrier DNA, causing increased synthesis of the 633 nuc. transcript. This is not represented in the bar graphs; values above 100% are presented as 100%. The numbers at the tops of the bars are the molar ratios (competitor to transcription template) at which the measurement was made. Results were very reproducible and represent the average data (± 10%) from two to three experiments using different extract and competitor preparations. The ALL lane indicates the competition when all the Hae III fragments were mixed for competition (see text). The nonspecific competitor is described in the text and the legend to Figure 2.
Competition by Hae III fragments of the LTR
The Acc I-Hind III fragment was cleaved with Hae III, generating the five Hae III fragments indicated in Figure 1 (specific restriction sites are given in Materials and Methods). As shown in Figure 1, these fragments isolate several of the identified cis-acting sequences within specific fragments. None of the individual Hae III fragments showed significant competition at 7.5-fold molar excess (Fig. 2A), nor at molar excesses as high as 30-fold (not shown). When the five Hae III fragments were added together to the reaction mix (“All” lanes, Fig. 2A), nominal competition (approximately 30%, see also Fig. 3) occurred at 7.5-fold molar excess of competitor to template. The individual Hae III fragments, as well as the non-specific competitor, actually caused a slight increase in the level of specific transcription when added to the reactions (Fig. 2A). This effect has been observed by others (Ghazal et al., 1988; Wildeman et al., 1984) and appears to be indicative of fragments which either have diminished or no competitive ability. Such fragments act similarly to carrier DNA, which is often added to in vitro transcription reactions to increase activity.
The results indicate that none of the individual Hae III fragments contain enough sequence to prevent transcription complex formation on the intact LTR of the transcription template. The finding that the combined fragments competed only nominally suggests that cis-acting sequences within two or more Hae III fragments must lie together on an intact piece of DNA in order to allow efficient factor binding capable of producing a competitive complex. Additionally, the inefficient competition of the combined fragments argues that the efficient competition of the intact Acc I to Hind III competitor is not due to simple mass effects (i.e., a 7.5-fold molar excess of Acc I to Hind III fragment cleaved with Hae III is a much less efficient competitor than a 7.5-fold molar excess of intact Acc I to Hind III fragment, Figure 2A and 3).
Competition with LTR fragments overlapping Hae III sites
In order to define the minimum sequences required for effective competition, and to rule out the possibility that Hae III cleavage disrupted important sites in the LTR, we prepared larger competitor fragments which spanned Hae III cleavage sites (Fig. 1). Cleaving the Acc I-Hind III fragment with Ava I separates the 3′ half of the LTR (−158 to +80), containing all the well defined cis-acting elements, from the 5′ half, containing the negative regulatory element (NRE) the binding sites for AP1, “Site A” and “Site B”, the NFAT-1 binding site, and sequences homologous to those observed in the IL2 and IL2 receptor promoter. Both subfragments (Acc I to Ava I and Ava I to Hind III) had some competitive ability at a 15-fold molar excess of competitor (Figs. 2B and 3); however, neither inhibited transcription as well as the intact LTR competitor (Acc I to Hind III). Each Ava I sub-fragment competitor actually caused increased specific transcription at low molar ratios of competitor/template (2.5 to 5.0). This effect is seen with nonspecific or carrier DNA, as discussed above. However, at higher molar ratios the level of specific transcription decreases. Such decreases are not seen with the true nonspecific competitor (Figs. 2A and 3). These data indicate that specific competition prevails at higher concentrations of the Ava I subfragment competitors. This effect is clearly seen at a molar ratio of 15 for the Acc I to Ava I fragment which reproducably caused a 60% to 70% drop in specific transcription compared to the control with no competitor (i.e., 30% of the 633 nucleotide transcript remains, Figs. 2B and 3). The Ava I to Hind III competitor was less effective. We estimate from the results of several experiments that it causes no more than a 30% decrease in specific transcription at a 15-fold molar excess (i.e., 70% of the 633 nucleotide transcript remained, Fig. 3). The simultaneous presence of both Ava I subfragments in the reaction mix did not significantly increase the level of competition over that of the Acc I to Ava I fragment alone (not shown).
