

Abstract
The promoters and enhancers of cell type-specific genes are often conserved in evolution, and hence one might expect that a given enhancer has evolved to work best with its own promoter. While this expectation may be realized in some cases, we have not found evidence for it. A total of 27 combinations of different promoters and enhancers were tested by transfection into cultured cells. We found that the relative efficiency of the enhancers is approximately the same, irrespective of the type of promoter used, i.e., there was no strong preference for any given enhancer/promoter combination. Notably, we do not see particularly strong transcription when the immunoglobulin kappa enhancer (or the immunoglobulin heavy chain enhancer) is used to activate a kappa gene promoter. We propose that a generally permissive enhancer/promoter interaction is of evolutionary benefit for higher eukaryotes: by enhancer shuffling, genes could be easily brought under a new type of inducibility/cell type specificity.
Regulatory regions for eukaryotic RNA polymerase II transcription units are located both upstream and downstream of the RNA start site and are usually defined as (proximal) promoters and (remote) enhancers. The difference between an enhancer and a promoter is largely operational, since both are composed of a number of short DNA sequence motifs that serve as recognition sequences for transcription factors. Enhancers can act in either orientation and over long distances, whether tested on homologous or heterologous promoters (reviewed in Serfling et al., 1985). Some of these cis-acting DNA sequence motifs (= modules, elements) are preferentially found in either a promoter or an enhancer position, whereas others, like the octamer motif, can be found in close proximity to the transcriptional start site as well as at remote positions. The activity of a particular cis-acting element varies between different cell types. The general picture emerging from studies in several laboratories is that the concentration of the active form of a transcription factor, as well as the number and affinity of corresponding binding sites on the DNA, influence the level of transcription (for reviews, see Maniatis et al., 1987; Müller et al., 1988a; Mitchell and Tjian, 1989; Johnson and McKnight, 1989). In principle, a single binding site in conjunction with a corresponding transcription factor can be subject to cell type-specific (or in-ducible) regulation (Dreyfus et al., 1987; Wirth et al., 1987; Gerster et al., 1987). The interplay between the various factors is as yet poorly understood, but it influences gene activity as well (see, for example, Hu et al., 1990; Murre et al., 1990; Schatt et al., 1990; for reviews see Mitchell and Tjian, 1989; Johnson and McKnight, 1989; Abel and Maniatis, 1990; Jones, 1990).
The enhancer of immunoglobulin heavy chain (IgH) genes was the first example found of an enhancer associated with a cellular gene, and also the first component identified in the then enigmatic phenomenon of cell type-specific gene expression (Banerji et al., 1983; Gillies et al., 1983, Neuberger, 1983). The IgH enhancer shows a strict cell type specificity: it is active in B lymphocytes but not, for example, in epithelial or fibroblast cells. Subsequently, it was found that not only the Ig enhancers but also the promoters of Ig heavy and light chain genes are preferentially active in lymphoid cells (Falkner et al., 1984; Picard and Schaffner, 1985; Mason et al., 1985; Grosschedl and Baltimore, 1985; Foster et al., 1985; Mizushima-Sugano and Roeder, 1986). It is well-established that a functional enhancer and a promoter act synergistically to bring about strong transcription. There are also reports that there is an additional level of specificity to this enhancer-promoter interaction. For example, Garcia et al. (1986) have proposed that the cell type-specific immunoglobulin gene enhancers are preferentially active in combination with their own promoters. Such preferences could have been the result of coevolution of an enhancer and its corresponding promoter. We have addressed the question of preferential activity by analyzing the functional cooperation between given promoter and enhancer sequences with a number of constructions. We wanted to determine whether it was possible, for example, to create a particularly strong transcription unit by having the same DNA motif act from both an upstream and a downstream position. The results presented in this paper, which were obtained by measuring expression of transiently transfected cell lines, do not indicate a marked preference for a particular enhancer/promoter combination. Rather, different promoters were always activated to about the same relative level by the various enhancers tested. This ability to combine various enhancers and promoters may have facilitated the evolution of new regulatory pathways.
Materials and methods
Construction of plasmids
All clones were constructed according to standard recombinant DNA protocols (Maniatis et al., 1982).
The plasmid constructs containing the Ig kappa-promoter and its derivatives are based on the pK plasmid (Picard and Schaffner, 1985) in which different promoter and enhancer elements were placed upstream and downstream, respectively, of a truncated β-globin gene (Fig. 1A). In the KapP series a promoter fragment (Ddel-HinfI) from a mouse κ-light chain gene, including the octamer and TATA box sequences as well as the cap site (nucleotides −180 to + 22) was fused to the β-globin reporter gene.
Figure 1.
