

Presently, there exist two distinct human retro-virus families: the human T-cell leukemia (HTLV) and the human immunodeficiency viruses (HIV). Viruses from both groups infect human T-4 lymphocytes, with HTLV-1 infection leading to immortalization, while infection with HIV results in cell death. Unlike the prototype murine and avian retroviruses, the human retroviruses encode numerous non-structural proteins with diverse regulatory functions. Two HIV proteins, referred to as Tat and Rev, are essential positive regulators of gene expression. Both regulate virus gene expression through interaction with RNA target elements present within the 5′ untranslated leader sequence and envelope gene, respectively. Most recent studies suggest that these interactions in themselves are not sufficient to confer regulation without the presence of additional host cell factors.
Tat
The HIV-1 Tat protein (Fig. 1), which is conserved amongst HIV-2 and the simian immunodeficiency viruses, is encoded by two exons, one which precedes the env gene and codes for a 76 amino acid protein, and the second within env, coding for an additional 12 amino acids (Arya et al., 1985; Sodroski et al., 1985). From mutational analysis it is clear that the first 58 amino acids confer full activity (Siegel et al., 1986). Mutational analysis also suggests the presence of at least three distinct functional domains. Present at the amino terminus is a small group of acidic amino acids. This region has been proposed to have a periodicity of acidic, polar, and hydrophobic residues (Rappaport et al., 1989), a feature common to the activation domain of several well characterized transcription factors. Whether this region of Tat serves a similar function awaits further study.
Figure 1.
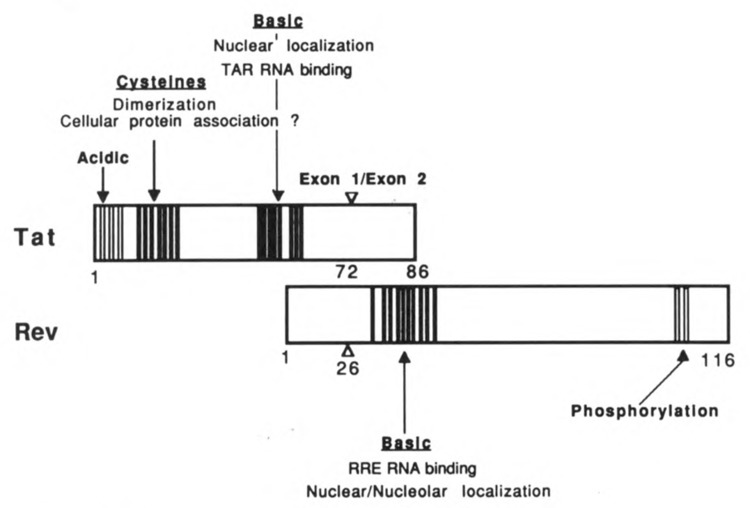
Location of functional domains within the HIV-1 Tat and Rev proteins.
The highly conserved cluster of 7 cysteine residues, which are present within a span of 20 amino acids, are thought to constitute a second functional domain. In vitro studies suggest that these residues bind metal ions and promote dimerization of Tat (Frankel et al., 1988). As mutation of all but one of these residues abolishes function (Ruben et al., 1989; Sadaie et al., 1988), the ability of the cysteines to coordinate with metal is likely to serve an important function. However, the ability of both Tat peptides and Tat proteins, lacking the cysteine residues, to interact with RNA (see below) indicates that the physiological function of metal-linked dimerization is something other than RNA binding.
The carboxy terminal domain of Tat encompasses a group of positively charged amino acids. These residues serve at least two essential functions. First, residues 48–52 (GRKKR) encode a nuclear signal motif (Hauber et al., 1989; Ruben et al., 1989) which is sufficient to direct heterologous, normally cytoplasmic, proteins to the nucleus (Ruben et al., 1989). The basic amino acids are also required for interaction of Tat with its target RNA (Roy et al., 1990).