The Sau3A I to Sau3A I fragment of the LTR (Fig. 1) was also used as a competitor, because it spans the Ava I site and a Hae III site which was not spanned by the Ava I subfragments. This fragment also showed much less competitive efficiency compared with the intact LTR competitor (the Acc I to Hind III fragment). In repeated experiments only a 20% to 30% decrease in specific transcription could be detected at a 15-fold excess of competitor to template (Figs. 2B and 3). These data support the argument that the lack of competition by the Hae III fragments was not due to cleavage at sites essential for factor-DNA interactions.
Although the 3′ end LTR sequences, from Ava I to Hind III, are sufficient for transcription in vitro (Dinter et al., 1987; Patarca et al., 1987; our unpublished observations), our in vitro transcription competition data suggest that transcription complex formation on these sequences cannot compete successfully with transcription complex formation on an intact LTR. This observation combined with the moderate competive ability of Acc I-Ava I fragment strongly suggests that sequences within the 5′ half of the LTR (from the Ava I site to the 5′ end of the LTR), contribute to the formation of transcription complexes for in vitro transcription in normal HeLa cell extracts. It could be argued that the 3′ end LTR fragment (Ava I to Hind III) may not be a good competitor due to end effects, i.e., DNA of any kind may be required on its 5′ side to form a stable complex. We have added up to 700 base pairs of nonspecific sequences to the 5′ side of the Ava I to Hind III competitor and detected only a marginal increase in competitive ability (not shown).
In vivo competition experiments
In order to validate that the in vitro transcription competition results were not an in vitro artifact, we duplicated selected experiments using transfection competition experiments. The reporter plasmid pXLTR-CAT, containing the intact LTR running the CAT gene (see Materials and Methods), was transfected into Jurkat cells. The activity of the LTR was measured by chloramphenicol acetyltransferase (CAT) activity. As competitors, the intact LTR fragment (Hinf I to Hind III) was cloned into pGEM3Zf+, and the 5′ half fragment (Hinf I to Ava I) and the 3′ half fragment (Ava I to Hind III) were cloned into pGEM7Zf−. Five to 40 fold molar excesses of competitor plasmids or pGEM3Zf+ alone were cotransfected with a constant amount (0.5 μg) of pXLTR-CAT Forty-eight hours after transfection the cells were harvested and CAT activity was quantitated. Figure 4 shows the results of the experiment using stimulated Jurkat cells; similar relative results were obtained with unstimulated cells. The data are presented as the ratio of the CAT activities measured in the presence of the specific competitor over the CAT activity in the presence of the equivalent amount of nonspecific competitor (the pGEM3Zf+ vector) plotted against the molar ratio of competitor to reporter. In repeated experiments (at least 6 times), the data supported the in vitro results; neither the 3′ half nor 5′ half of the LTR can compete better than the nonspecific competitor, whereas the intact LTR does compete. In both the in vivo and in vitro experiments, moderate amounts of the 5′ half competitor produced increases in expression over that observed in the absence of competition, while very high concentrations of competitor caused a decrease in expression only in the in vitro systems. The reason for this difference is not known, but we suspect that the necessary relative concentration of intranuclear competitor needed to cause inhibition cannot be attained by transfection.