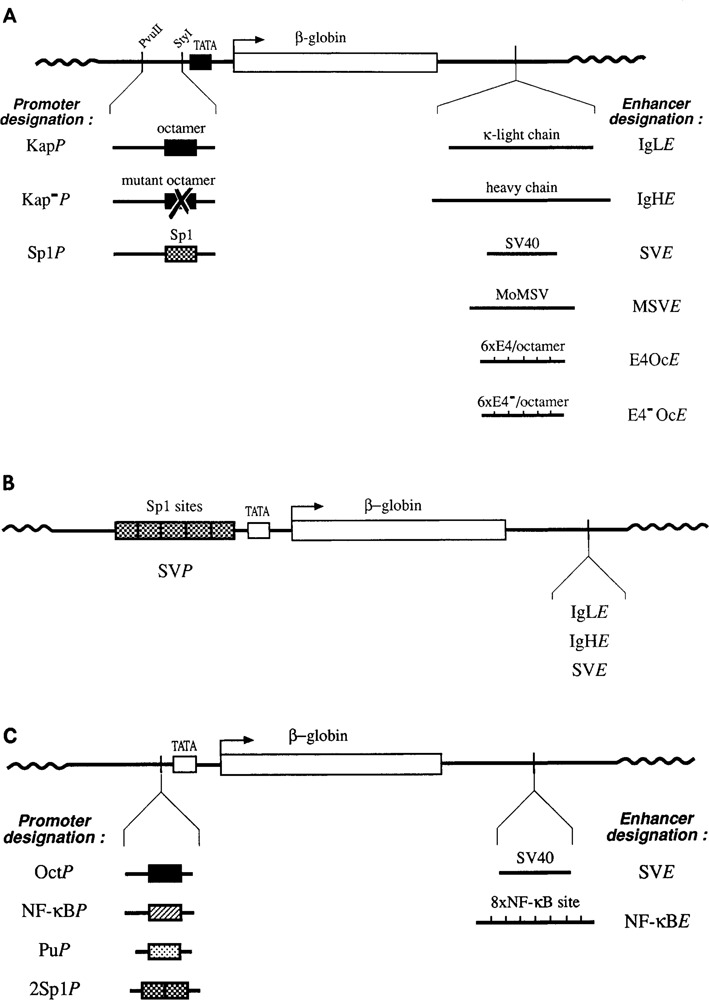
Schematic representation of the recombinant DNAs. A. DNA constructs with the immunoglobulin kappa light chain promoter and its derivatives. A 202 bp fragment of a mouse k chain promoter or its derivatives (black region) was fused to a truncated rabbit β-globin gene (white region). Promoter and enhancer sequences used in these constructs are designated with the letters P and E, respectively. Plasmid DNA is shown by a wavy line. The constructions are not drawn to scale. B. DNA constructs with the SV40 promoter. The 21 bp repeats from the early promoter region of SV40 containing 5 functional Spl binding sites (stippled boxes) were placed in front of the β-globin gene (white region). The enhancers used are identical to some of those shown in A. C. DNA constructs containing the OVEC reporter gene. OVEC is a reporter gene containing the coding sequence of the rabbit β-globin gene (as are the constructs in A and B) and convenient cloning sites for promoter and enhancer elements (Westin et al., 1987). DNA oligonucleotides containing a single or a duplicated binding site for different transcription factors were inserted upstream of the β-globin TATA box (white). These promoter constructions were tested with the SV40 enhancer or a multimeric NF-κB enhancer.
In the two promoter variants Kap(-)P and SplP, the octamer motif of the promoter was replaced by its mutated variant or by the binding site for Spl, respectively, preserving the original TATA box. To this end, a 52 bp PvuII-Styl fragment in KapP was replaced by synthetic sequences, as shown below:
| KapP | wild type octamer | 5′ ATTTGCATTAAACGTA 5′ |
| Kap(−)P | mutated octamer | 5′ATGTTCAGTACAAGTC 5′ |
| SplP | Spl binding site (HSV) | 5′GGGCGGGGCCCCGCCCCG 5′ |
The enhancer elements were inserted downstream of the test gene: IgLE, a 480 bp Alul fragment derived from the mouse K-light chain enhancer (Picard and Schaffner, 1984); IgHE, a 690 bp Xbal-EcoRI fragment from the mouse heavy chain enhancer (Banerji et al., 1983); SV40E, a 200 bp segment including the two 72 bp repeats of the SV40 enhancer (Banerji et al., 1981); MSVE, a 350 bp Bam HI fragment from the Moloney sarcoma virus LTR (Levinson et al., 1982).
The synthetic enhancers used in the plas-mid series, E4 OcE and E4(-)OcE, consist of six repeated 51 bp fragments from the IgH enhancer, each consisting of the octamer sequence plus a functional or mutated E4 motif, respectively (Gerster et al., 1987). The reference plasmid REF was described previously (Picard and Schaffner, 1985).
The SVP constructs (Fig. 1B) contain an SV40 fragment consisting of the three 21 bp early promoter repeats (five Spl binding sites), was placed in front of the TATA box of the β-globin gene, whose first intron in this case was preserved.