Interaction of Tat with TAR RNA: mechanistic insights
The mechanism of Tat function has been the subject of much debate and at present is not completely understood. The target sequence for Tat, referred to as TAR (Fig. 2), is present between nucleotides +1 to +60 (Rosen et al., 1985; Hauber and Cullen, 1988), placing TAR in both DNA and RNA. Function of TAR is position-and orientation-dependent, with maximum activity achieved when it is present at the extreme 5′ terminus of the mRNA transcript. Several studies, including direct demonstration that Tat interacts with TAR containing RNA (Roy et al., 1990; Weeks et al., 1990) and not DNA, support the hypothesis that TAR functions as an RNA target. It has also been shown that TAR can be replaced with heterologous RNA target elements (Southgate et al., 1990; Selby et al., 1990) or DNA (Berkhout et al., 1990), and that transactivation can be achieved using chimeric Tat fusion proteins, whereby the fusion partner interacts with the replacement target sequence. At initial glance, these findings suggest that only Tat need be present, and that TAR functions to bring Tat in proximity with other functional sequences near the promoter. However, since the level of transactivation achieved in these studies is generally quite small (i.e., less than 10% of wild-type), the data suggest that cellular factors which recognize TAR, or other regions of the LTR, play an important role in transactivation. Consistent with this prediction, evidence for interaction of multiple cellular factors with TAR (Jones et al., 1988; Gatignol et al., 1989; Gaynor et al., 1989) and surrounding sequences (Garcia et al., 1987) has been obtained. Since mutations which alter the terminal loop in the TAR element disrupt both transactivation (Feng and Holland, 1988) and the ability of a 68 kD cellular factor to bind (Marciniak et al., 1990), the studies suggest that this factor may play an important role in transactivation. In vitro findings showing that addition of the p68 protein enhances transactivation by Tat lends further support to this hypothesis (Marciniak et al., 1990a).
Figure 2.
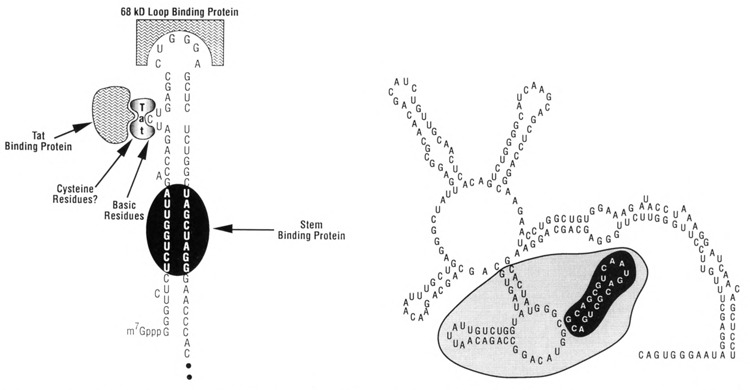
RNA target sequences recognized by Tat and Rev. Some of the many protein interactions occurring with the TAR RNA structure (left) are indicated. The predicted secondary structure assumed by the Rev response element, RRE (Malim et al., 1989b), is shown (right), with the area required for interaction with Rev protein shaded.
A clear picture of how the interaction of Tat and/or cellular factors with TAR RNA regulates HIV gene expression has yet to emerge. Studies demonstrating transcriptional and post-transcriptional effects have suggested that Tat can function at several levels to control gene expression.
The best example of Tat functioning at the posttranscriptional level can be derived from studies in Xenopus oocytes (Braddock et al., 1989). When heterologous RNA transcripts containing the TAR region are injected into the nucleus of Xenopus oocytes, transactivation is obtained in the presence of Tat, even in the presence of transcriptional inhibitors. Moreover, Tat has no effect when co-injected with TAR RNA in the cytoplasm. Thus, in Xenopus the function of Tat is independent of transcription and may affect the translational competence of nuclear TAR RNA. However, when purified functional Tat protein is added to mammalian cells in the presence of transcription inhibitors, no transactivation is observed (Gentz et al., 1989). Therefore, the data do not support a mechanism whereby Tat interacts directly with TAR RNA to affect its transport, stability, or translation. The discrepancies between these two systems further suggest that Tat can function at several levels and imply that factors involved in transactivation in Xenopus may be unique to this system and different from those present in mammalian cells.
Earlier results obtained in human and non-human cell lines have suggested that Tat functions to prevent premature termination of transcription (Kao et al., 1987). In these studies the overall rate of transcription initiation was found to be constant, in contrast to other reports suggesting that Tat functions to enhance transcription initiation (Cullen et al., 1986; Muesing et al., 1987). Since removal of TAR has little effect on the basal activity of the LTR, indicating that it is not a negative element, the genetics of this system do not fully support the anti-termination model but do not rule out a role for Tat in elongation. Furthermore, evidence that Tat enhances elongation has been obtained both in vivo (Laspia et al., 1989; Laspia et al., 1990) and in vitro (Marciniak et al., 1990a). In these studies an effect on transcription initiation cannot be ruled out.