Figure 4.
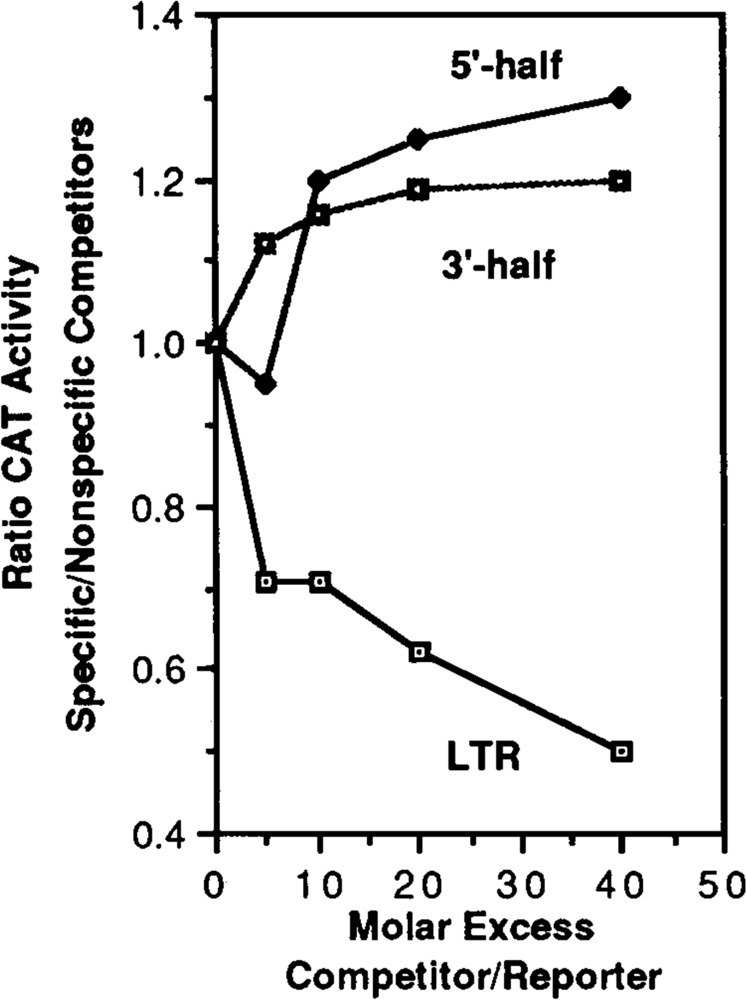
Transfection analysis of the competitive ability of the intact LTR in comparison to the 3′ and 5′ halves of the LTR. The pXLTR-CAT plasmid was transfected into activated Jurkat cells along with increasing amounts of competitor plasmids representing the intact LTR (Hinf I to Hind III), the 5′ half (Hinf I to Ava I), the 3′ half (Ava I to Hind III) or a nonspecific competitor. The data are presented as the ratio of reporter gene (CAT) activity measured in the presence of the specific competitor over the reporter gene activity in the presence of the equivalent molar ratio of nonspecific competitor (the pGEM3Zf+ vector) plotted against the molar ratio of competitor to reporter. Similar results were obtained using unactivated Jurkat cells.
Linker scanning analysis of transcriptional activity of the LTR
The in vitro transcription competition data suggest that besides the well characterized promoter elements within the 3′ half of the LTR, between −158 and +80 (Ava I to Hind III; Fig. 1), significant elements must also reside in the 5′ half between −159 and the 5′ end of the LTR, −453. To locate these putative elements we prepared and tested the in vivo transcriptional activity of linker scanning mutants across the LTR. The wild type test plasmid, pXLTRCAT (Fig. 5), contains the U3 and R region of the LTR from HIV strain HXB2 placed upstream of the CAT gene. An Xba I site has been placed 61 bp upstream of the 5′ end of the LTR. This site and the Hind III site, joining the LTR to the CAT gene, were used in the PCR directed construction of the linker scanning mutants (see Materials and Methods, Zeichner et al., submitted). As shown in Figure 5, the set of linker scanning mutants consecutively replace 18 bp of wild type sequence with an 18 bp Nde I-Xho I-Sal I (NXS) polylinker throughout the entire LTR. The mutants begin at the 5′ end of the LTR (nucleotide −453) and continue to nucleotide +15. The mutants are named numerically (Fig. 5), indicating the first and last LTR nucleotide replaced by the linker in the 5′ to 3′ direction. For example, mutant −453/−436 NXS has LTR nucleotide −453 replaced with the first base of the NXS polylinker and LTR nucleotide −436 replaced with the eighteenth, and last, nucleotide of the NXS polylinker. Figure 5 indicates the positions of various mutations with respect to known or putative regulatory regions.
Figure 5.
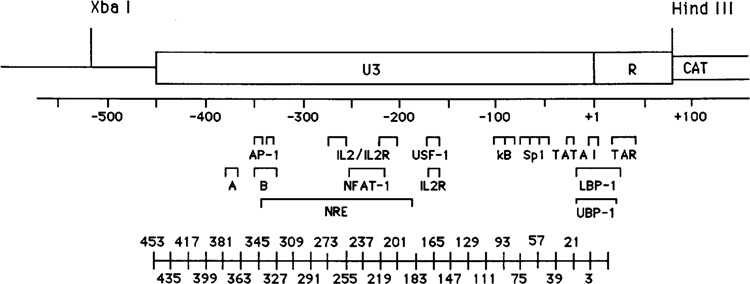
Map of the U3 and R regions of the HIV-1 LTR as they appear in pXLTRCAT. Nucleotide numbers are relative to the transcription start site, +1. Significant known and potential transcriptional control regions are indicated. The Xba I and Hind III sites were used in the construction of the linker scanning mutants. The consecutive positions of the NXS linker scanning mutations are diagrammed under their respective positions in the LTR.