The two plasmids containing the rabbit β-globin promoter are described in Gerster et al. (1987). The OVEC system (Westin et al., 1987) was employed to construct the series of plasmids containing single transcription factor binding sites. Oligonucleotides with the octamer (oct) or NF-kB binding sites or the Pu box (Pu) were synthesized with Sad and Sail protruding ends and inserted in front of the TATA box. The Spl oligonucleotide (2 Spl) contains a dimer of a binding site found in the immediate early gene 3 of herpes simplex virus. It was synthesized with blunt ends and ligated into the Sail site, which was filled in by T4 polymerase. The sequences of the oligonucleotides are shown in Table 1. The reference plasmid used for this series of plasmids was OVEC-REF (Westin et al., 1987).
Table 1.
Sequences of oligonucleotides.
| Oct: | 5′ CGAGCCCGCGGTAATTTGCATTTCTACTAG |
| TCGAGCTCGGGCCCCATTAAACGTAAAGATGATCAGCT 5′ | |
| NF-κB: | 5′ CGAGAACAGAGGGGACTTTCCGAGAGGCC |
| TCGAGCTCTTGTCTCCCCTGAAAGGCTCTCCGGCAGCT 5′ | |
| Pu: | 5′ CGAGAGTTCCTCTTTCAGAG |
| TCGAGCTCTCAAGGAGAAAGTCTCAGCT 5′ | |
| 2Sp1: | 5′ CCGGCCCCGCCCATCCCCGGCCCCGCCCATCC |
| GGCCGGGGCGGGTAGGGGCCGGGGCGGGTAGG 5′ |
Cell growth, transfection, and RNA analysis
The human lymphoblastoid cell line BJA-B and the myeloma cells X63Ag8 were grown in RPMI and DMEM media, respectively. Both media were supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin. The cells were transfected with 5 or 10 μg test plasmid and 2 μg reference by the DEAE-dextran procedure followed by a DMSO boost. Cytoplasmic RNA was isolated 40–42 hr after transfection and purified as described previously (De Villiers and Schaffner, 1983). The specific transcripts were quantitated by ribo-nuclease mapping (Melton et al., 1984, with some modifications, see Picard and Schaffner, 1985) or SI nuclease analysis (de Villiers and Schaffner, 1983). For SI nuclease analysis of the transcripts from the KapP constructs and their derivatives, as well as for the SVP constructs, a BamHI-labeled SacI-BamHI fragment (nucleotides −195 to +215) was used as probe. The reverse SP6 polymerase transcript of the same fragment was used for ribonuclease mapping (Picard and Schaffner, 1985).
Results
Transcriptional activation of the lg kappa promoter by different enhancers in B cells
We have tested the activity of a β-globin reporter gene driven by an immunoglobulin kappa gene promoter in B-type lymphoid cells (BJA-B; Fig. 1A). The reporter gene, together with a reference gene as a standard for transfection efficiency, was transfected into these cells by the DEAE dextran method. After 40–42 hr specific transcripts from the kappa promoter were analyzed by SI nuclease or ribonuclease mapping. The gene was either expressed without enhancer, or with one of the following six enhancers: 1. The homologous immunoglobulin kappa enhancer located within the J/C intron (Queen and Baltimore, 1983; Picard and Schaffner, 1984). We have previously tested this enhancer with the β-globin promoter and the SV40 early promoter, where it shows only about 5% of the activity of the strong enhancer from immunoglobulin heavy chain genes (Picard and Schaffner, 1984). Others have reported that a preferential interaction between the kappa enhancer and kappa promoter results in a transcriptional efficiency approaching that seen with strong enhancers (Garcia et al., 1986). 2. The IgH enhancer, which is also B cell-specific, as mentioned above (Banerji et al., 1983; Gillies et al., 1983; Neuberger, 1983). 3. The SV40 enhancer, the first described and thoroughly characterized enhancer (Banerji et al., 1981; Moreau et al., 1981). Under natural circumstances, it drives expression from the SV40 early promoter that contains multiple sites for the transcription factor Spl and a weak TATA box (Mathis and Chambon, 1981; Benoist and Cham-bon, 1981; Dynan and Tjian, 1983). 4. The enhancer from the Moloney retrovirus (MSV) (Levinson et al., 1982). 5. A synthetic enhancer composed of 6 identical tandem segments of 50 bp from the immunoglobulin heavy chain enhancer, containing both an octamer motif and an Ephrussi 4 motif (Gerster et al., 1987). 6. The same synthetic enhancer with the Ephrussi 4 motif mutated, leaving only the octamer motif intact. Both synthetic enhancers retain the lymphocyte-specificity of the complete IgH enhancer (Gerster et al., 1987).
As seen in Figure 2A, all the enhancers tested are active; however, the kappa light chain enhancer (IgLE) is only about 10% as active as the heavy chain enhancer and only 5% as active as the SV40 enhancer. Similar results are obtained in J-558 myeloma cells (not shown). These data indicate that the kappa enhancer, which is known to be weakly active with non-homologous promoters, does not perform significantly better with a corresponding kappa promoter. A possible solution to the paradox of the low activity of this kappa enhancer has recently been found with the identification of a second enhancer located downstream of the constant region of the kappa gene (Meyer and Neuberger, 1989). The same order of relative activity is also observed with a different promoter, namely the SV40 early promoter, whose major components are 5 binding sites for the Spl factor (Fig. 2B).