The ability of Tat to elicit clear transcriptional effects through interaction with an RNA target poses several mechanistic possibilities. If we view TAR as an RNA enhancer (Sharp and Marciniak, 1989), a function which has yet to be identified in other systems, the interaction of Tat with TAR may facilitate formation of a transcription factor complex at the promoter, the enhancer, or both. Similar cooperative mechanisms are known to exist for DNA-associated promoter and enhancer factors, and are believed to enhance transcriptional activity. If the interaction of Tat with TAR has the same effect, then the requirement that TAR be at the 5′ terminus of the transcript suggests that the Tat-TAR interaction must occur close to the promoter. Thus, although producing the same effect as a DNA enhancer, the RNA-mediated enhancement of transcription would have less flexibility.
Alternatively, the interaction of Tat with TAR may serve a function analogous to that of phage lambda N protein, whereby N can engage RNA polymerase to prevent premature termination of transcription (Horwitz et al., 1987). Furthermore, the target for N, referred to as the nut site, is present downstream from the start of transcription initiation and is thought to be recognized from an RNA target (Lazinski et al., 1989).
If either of the above scenarios is correct, it remains likely that the interaction of Tat with TAR promotes interaction of cellular factors with TAR itself or with Tat. In support of the latter possibility, recent studies show that Tat associates with at least one nuclear protein, designated TBP-1 (Nelbock et al., 1990). Although the role of the TBP-1 interaction has yet to be determined, it is possible that interaction of Tat with TBP-1 and/or other cellular factors forms part of a transcription initiation or elongation complex involving multiple interactions between protein, DNA, and RNA.
A role for Tat in modulation of cellular gene expression?
Most recent studies suggest that the action of Tat is not confined to regulation of virus gene expression and support possible roles for Tat in regulation of cellular processes. Studies with transgenic mice expressing Tat provide clear evidence for one such additional function (Vogel et al., 1988). Several of these animals develop a syndrome similar to Kaposi’s sarcoma, a malignancy prevalent among HIV-infected individuals. Moreover, Tat has also been found to enhance the growth of Kaposi’s sarcoma-derived tissue in culture (Ensoli et al., 1990). This latter observation, together with the absence of Tat in affected cells, supports the notion that Tat may behave as a growth factor or cytokine. Although this has yet to be proven, and evidence for in vivo circulation of Tat has not been obtained, in vitro studies show that Tat is readily taken up by cells when placed in the extracellular environment (Frankel and Pabo, 1988; Gentz et al., 1989). In other in vitro studies, addition of Tat to culture medium has been found to suppress antigen-induced proliferation of T-lymphocytes (Viscidi et al., 1989). As this finding closely parallels that observed in infected individuals, it has been suggested that circulating Tat, if it does indeed exist, may contribute to the immu-nosuppression associated with AIDS.
Rev, an antirepressor protein
The Rev protein (Fig. 1) represents another essential HIV regulatory protein which functions through an RNA target element. A clue to Rev’s existence first came to light from a phenotypic observation that proviral mutants containing frameshift deletions in the region now known to encode Rev, did not yield detectable levels of env or gag gene products (Sodroski et al., 1986). It is now believed that Rev mediates the export of nuclear entrapped viral structural mRNA to the cytoplasm (Felber et al., 1989; Emerman et al., 1989; Malim et al., 1989b). If HIV gene expression is divided into early and late phases, analogous to that observed with DNA tumor viruses, the following scenario may be envisioned. In the absence of Rev, virus would be unable to progress to the structural phase of gene expression (late) and would remain trapped in the regulatory phase of replication (early). Indeed, evidence exists that early in infection one sees expression of the regulatory proteins Tat, Rev, and Nef, which is then followed by the appearance of incompletely spliced transcripts encoding the structural gene products (Kim et al., 1989).