The transcriptional activities of wild type and mutant LTRs were determined by CAT transient expression analysis. In these experiments one ng of wild type or mutant LTR plasmids were transfected in parallel using Lipofectin into HeLa cells. The cells were harvested after 48 hours and CAT activity determined. The CAT activities of mutant LTRs were calculated relative to the activity of the wild type LTR (wild type activity is designated 1.00). Figure 6 shows the graphic representation of the relative activities of the linker-scanning mutants; the data are the results of at least three separate transfection experiments. Mutations in the region between −111 and +15 had dramatic effects on the transcriptional activity. This was predictable, since this region contains the well characterized elements of the HIV promoter (Fig. 5). For example, −39/−22 NXS, which mutates the TATA sequences, decreases expression to about 18% of wild type in HeLa cells; −76/−58 NXS mutates the two 5′-mostSpl sites, decreasing expression to 20% of wild type. Mutants −111/−94 NXS and −93/−76 NXS each alter an NF-kB site and decrease expression to 12% and 5% of wild type, respectively. There were also some mutants that produced modest increases in expression, notably −129/−112 NXS. Further, the data indicated three previously un-described regions which mediate positive regulatory activities. These lie between −453 and −328, −273 and −220, −183 and −130. It should be pointed out that these alterations in activity were seen in HeLa cells, the effects of some of the mutations are different in other cell types (Zeichner et al., submitted).
Figure 6.
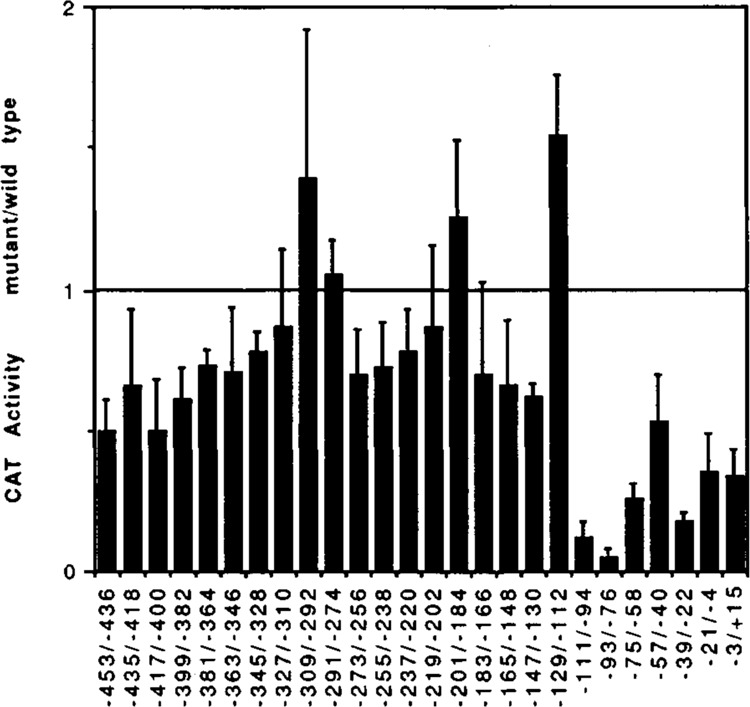
Results of transient transfection analysis of the transcriptional activity of the linker-scanning mutants. The CAT activity of each NXS mutant LTR is expressed relative to the activity of the wild type LTR (wild type activity = 1.00). The results shown are the average of 3 separate experiments. For each experiment triplicate transfections were done using the wild type plasmid to assure accuracy of this measurement.
Discussion
The results of our studies suggest that the 5′ half of the U3-R (−453 to −158 (the Ava I site) contains significant transcriptional control elements which appear to function to improve transcription complex formation as well as modulate transcriptional activity in HeLa cells. These conclusions are based on several observations: 1) the finding that effective competition of in vitro transcription from the U3-R reporter requires a competitor which consists of an intact DNA representing virtually the entire U3-R; 2) the finding that the 5′ half (−453 to −159), but not the 3′ half (−158 to +80), of U3-R was a modestly effective competitor; 3) the finding that neither the 5′ nor the 3′ half alone competes well during in vivo transfection competition experiments, but the whole LTR does compete well during the in vivo competition experiments; 4) the location, by transfection analysis of linker scanning mutations of the LTR, of three regions within the 5′ half of the LTR (between −453 and −328, −273 and −220, −183 and −130) which significantly lower LTR activity when mutated. The linker scanning data showed that some of the sequences in the LTR mediated questionably significant levels of negative regulatory effects.