Figure 2.

Transcriptional activation of two genuine promoters by different enhancers. A. S1 nuclease analysis of Igκ promoter activity. Human B-type lymphoblastoid cells (BJA-B) were transfected with seven constructs including the κ-light chain promoter alone (designated “0,” lane 1) or in combination with the six enhancers shown in Figure 1A (lanes 2–7, designation according to Fig. 1A). The products of S1 nuclease protection were analyzed on a denaturing 6% polyacrylamide gel in parallel with size markers (M) of HpaII digested pBR322 DNA. The bands corresponding to the correctly initiated and the reference transcripts are indicated with “ct” and “ref,” respectively. B. Activity of the SV40 promoter. The same procedure as in A was used for transcriptional analysis of the SV40 promoter constructs depicted in B. The activation of this promoter by the SV40-, IgH (heavy)- and IgL(κ)-enhancers is shown in lanes 3, 4 and 5, respectively. “0,” SV40 promoter construct without enhancer. All other designations are as in A.
A promoter with an Sp1 site replacing the octamer sequence responds to the various enhancers like the genuine kappa promoter
In the next series of experiments, we wanted to see whether a given upstream factor would have a preference for a particular enhancer. In addition to the kappa promoter, we also tested a derivative in which the octamer motif was replaced by consensus binding sites for Spl factor (Fig. 1A). Both the kappa promoter and the Spl promoter were tested in parallel with the four natural enhancers, namely the Ig kappa light and heavy chain gene enhancers and the SV40 and MSV viral enhancers. The results of the transfections are shown in Figure 3. As is evident, the Spl-containing promoter is weaker than the kappa promoter in the BJA-B cells. More importantly, it is also obvious that the relative efficiency of the Spl vs. wild-type kappa promoter is about the same for each of the four enhancers tested. These results indicate that there is neither a preference of the immuno-globulin enhancers for the Ig kappa promoter, nor a preference of the viral enhancers for the Spl-containing promoter, as was already indicated from the experiments show in Figure 2B.
Figure 3.

Transcriptional activation of the Igk promoter and its derivatives by different enhancers. The transcriptional activity of four series of constructs including the wild-type promoter (KapP) and its derivatives (SplP or Kap(-)P, see Figure 1A) in combination with the enhancers from IgL(K) (A); IgH (heavy chain) (B); SV40 (C) or MoMSV (D) were analyzed by SI nuclease mapping. The construct designations are according to Figure 1A. “0,” IgK promoter without enhancer. All other designations areas in Figure 2. Mappings shown in A and B were run on the same gel along with the same size marker lane. The bands between the “ref”- and “ct” bands are from read-through transcription of the reference plasmid. To improve presentation of the data, different autoradiogram exposures are shown for A and B, and the size marker is now shown at the sides of both A and B.
Transcriptional activity of different promoter constructions driven by the SV40 enhancer or a multimer of the NF-κB binding site
In a final series of experiments, we constructed a series of promoters where one or two binding sites for a single transcription factor were placed upstream of the β-globin TATA box in the OVEC reporter plasmid (Westin et al., 1987). These promoters contain the binding site for either the octamer factors (Wirth et al., 1987; Muller et al., 1988b), NF-κB, a transcription factor that is constitutively active in B lymphocytes, the so-called Pu box factor that confers lymphoid-specific activity (Moreau-Gachelin et al., 1990; Karim et al., 1990), or the Spl factor (reviewed in Dynan and Tjian, 1985; Kadonaga et al., 1986). All of these promoter constructs were tested with either the generally active SV40 enhancer or with eight tandem copies of the binding site for NF-κB (Sen and Baltimore, 1986; Atchison and Perry, 1987; Lenardo et al., 1987; cloning of NF-κB, see Gosh et al., 1990; Kieran et al., 1990; for review, see Lenardo and Baltimore, 1989). In addition, a construction containing the entire β-globin promoter was included in the analysis.
The constructs were transfected into X63Ag8 myeloma cells together with the reference plasmid OVEC-REF, and the amount of specific transcripts was determined by ribonuclease mapping. When transcription is driven from a solitary TATA box, no synthesis of reporter gene mRNA is detected (Fig. 4A, lane 1). Low levels of transcription are observed with the β-globin promoter, as well as with those constructions containing the NF-κB or Pu box monomer in front of the TATA box (Fig. 4A, lanes 3, 4, and 6). With the octamer or the Spl upstream promoter sites, a higher level of specific transcription is observed (Fig. 4A, lanes 2 and 5).
Figure 4.

Activity of β-globin promoter derivatives with either of two enhancers. The level of transcription was measured from different promoters without an enhancer (A), with the SV40 enhancer (B), or with eight NF-κB sites (C). The constructs used are schematically drawn in Figure 1C. The following promoters were tested: TATA box alone (lane 1); the octamer motif (lane 2); the NF-kB binding site (lane 3); the Pu box (lane 4); the GC box, recognized by the Spl transcription factor (lane 5); and the entire β-globin promoter (lane 6). The constructs were transfected into X63Ag8 myeloma cells and the amount of β-globin mRNA quantitated by RNase mapping, ct indicates correctly initiated transcripts, and ref indicates the transcripts from OVEC-REF (Westin et al., 1987) which was included as an internal reference for transfection efficiency. HpaII digested pBR322 DNA was used as size marker (lane 7).