The Rev protein, like Tat, is conserved among various members of the lentivirus family. The genes encoding Rev and Tat overlap, with each being produced from a different reading frame. Rev is a 19 kDa nuclear phosphoprotein (Cullen et al., 1988) found almost exclusively in the nu-cleolus (Cochrane et al., 1990b). Although the role of nucleolar localization remains obscure, studies from other systems, which demonstrate that nucleolar proteins shuttle to the cytoplasm, would be consistent with Rev’s involvement in mediating transport of HIV structural mRNAs.
There is general agreement that Rev, through an interaction with an RNA structure, RRE (Fig. 2), serves to activate gene expression (Daly et al., 1989; Zapp and Green, 1989; Cochrane et al., 1990a). However, it probably does so indirectly by overcoming negative effects exerted by other regions of the genome. In earlier studies the existence of cis-acting negative elements (termed CRS sequences), dispersed throughout the HIV genome, which suppress gene expression in Rev’s absence were identified (Rosen et al., 1988; Hadzopoulou-Cladaras et al., 1989). Other studies imply that inefficient splicing of HIV transcripts, which results in nuclear accumulation of unspliced precursors, may be responsible for the lower level of expression of the structural genes (Malim et al., 1989b). In an artificial Rev-dependent system involving hybrid globin RRE transcripts, lacking either a functional splice donor or acceptor sequence, Rev has been shown to “rescue” the nuclear entrapped RNA and mediate its transport to the cytoplasm (Chang and Sharp, 1989). However, Rev can also act on mRNA that lacks functional splice sites. To reconcile these potential differences, it can be hypothesized that factors which bind to inefficient or mutated splice sites (exemplified by the hybrid globin RRE RNA) and possibly cryptic splice sites present within the CRS elements, elicit nuclear entrapment of HIV structural mRNA. The mechanism for nuclear entrapment of the HIV structural mRNA represents an area clearly in need of further study.
Whatever the mechanism of entrapment may be, there is agreement that the Rev response element, termed RRE, through an interaction with Rev likely functions independently of these negative elements to mediate transport of the nuclear entrapped mRNA to the cytoplasm. The RRE element (Malim et al., 1989b), originally referred to as CAR, for cis-acting antirepression sequence (Dayton et al., 1988), forms a complex RNA secondary structure (Malim et al., 1989b). Mutational analysis supports the existence of secondary structure within this region and further suggests that secondary structure, as opposed to primary nucleotide sequence, is the major determinant for interaction with Rev (Olsen et al., 1990; Malim et al., 1990; Heaphy et al., 1990). In support of this hypothesis, compensatory mutations that maintain secondary structure, but alter primary nucleotide sequence in the stem loop structures required for Rev interaction, interact with Rev and are functional in vivo. Similar findings have been obtained with HIV-2 Rev protein which interacts with RNA secondary structures generated within HIV-2 RRE RNA (Dillon et al., 1990). Further investigations should answer whether some degree of primary nucleotide sequence, in addition to secondary structure, is required for the Rev-RRE interaction.
Transdominant Rev mutants