Table 1.
Means and standard deviations of CAT activity generated by the NXS LTR mutants.
Results of transient transfection analysis of the transcriptional activity of the linker-scanning mutants. The results presented in Figure 6 are tabulated. The table lists the NXS mutants together with the CAT activity relative to the CAT activity of the wild type plasmid.
| Mutant | CAT activity mutant/wild type | ±SD |
|---|---|---|
| −453/−436 NXS | 0.50 | 0.11 |
| −435/−418 NXS | 0.66 | 0.27 |
| −417/−400 NXS | 0.50 | 0.18 |
| −399/−382 NXS | 0.61 | 0.11 |
| −381/−364 NXS | 0.73 | 0.06 |
| −363/−346 NXS | 0.71 | 0.23 |
| −345/−328 NXS | 0.78 | 0.07 |
| −327/−310 NXS | 0.87 | 0.27 |
| −309/−292 NXS | 1.39 | 0.53 |
| −291/−274 NXS | 1.05 | 0.12 |
| −273/−256 NXS | 0.70 | 0.16 |
| −255/−238 NXS | 0.72 | 0.16 |
| −237/−220 NXS | 0.78 | 0.15 |
| −219/−202 NXS | 0.87 | 0.29 |
| −201/−184 NXS | 1.25 | 0.28 |
| −183/−166 NXS | 0.70 | 0.33 |
| −165/−148 NXS | 0.66 | 0.23 |
| −147/−130 NXS | 0.62 | 0.05 |
| −129/−112 NXS | 1.54 | 0.22 |
| −111/−94 NXS | 0.12 | 0.06 |
| −93/−76 NXS | 0.05 | 0.03 |
| −75/−58 NXS | 0.26 | 0.05 |
| −57/−40 NXS | 0.53 | 0.17 |
| −39/−22 NXS | 0.18 | 0.03 |
| −21/−4 NXS | 0.35 | 0.14 |
| −3/+15 NXS | 0.34 | 0.09 |
In the in vitro transcription competition experiments, the extent of the LTR necessary for efficient competition was larger than expected. Transcription templates containing only the 3′ half of the LTR (e.g., from the Ava I to the Hind III site) function in in vitro transcription assays as efficiently as templates containing the entire LTR (Dinter et al., 1987; Patarca et al., 1987; our unpublished observations). This may be interpreted as indicating that sequences within the region between −453 and −158 have no effect on in vitro transcription. However, our data clearly indicate that sequences involved in mediating transcription complex formation in vitro do exist in this region. Supporting our conclusion, Garcia et al. (1987) have shown that the DNase I protection pattern over the region of the 3′ LTR changes when the 5′ LTR sequences are present on the probe. The basis for the inability to detect effects of deletions involving the 5′ half of the LTR by in vitro transcription analysis of LTR deletion mutants is not understood. Although in vitro transcription extracts appear to be able to form complexes on the intact LTR involving some factors bound to the 5′ end, they may lack specific factors needed to facilitate the function of these factors. By this mechanism the transcription from an intact LTR and from an LTR deleted of its 5′ half would appear to be equal. Hence deletion of the 5′ half sequences would show no apparent effect by direct in vitro transcription analysis. The need for both the 5′ and 3′ halves in transcription complex formation would only be detected by competition analyses.
The requirement for a very large region of the HIV LTR for competitive complex formation contrasts with in vitro transcription competition analyses of some other promoters. For example, using the cytomegalovirus (CMV) enhancer, Ghazal et al. (1988, 1987) found that small oligonucleotide competitors containing known factor binding sites inhibited in vitro transcription from a CMV promoter-CAT gene transcription template. Our competition data using the Hae III fragments (each including one or another factor binding site (e.g., API, NF-kB, SP1; see Fig. 1), show that competition for specific factors causes no significant inhibition of LTR-mediated transcription. Hence the HIV LTR may utilize a different transcriptional strategy. The promoter may be responsive to a variety of different transcription complexes formed on the LTR; some of these factor-DNA interactions appear to involve sequences in the 5′ portion of the LTR. The many characterized factor binding sites, as well as the number of possible interactions which could occur between factors, provide potential for the formation of alternative transcription complexes on the LTR. Formation and alteration of complexes may mediate the various phases of the viral infection. In support of this variable factor transcription complex model, Leonard et al. (1989) have shown that HIV with the NF-kB sites deleted, or with mutations in one or two of the Spl sites (see Fig. 1), replicate efficiently in human T lymphocytes. These data suggest that the transcription complex formed on the LTR may have the ability to compensate for some deletions and mutations, as our model predicts.