The expression level driven by the two enhancers differs among the various promoters. A very low level of expression is seen from a crippled promoter consisting of just a TATA box, even if the SV40 enhancer or the8xNF-κB enhancer are present in a downstream position (Fig. 4B and C, lane 1). In contrast, insertion of the various cis-acting motifs in front of the TATA box results in strong transcription with either of the two enhancers linked to the gene. The strongest stimulation is seen with the octamer- and Spl-containing promoters (Fig. 4B and C, lanes 2 and 5). These are also the promoters which by themselves give the highest basal activity (see Fig. 4A). When directly compared, both enhancers analyzed activate a particular promoter to about the same extent (compare Fig. 4B and C, lanes 2–6). Although there may be small variations in this set of experiments (for example, it appears that transcription is some 2–3 times more efficient when the octamer-containing promoter is linked to the “NF-κB enhancer” as compared to the SV40 enhancer), we do not observe a marked preferential interaction for any of the enhancer/promoter combinations.
Discussion
No general evidence for enhancer/ promoter preference
It is well established that an enhancer and a promoter can act synergistically to yield a level of transcription greater than the sum of the levels achieved by each element individually. We have tested whether there are specific preferences between certain enhancers and promoters. By transfection into cultured cell lines, we have analyzed a total of 27 enhancer/promoter combinations, and we see no preferential interaction (more than a factor of 2–3 fold) between any given enhancer and promoter. Since each enhancer activates all of the promoters tested, our results show that there is a great flexibility in the interaction between mammalian transcriptional control elements. When we initiated our studies, we expected to find cases of exclusive - or at least particularly high -activity with a naturally occurring enhancer/ promoter combination. After all, coevolution of components involved in the same regulatory pathway is a common biological phenomenon. Not unexpectedly, regulatory sequences of genes transcribed by different RNA polymerases can have diverged such that they are not compatible, such as a thymidine kinase promoter (RNA polymerase II) and an rDNA enhancer (RNA polymerase I) (Pape et al., 1989). Also, the highly specialized U2 snRNA genes, even though transcribed by RNA polymerase II, cannot be activated by an SV40 enhancer (Tanaka et al., 1988).
In addition to some reports on preferential activity of certain enhancer/promoter combinations (see below), one set of observations seems to strengthen the argument for coevolution resulting in made-to-measure enhancer/promoter combinations. In several of the cases analyzed so far, including the Ig heavy chain locus, binding sites for a given transcription factor are present both in a enhancer and in a proximal promoter region (see, for example, Gerster et al., 1987; Landschulz et al., 1988; Wall et al., 1988). In the Ig heavy chain genes, the octamer motif ATGCAAAT is present both in the promoter and, in inverse orientation, the enhancer. Thus it was tempting to speculate that, for example, dimerization of octamer factors would link up the enhancer and promoter. A precedent for such an interaction is found in prokaryotes: cooperative repressor binding can bring together operator sites over distances of more than one hundred bp, with concomitant looping out of the intervening DNA (for reviews see Ptashne, 1986; Lobell and Schleif, 1990). However, so far no evidence has been obtained for cooperative binding of a factor over the long distances that can separate enhancers and promoters, even though enhancer and promoter have been shown to functionally interact when brought in close proximity via a protein bridge (Müller et al., 1989; for review see Muller and Schaffner, 1990). Furthermore, our present experiments fail to show a significant improvement in activity when the enhancer and promoter contain the same factor binding site. A similar lack of preference is also found in HeLa cells when an enhancer with multiple glucocorticoid-responsive elements (GREs) is tested with GRE promoters versus an Spl promoter (Schatt et al., 1990). Therefore, it may be that the interaction between factors bound to enhancers and promoters is not direct, as in prokaryotic repressor binding, but rather mediated by protein/protein interaction with additional factors and/or RNA polymerase itself (reviewed in Lewin, 1990).
The DNA constructions tested by us include combinations of the Ig kappa promoter with Ig kappa and Ig heavy chain enhancers, and of the SV40 early promoter with its enhancer. None of them shows preferential activity. Notably, we do not see particularly high gene expression when the immunoglobulin kappa enhancer (or the immunoglobulin heavy chain enhancer) is used to activate a kappa gene promoter. Meyer and Neuberger (1989) have also found weak activity of the kappa intron enhancer with a β-globin promoter, and they pointed out the existence of a second, stronger kappa gene enhancer far downstream of the constant region which was required for strong activation. However, the Ig kappa promoter/intron enhancer combination was previously reported to be preferentially active (Garcia et al., 1986). The difference between our results and those of Garcia et al. could be due to differences in the particular DNA constructs or B cell lines used. Some data on the expression of the herpes virus thymidine kinase gene (Parslow et al., 1987) and the alcohol dehydrogenase gene in Drosophila (Fischer and Maniatis, 1988) can be explained by selective enhancer/promoter interaction. Recently, Vincent et al. (1990) have inserted multiple copies of a synthetic binding site for the “engrailed” protein upstream of different promoters. These combinations have a more restricted and specific ability to enhance transcription when assayed in transgenic fly embryos as compared to a more permissive enhancer/promoter interaction in transiently transfected tissue culture cells. Nevertheless, others have selected for a great variety of stage-and cell type-specific enhancers in Drosophila by using one common promoter (Bellen et al., 1989; Wilson et al., 1989). Even though only one promoter was tested, these data are also compatible with a permissive enhancer/promoter interaction.