The ability of Rev to interact with RRE RNA is not sufficient in itself to restore gene expression. There exist Rev mutants which bind to RRE RNA, yet do not restore gene expression (Olsen et al., 1990). Co-transfection studies have shown that mutation of amino acids 78 and 79 of Rev produce a protein which functions as a trans-dominant suppressor of Rev function (Malim et al., 1989a). Studies with additional mutants in this region, as well as mutations within the same region of the HTLV-1 Rex protein, suggest that this region in both proteins functions as an activation domain. RNA binding studies with these HIV transdominant suppressor mutations demonstrate that they form a stable interaction with RRE RNA, comparable to that obtained with authentic Rev protein (Olsen et al., 1990). Thus, the transdominant suppression is not attributable to failure to interact with RRE RNA. The ability of the transdominant Rev proteins to restore gene expression, yet interact with RRE RNA, indicates that binding alone is insufficient for function. This suggests that binding of additional cellular factors to either Rev or the Rev-RRE complex is necessary for function. The accumulated data therefore suggest that at least two steps are required for Rev function: association of Rev with RRE RNA, followed by interaction of cellular factors with the Rev-RRE complex to mediate the productive export of structural mRNA from the nucleus to the cytoplasm. As with Tat, identification and elucidation of function of cellular factors required for Rev function will likely aid in our understanding of HIV gene expression and provide further insight into novel regulatory pathways.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Arya S. K., Guo C., Josephs S. F., and Wong-Staal F. (1985), Science 229, 69–73. [DOI] [PubMed] [Google Scholar]
- Berkhout B., Gatignol A., Rabson A. B., and Jeang K.-T. (1990), Cell 62, 757–767. [DOI] [PubMed] [Google Scholar]
- Chambers M., Wilson A., Esnouf W., Adams M. P., Kingsman S. E., Kingsman A. J., and Braddock S. M. (1989), Cell 58, 269–279. [DOI] [PubMed] [Google Scholar]
- Chang D. D. and Sharp P. A. (1989), Cell 59, 789–795. [DOI] [PubMed] [Google Scholar]
- Cochrane A. W., Chen C.-H., and Rosen C. (1990a), Proc Natl Acad Sci USA 87, 1198–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane A. W., Perkins A., and Rosen C. A. (1990b), J Virol 64, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen B. R. (1986), Cell 46, 973–982. [DOI] [PubMed] [Google Scholar]
- Cullen B. R., Hauber J., Campbell K., Sodroski J. G., Haseltine W. A., and Rosen C. A. (1988), J Virol 62, 2498–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly T., Cook K., Gray G., Maione T., and Rusche J. (1989), Nature 342, 816–819. [DOI] [PubMed] [Google Scholar]
- Dayton A. I., Terwilliger E. F., Potz J., Kowalski M., Sodroski J. G., and Haseltine W. A. (1988) J Acquir Immune Defic Syndr 1, 441–452. [PubMed] [Google Scholar]
- Dillon P. J., Nelbock P., Perkins A., and Rosen C. A. (1990), J Virol 64, 4428–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerman M., Vazeux R., and Peden K. (1989), Cell 57, 1155–1165. [DOI] [PubMed] [Google Scholar]
- Ensoli B., Barillari G., Salahuddin S. Z., Gallo R. C., and Wong-Staal F. (1990), Nature 345, 84–86. [DOI] [PubMed] [Google Scholar]
- Felber B. K., Hadzopoulou-Cladaras M., Cladaras C., Copeland T., and Pavlakis G. N. (1989), Proc Natl Acad Sci USA 86, 1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S. and Holland E. C. (1988), Nature 334, 165–167. [DOI] [PubMed] [Google Scholar]
- Frankel A. D. and Pabo C. O. (1988), Cell 55, 1189–1193. [DOI] [PubMed] [Google Scholar]
- Frankel A. D., Bredt D. S., and Pabo C. O. (1988), Science 240, 70–73. [DOI] [PubMed] [Google Scholar]
- Garcia J. A., Wu F. K., Mitsuzasu R., and Gaynor R. B. (1987), EMBO J 6, 3761–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatignol A., Kumar A., Rabson A., and Jeang K. T. (1989), Proc Natl Acad Sci USA 86, 7828–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor R., Soultanakis E., Kuwabara M., Garcia J., and Sigman D. S. (1989), Proc Natl Acad Sci USA 86, 4858–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentz R., Chen C.-H., and Rosen C. A. (1989), Proc Natl Acad Sci USA 86, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadzopoulou-Cladaras M., Felber B. K., Cladaras C., Athanassopoulos A., Tse A., and Pavlakis G. N. (1989), J Virol 63, 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauber and B. Cullen J. (1988), J Virol 62, 673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauber J., Malim M. H., and Cullen B. R. (1989), J Virol 63, 1181–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy S., Dingwall C., Ernberg I., Gait M. J., Green S. M., Karn J., Lowe A. D., Singh M., and Skinner M. A. (1990), Cell 60, 685–693. [DOI] [PubMed] [Google Scholar]