Acknowledgments
This work was supported by Public Health Service Grant GM36993 awarded to J. C. A. S. L. Z. was a fellow of the Institute for Pediatric Services of the Johnson and Johnson Baby Products Company and has been supported by PHS Training Grant NS07180, by the Children’s Hospital of Philadelphia Medical Associates Research and Education Fund, and by Public Health Service Clinical Investigator Award AI0907. J.Y.H.K. is a trainee of the University of Pennsylvania School of Medicine Medical Scientists Training Program. We thank Sherri Adams and Tom Kadesch for helpful discussions and for their comments on the manuscript; Roberto Weinmann for helpful discussions and reagents; Jane Picardi and Tobi Goldberg for excellent experimental assistance.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Alwine J. C. (1985), Mol Cell Biol 5, 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya S. K., Guo C., Josephs S. F., and Wong-Staal F. (1985), Science 229, 69–73. [DOI] [PubMed] [Google Scholar]
- Bohnlein E., Lowenthal J. W., Siekevitz M., Ballard D. W., Franza B. R., and Greene W. C. (1988), Cell 53, 827–836. [DOI] [PubMed] [Google Scholar]
- Dayton A. I., Sodroski J. G., Rosen C. A., Goh W. C., and Haseltine W. A. (1986), Cell 44, 941–947 [DOI] [PubMed] [Google Scholar]
- Dinter H., Chiu R., Imagawa M., Karin M., and Jones K. A. (1987), EMBO J 6, 4067–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgner P. L., Gadek T. R., Holm M., Roman R., Chan H. W., Wenz M., Northrop J. P., Ringold G. M., and Danielsen M. (1987), Proc Natl Acad Sci USA 84, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franza B. R. Jr., Josephs S. F., Gilman M. Z., Ryan W., and Clarkson B. (1987), Nature 330, 391–395. [DOI] [PubMed] [Google Scholar]
- Franza B. R. Jr., Rauscher F. J., Josephs S. F., and Curran T. (1988), Science 239, 1150–1153. [DOI] [PubMed] [Google Scholar]
- Garcia J. A., Wu F. K., Matsuyasu R., and Gaynor R. B. (1987), EMBO J 6, 3761–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazal P., Lubon H., and Hennighausen L. (1988), Mol Cell Biol 8, 1809–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazal P., Lubon H., Fleckenstein B., and Hennighausen L. (1987), Proc Natl Acad Sci USA 84, 3658–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman C. M., Moffat L. F., and Howard B. H. (1982), Mol Cell Biol 2, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrich D., Garcia J. A., Wu F. K., Mitsuyasu R., Gonzalez J., and Gaynor R. B. (1989), J Virol 63, 2585–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobovits A., Smith D. H., Jakobovits E. B., and Capon D. J. (1988), Mol Cell Biol 8, 2555–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. A., Kadonaga J. T., Luciw P. A., and Tjian R. (1986), Science 232, 755–759. [DOI] [PubMed] [Google Scholar]
- Jones K. A., Luciw P. A., and Duchange N. (1988), Genes Dev 2, 1101–1114. [DOI] [PubMed] [Google Scholar]
- Jones K. A. (1989), New Biol 1, 127–135. [PubMed] [Google Scholar]
- Kaufman J. D., Valandra G., Roderiquez G., Bushar G., Giri C., and Norcross M. A. (1987), Mol Cell Biol 7, 3759–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J., Parrott C., Buckler-White A. J., Turner W., Ross E. K., Martin M. A., and Rabson A. D. (1989), J Virol 63, 4919–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Stenzel M., Sodorski J. G., and Hazeltine W. A. (1989), J Virol 63, 4115–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Touzjian N., Stenzel M., Dorfman T., Sodroski J. G., and Haseltine W. A. (1990), J Virol 64, 5226–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay B. and Reeder R. H. (1986), Cell 47, 913–920. [DOI] [PubMed] [Google Scholar]