Evolution by enhancer shuffling?
There could well be cases of preferential enhancer/promoter interaction. Even subtle differences in responsiveness could be important during embryogenesis, when gene dosage appears to be most critical. Nevertheless, our data with quantitative transcription mapping in transfected B cell lines imply that generally a given promoter can respond to any enhancer (assuming, of course, that they are active at all in the particular cell type). This raises the question of the possible biological consequence(s) of this phenomenon. One might argue that all enhancers and promoters are channeling their actions through some common element, e.g., RNA polymerase II, and therefore cannot diverge from a ubiquitous function. However, in procaryotes there is strong selectivity: operators in upstream and promoter-proximal positions must bind the same factor to interact via DNA looping (for the ara operon, see Lobell and Schleif, 1990; for the lac operon, see Kramer et al., 1987). If in eukaryotes a particular enhancer is able to activate almost any promoter brought under its domain of influence, we speculate that this may facilitate the generation of new regulatory pathways, thus accelerating evolution. We also note a possible similarity of this phenomenon to the so-called exon shuffling (Gilbert, 1978), in which a compatibility of heterologous 5′ and 3′ splice sites makes possible the production of new combinations of protein domains, as a result of translocation events.
In the realm of transcription, multiple enhancers controlling the same gene may have been acquired by enhancer shuffling. For example, expression of the alpha fetoprotein gene in multiple cell types is governed by three separate enhancers spread over a sequence of 4 kb (Camper and Tilghman, 1989). In Drosophila embryos, separate DNA regions control expression in ecto/mesoderm and neural precursors cells (Hiromi and Gehring, 1987; Pick et al., 1990). Another possible example is lysozyme, an antibacterial enzyme, produced in chicken macrophages and also deposited in enormous quantities in egg white. Expression in macrophages and in oviduct cells is controlled by separate enhancers with a common promoter (Theisen et al., 1986; Steiner et al., 1987). It is also interesting to note that in different animal classes, various proteins such as lactate dehydrogenase, enolase, and a small heat shock protein have been recruited to become the major lens crystallin proteins (De Jong et al. 1989). One explanation for this new function could be a translocation event involving the particular gene and a lens cell-specific enhancer. In special cases, however, such mixing and matching phenomena may have adverse effects, resulting in ectopic expression of a proto-oncogene and thus contributing to malignant transformation (reviewed in Lang and Spandidos, 1988).
Reprogramming of genes during evolution may happen frequently in many organisms, including Drosophila, where the spacer DNA between genes contains a great variety of enhancers with particular temporal and cell type specificities (Wilson et al., 1989; Bellen et al., 1989). Thus, in higher eukaryotes the most convenient way to extend or restrict the specificity of a given gene may be by adding or deleting remote enhancer sequences.
Acknowledgments
We are grateful to Michael Schatt, Dr. David Arnosti, and Dr. Keith Harshman for critical reading of the manuscript and for valuable discussions. We also thank Silvia Oberholzer for help in the preparation of the manuscript, Rudolph Meszlenyi for synthesizing the oligonucleotides, and Fritz Ochsenbein for graphic artwork. This work was supported by the Canton of Zurich and a grant from the Swiss National Science Foundation, No. 31-25650.88.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Abel T. and Maniatis T. (1990), Nature 341, 24–25. [DOI] [PubMed] [Google Scholar]
- Atchison M. L. and Perry R. P. (1987), Cell 48, 121–128. [DOI] [PubMed] [Google Scholar]
- Banerji J., Rusconi S., and Schaffner W. (1981), Cell 27, 299–308. [DOI] [PubMed] [Google Scholar]
- Banerji J., Olson L., and Schaffner W. (1983), Cell 33, 729–740. [DOI] [PubMed] [Google Scholar]
- Benoist C. and Chambon P. (1981), Nature 290, 310–315. [DOI] [PubMed] [Google Scholar]