- Horwitz R. J., Li J., and Greenblat J. (1987), Cell 51, 631–641. [DOI] [PubMed] [Google Scholar]
- Jones K. A., Luciw P. A., and Duchange N. (1988), Genes Dev 2, 1101–1114. [DOI] [PubMed] [Google Scholar]
- Kao S. Y., Caiman A. E., Luciw P. A., and Peterlin B. M. (1981), Nature 330, 489–493. [DOI] [PubMed] [Google Scholar]
- Kim S., Byrn R., Groopman J., and Baltimore D. (1989), J Virol 63, 3708–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laspia M., Rice A., and Mathews M. (1989), Cell 59, 283–292. [DOI] [PubMed] [Google Scholar]
- Laspia M. F., Rice A. P., and Mathews M. B. (1990), Genes Dev 4, 2397–2408. [DOI] [PubMed] [Google Scholar]
- Lazinski D., Grzsadzielska E., and Das A. (1989), Cell 59, 207–218. [DOI] [PubMed] [Google Scholar]
- Malim M. H., Bohnlein S., Hauber J., and Cullen B. R. (1989a), Cell 58, 205–214. [DOI] [PubMed] [Google Scholar]
- Malim M. H., Hauber J., Le S.-Y., Maizel J. V., and Cullen B. R. (1989b), Nature 338, 254–257. [DOI] [PubMed] [Google Scholar]
- Malim M. H., Tiley L. S., McCarn D. E., Rusche J. R., Hauber J., and Cullen B. R. (1990), Cell 60, 675–683. [DOI] [PubMed] [Google Scholar]
- Marciniak R. A., Garcia-Blanco M. A., and Sharp P. A. (1990), Proc Natl Acad Sci USA 87, 3624–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak R. A., Calnan B. J., Frankel A. D., and Sharp P. A. (1990a), Cell 63, 791–802. [DOI] [PubMed] [Google Scholar]
- Muesing M. A., Smith D. H., and Capon D. J. (1987), Cell 48, 691–701. [DOI] [PubMed] [Google Scholar]
- Nelbock P., Dillon P. J., Perkins A., and Rosen C. A. (1990), Science 248, 1650–1653. [DOI] [PubMed] [Google Scholar]
- Olsen H., Nelbock P., Cochrane A., and Rosen C. (1990), Science 247, 845–848. [DOI] [PubMed] [Google Scholar]
- Olsen H. S., Cochrane A. W., Dillon P. J., Nalin C. M., and Rosen C. A. (1990), Genes Dev 4, 1357–1364. [DOI] [PubMed] [Google Scholar]
- Rappaport J., Lee S. J., Khalili K., and Wong-Staal F. (1989), New Biol 1, 101–110. [PubMed] [Google Scholar]
- Rosen C. A., Sodroski J. G., and Haseltine W. A. (1985), Cell 41, 813–823. [DOI] [PubMed] [Google Scholar]
- Rosen C. A., Terwilliger E., Dayton A., Sodroski J. G., and Haseltine W. A. (1988), Proc Natl Acad Sci USA 85, 2071–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Delling U., Chen C.-H., Rosen C. A., and Sonnenberg N. (1990), Genes Dev 4, 1365–1374. [DOI] [PubMed] [Google Scholar]
- Ruben S., Perkins A., Purcell R., Joung K., Sia R., Burghoff R., Haseltine W. A., and Rosen C. A. (1989), J Virol 63, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M. R., Benter T., and Wong-Staal F. (1988), Science 239, 910–913. [DOI] [PubMed] [Google Scholar]
- Selby M. J. and Peterlin B. M. (1990), Cell 62, 769–776. [DOI] [PubMed] [Google Scholar]
- Siegel L. J., Ratner L., and Josephs S. F. (1986), Virology 148, 226–231. [DOI] [PubMed] [Google Scholar]
- Sodroski J., Patarca R., Rosen C., Wong-Staal F., and Haseltine W. (1985), Science 229, 74–77. [DOI] [PubMed] [Google Scholar]
- Sodroski J., Goh W. C., Rosen C., Dayton A., Terwilliger E., and Haseltine W. A. (1986), Nature 321, 412–417. [DOI] [PubMed] [Google Scholar]
- Southgate C., Zapp M. L., and Green M. R. (1990), Nature 345, 640–642. [DOI] [PubMed] [Google Scholar]
- Viscidi R. P., Mayor K., Lederman H. M., and Frankel A. D. (1989), Science 246, 1606–1608. [DOI] [PubMed] [Google Scholar]
- Vogel J., Hinrichs S. H., Reynolds R. K., Luciw P. A., and Jay G. (1988), Nature 335, 606–611. [DOI] [PubMed] [Google Scholar]
- Weeks K. M., Ampe C., Schultz S. C., Steitz T. A., and Crothers D. M. (1990), Science 249, 1281–1285. [DOI] [PubMed] [Google Scholar]
- Zapp M. and Green M. (1989), Nature 342, 714–716. [DOI] [PubMed] [Google Scholar]