- Muessing M. A., Smith D. H., and Capon D. J. (1987), Cell 48, 691–701. [DOI] [PubMed] [Google Scholar]
- Nabel G. and Baltimore D. (1987), Nature 326, 711–713. [DOI] [PubMed] [Google Scholar]
- Nabel G. J., Rice S. A., Knipe D. M., and Baltimore D. (1988), Science 239, 1299–1302. [DOI] [PubMed] [Google Scholar]
- Okamoto T. and Wong-Staal F. (1986), Cell 47, 29–35. [DOI] [PubMed] [Google Scholar]
- Okamoto T., Reitz M. S. Jr., Clarke M. F., Jagodzinski L. L., and Wong-Staal F. (1986), J Biol Chem 261, 4615–4619. [PubMed] [Google Scholar]
- Orchard K., Perkin N., Chapman C., Harris J., Emery V., Goodwin G., Latchman D., and Collins M. (1990), J Virol 64, 3234–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patarca R., Heath C., Goldenberg G. J., Rosen C. A., Sodroski J. G., Haseltine W. A., and Hansen U. M. (1987), AIDS Res Hum Retroviruses 3, 41–55. [DOI] [PubMed] [Google Scholar]
- Pavlakis G. N. and Felber B. K. (1990), New Biol 2, 20–31. [PubMed] [Google Scholar]
- Peterlin B. M., Luciw P. A., Barr P. J., and Walker M. D. (1986), Proc Natl Acad Sci USA 83, 9734–9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner L., Fisher A., Jagodzinski L. L., Mitsuya H., Liou R.-S., Gallo R. C., and Wong-Staal F. (1987), AIDS Res Hum Retroviruses 3, 57–69. [DOI] [PubMed] [Google Scholar]
- Rice A. P. and Matthews M. B. (1988), Nature 332, 551–553. [DOI] [PubMed] [Google Scholar]
- Rosen C. A., Sodroski J. G., and Haseltine W. (1985), Cell 41, 813–823. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P., Wildeman A., and Chambon P. (1985), Nature 313, 458–463. [DOI] [PubMed] [Google Scholar]
- Shapiro D. J., Sharp P. A., Wahli W. W., and Keller M. J. (1988), DNA 7, 47–55. [DOI] [PubMed] [Google Scholar]
- Shaw G. M., Hahn B. H., Arya S. K., Groopman J. E., Gallo R. C., and Wong-Staal F. (1984), Science 226, 1165–1171. [DOI] [PubMed] [Google Scholar]
- Shaw J. P., Utz P. J., Durand D. B., Toule J. J., Emmel E. A., and Crabtree G. R. (1988), Science 241, 202–203. [DOI] [PubMed] [Google Scholar]
- Siekevitz M., Josephs S. F., Dukovich M., Peffer N., Wong-Staal F., and Greene W. C. (1987), Science 238, 1575–1578. [DOI] [PubMed] [Google Scholar]
- Smith M. R. and Green W. C. (1989), Proc Natl Acad Sci USA 86, 8526–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodroski J., Patarca R., Rosen C., Wong-Staal F., and Haseltine W. (1985a), Science 229, 74–77. [DOI] [PubMed] [Google Scholar]
- Sodroski J., Rosen C., Wong-Staal F., Salahuddin S. Z., Popovic M., Arya S., Gallo R., and Haseltine W. A. (1985b), Science 227, 171–173. [DOI] [PubMed] [Google Scholar]
- Tong-Starksen S. E., Luciw P. A., and Peterlin B. M. (1987), Proc Natl Acad Sci USA 84, 6845–6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varmus H. (1988), Genes Dev 2, 1055–1062. [DOI] [PubMed] [Google Scholar]
- Wildeman A. G., Sassone-Corsi P., Grundstrom T., Zenke M., and Chambon P. (1984), EMBO J 3, 3129–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Garcia J. A., Harrich D., and Gaynor R. B. (1988a), EMBO J 7, 2117–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Garcia J., Mitsuyasu R., and Gaynor R. (1988b), J Virol 62, 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret K. S., Liu J.-K., and DiPersio C. M. (1990), Proc Natl Acad Sci USA 87, 5469–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]