- Bellen H. J., O’Kane C. J., Wilson C., Grossniklaus U., Pearson R. K., and Gehring W. J. (1989) Genes Dev 3, 1288–1300. [DOI] [PubMed] [Google Scholar]
- Camper S. and Tilghman S. (1989), Genes Dev 3, 537–546. [DOI] [PubMed] [Google Scholar]
- Carter A. D., Felber B. K., Walling M., Jubier M.-F., Schmidt C. J., and Hamer D. H. (1984), Proc Natl Acad Sci USA 81, 7392–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran T. and Franza R. B. (1988), Cell 55, 395–397. [DOI] [PubMed] [Google Scholar]
- Dejong W. W., Hendriks W., Mulders J. W. M., and Bloemendal H. (1989), Trends Biol Sci 14, 365–368. [DOI] [PubMed] [Google Scholar]
- De Villiers J. and Schaffner W. (1983), in Techniques in the Life Sciences, B5, Techniques in Nucleic Acid Biochemistry B507 (Flavell R. A., ed.), Elsevier Scientific Publishers Ireland Ltd., pp. 1–20. [Google Scholar]
- Dreyfus M., Doyen N., and Rougeon F. (1987), EMBO J 6, 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durnam D. M., Hoffman J. S., Quaife C. J., Benditt E. P., Chen H. Y., Brinster R. L., and Palmiter R. D. (1984), Proc Natl Acad Sci USA 81, 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynan W. S. and Tjian R. (1983), Cell 35, 79–87. [DOI] [PubMed] [Google Scholar]
- Dynan W. S. and Tjian R. (1985), Nature 316, 774–778. [DOI] [PubMed] [Google Scholar]
- Falkner F. G., Neumann E., and Zachau H. G. (1984), Hoppe Seyler’s Z Physiol Chem 365, 1331–1343. [DOI] [PubMed] [Google Scholar]
- Fischer J. and Maniatis T. (1988), Cell 53, 451–461. [DOI] [PubMed] [Google Scholar]
- Foster J., Stafford J. and Queen C. (1985), Nature 315, 423–425. [DOI] [PubMed] [Google Scholar]
- Garcia J. V., Bich-Thuy L. T., Stafford J., and Queen C. (1986), Nature 322, 383–385. [DOI] [PubMed] [Google Scholar]
- Gerster T., Matthias P., Thali M., Jiricny J., and Schaffner W. (1987), EMBO J 6, 1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert W. (1978), Nature 271, 501. [DOI] [PubMed] [Google Scholar]
- Gillies S. D., Morrison S. L., Oi V. T., and Tonegawa S. (1983), Cell 33, 717–728. [DOI] [PubMed] [Google Scholar]
- Gosh S., Gifford A. M., Riviere L. R., Tempst P., Nolan G. P., and Baltimore D. (1990), Cell 62, 1019–1029. [DOI] [PubMed] [Google Scholar]
- Grosschedl R. and Baltimore D. (1985), Cell 41, 885–897. [DOI] [PubMed] [Google Scholar]
- Hiromi Y. and Gehring W. J. (1987), Cell 50, 963–974. [DOI] [PubMed] [Google Scholar]
- Hu Y-F., Lüscher B., Admon A., Mermod N., and Tjian R. (1990), Genes Dev 4, 1741–1752. [DOI] [PubMed] [Google Scholar]
- Johnson P. F. and McKnight S. L. (1989), Ann Rev Biochem 58, 799–839. [DOI] [PubMed] [Google Scholar]
- Jones N. (1990), Cell 61, 9–11. [DOI] [PubMed] [Google Scholar]
- Kadonaga J. T., Jones K. A., and Tjian R. (1986), Trends Biochem Sci 11, 20–23. [Google Scholar]
- Karin M., Haslinger A., Holtgreve H., Richards R. I., Krauter P., Westphal H. M., and Beato M. (1984), Nature 308, 513–519. [DOI] [PubMed] [Google Scholar]
- Karim F. D., Urness L. D., Thummel C. S., Klemsz M. J., McKercher S. R., Celada A., Van Beveren C., Maki R. A., Gunther C. V., Nye J. A., and Graves B. J. (1990), Genes Dev 4, 1451–1453. [DOI] [PubMed] [Google Scholar]
- Kieran M., Blank V., Logeat F., Vandekerckhove J., Lottspeich F., Le Bail O., Urban M. B., Kourilsky P., Baeuerle P. A., and Israel A. (1990), Cell 62, 1007–1018. [DOI] [PubMed] [Google Scholar]
- Krämer H., Niemöller M., Amouyal M., Revet B., Wilcken-Bergmann B., and Müller-Hill B. (1987), EMBO J 6, 1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landschulz W. H., Johnson P. F., Adashi E. Y., Graves B. J., and McKnight S. L. (1988), Genes Dev 2, 786–800. [DOI] [PubMed] [Google Scholar]
- Lang J. C. and Spandidos D. A. (1986), Anticancer Res 6, 437–450. [PubMed] [Google Scholar]
- Lenardo M., Pierce J. W., and Baltimore D. (1987), Science 236, 1573–1577. [DOI] [PubMed] [Google Scholar]
- Lenardo M. and Baltimore D. (1989), Cell 58, 227–229. [DOI] [PubMed] [Google Scholar]
- Levinson B., Khoury G., Van de Woude G., and Gruss P. (1982), Nature 295, 568–572. [DOI] [PubMed] [Google Scholar]
- Lewin B. (1990), Cell 61, 1161–1164. [DOI] [PubMed] [Google Scholar]
- Lobell R. B. and Schleif R. (1990), Science 250, 528–532. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E., and Sambrook J. (1982), Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Maniatis T., Goodbourn S., and Fischer J. A. (1987), Science 236, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Mason J., Williams G., and Neuberger M. (1985), Cell 41, 479–487. [DOI] [PubMed] [Google Scholar]
- Mathis D. J. and Chambon P. (1981), Nature 290, 310–315. [DOI] [PubMed] [Google Scholar]
- Melton D. A., Krieg P. A., Rebagliati M. R., Maniatis T., Zinn K., and Green M. R. (1984), Nucl Acids Res 12, 7035–7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K. B. and Neuberger M. (1989), EMBO J 8, 1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. and Tjian R. (1989), Science 245, 371–378. [DOI] [PubMed] [Google Scholar]
- Mizushima-Sugano J. and Roeder R. G. (1986), Proc Natl Acad Sci USA 83, 8511–8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau P., Hen R., Wasylyk B., Everett R., Gaub M., and Chambon P. (1981), Nucl Acids Res 9, 6047–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau-Gachelin F., Ray D., Tambourin P. and Tavitian A., Klemsz M. J., McKercher S. R., Celada A., Van Beveren C., and Maki R. A. (1990), Cell 61, 1165–1166. [DOI] [PubMed] [Google Scholar]
- Müller M. M., Gerster T., and Schaffner W. (1988a), Eur J Biochem 176, 485–495. [DOI] [PubMed] [Google Scholar]
- Müller M. M., Ruppert S., Schaffner W., and Matthias P. (1988b), Nature 336, 544–551. [DOI] [PubMed] [Google Scholar]
- Müller H-P., Sogo J. M., and Schaffner W. (1989), Cell 58, 767–777. [DOI] [PubMed] [Google Scholar]
- Müller H-P. and Schaffner W. (1990), Trends Genet 6, 300–304. [DOI] [PubMed] [Google Scholar]
- Murre C., McCaw P. S., Vaessin H., Caudy M., Jan L. Y., Jan Y. N., Cabrera C. V., Buskin J. N., Hauschka S. D., Lassar A. B., Weintraub H., and Baltimore D. (1989), Cell 58, 537–544. [DOI] [PubMed] [Google Scholar]
- Neuberger M. (1983), EMBO J 2, 1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare P., Goding C. R., and Haigh A. (1988), EMBO J 7, 4231–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter R. D., Chen H. Y., and Brinster R. L. (1982), Cell 29, 701–710. [DOI] [PubMed] [Google Scholar]
- Pape L. K., Windle J. J., Mougey E. B., and Sollner-Webb B. (1989), Mol Cell Biol 9, 5093–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parslow T., Jones S., Bond B., and Yamamoto K. (1987), Science 235, 1498–1501. [DOI] [PubMed] [Google Scholar]
- Picard D. and Schaffner W. (1984), Nature 307, 80–82. [DOI] [PubMed] [Google Scholar]
- Picard D. and Schaffner W. (1985), EMBO J 4, 2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick L., Schier A., Affolter M., Schmidt-Glenewinkel T., and Gehring W. J. (1990), Genes Dev 4, 1224–1239. [DOI] [PubMed] [Google Scholar]
- Ptashne M. (1986), Nature 322, 697–701. [DOI] [PubMed] [Google Scholar]
- Queen C. and Baltimore D. (1983), Cell 33, 741–748. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P., Ransone L., Lamph W., and Verma I. (1988), Nature 336, 692–695. [DOI] [PubMed] [Google Scholar]
- Schatt M. D., Rusconi S., and Schaffner W. (1990), EMBO J 9, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R. and Baltimore D. (1986), Cell 46, 705–716. [DOI] [PubMed] [Google Scholar]
- Serfling E., Jasin M., and Schaffner W. (1985), Trends Genet 1, 224–230. [Google Scholar]
- Steiner C., Muller M., Baniahmad A., and Renkawitz R. (1987), Nucl Acids Res 15, 4163–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Grossniklaus U., Herr W., and Hernandez N. (1988), Genes Dev 2, 1764–1778. [DOI] [PubMed] [Google Scholar]
- Theisen M., Stief A., and Sippel A. E. (1986), EMBO J 5, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J.-P., Kassis J. A., and O’Farrell P. H. (1990), EMBO J 9, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall L., de Boer E., and Grosveld F. (1988), Genes Dev 2, 1089–1100. [DOI] [PubMed] [Google Scholar]
- Westin G., Gerster T., Müller M. M., Schaffner G., and Schaffner W. (1987), Nucl Acids Res 15, 6787–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C., Pearson R. K., Bellen H. J., O’Kane C. J., Grossniklaus U., and Gehring W. J. (1989), Genes Dev 3, 1301–1313. [DOI] [PubMed] [Google Scholar]
- Wirth T., Staudt L., and Baltimore D. (1987), Nature 329, 174–178. [DOI] [PubMed] [Google Scholar]