

Abstract
Bacteriophage T7 capsid protein 10B has previously been proposed to arise by a translational frameshift near the 3′ end of the capsid gene 10A coding sequence, adding an additional 53 amino acid residues to the carboxyl-terminal end of the protein. Here we show by peptide mapping experiments as well as by direct partial sequence analysis of an overlapping “junction” peptide, that 10B is in fact related to 10A by a −1 switch in reading frame in a narrow region near the carboxy terminus of 10A. Peptide mapping experiments demonstrate that 10A and 10B have the same amino terminus as well as virtually identical methionine-labeled peptide maps. However, the predicted unique carboxyl-terminal peptide from 10B was also identified. An overlapping peptide was isolated from 10B which spans the junction region in which the proposed translational frameshift is thought to occur. Partial sequencing of this junction peptide confirms a −1 frameshift within the last few codons of 10A.
Gene 10 of bacteriophage T7 encodes a 344 amino acid (36,414 daltons) protein which is the major capsid protein of the phage coat and is known as 10A. A larger size capsid protein 10B (∼41,800 daltons), whose function is obscure, is made in smaller amounts (∼10% of 10A). Dunn and Studier (1983) proposed from DNA sequence analysis that a −1 translational frameshift occurring at a run of 4 U residues spanning codons 340–341 of 10A mRNA would add an additional 53 amino acids at the carboxyl end of the protein (see Fig. 1). Supporting evidence for the proposed translational frameshift can be summarized as follows (as reviewed in Dunn and Studier, 1983): gene 10 amber mutants lack 10B as well as 10A. Both 10A and 10B are made in vitro from purified gene 10 mRNA. A coding sequence past the 10A termination codon is required for 10B synthesis. Since 10B contains no tryptophan, it cannot arise by in-frame read-through of the 10A termination codon, because read-through would lead to incorporation of a tryptophan residue downstream (see Fig. 1).
Figure 1.
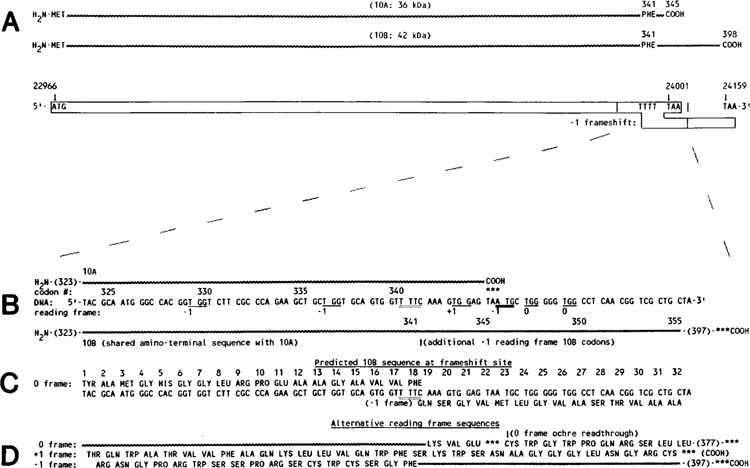
Bacteriophage T7 Gene 10. A. Schematic diagram of the two protein products 10A and 10B. B. DNA sequence at the proposed frameshift site. The DNA sequence where the −1 frameshift could occur (T-TTC) is indicated, and potential tryptophan (TGG) coding triplets are underlined. C. Predicted reading frame sequence of 10B through the proposed frameshift site. D. Alternate reading frame sequences at the proposed frameshift site; *** indicates stop codons.
In this study, we present peptide mapping experiments which demonstrate that both 10A and 10B show the same amino terminus, as well as very similar methionine-labeled peptide maps; however, a distinct carboxyl-terminal tryptic peptide was isolated from 10B which shows a pattern of amino acid labeling predicted by the DNA sequence for a −1 translational frame-shift. Further, a tryptic peptide was also isolated from 10B which appears to span the junction between the 10A and 10B sequences; partial sequencing of this peptide across the junction provides direct confirmation of a −1 frame-shift within the last few codons of the 704 coding region.
Materials and methods
Analysis of amino terminal and [35S]-methionine labeled gene 10 peptides by peptide mapping
Purified T7 RNA polymerase was used to prepare gene 10 mRNA from plasmid pAR436 DNA, a pBR322 derivative which carries a clone of gene 10 beginning at bp 22,858 of T7 DNA, ahead of the ϕ10 promoter, and ending at bp 24,273, after the Tϕ terminator (Dunn and Studier, 1983). The in vitro transcribed RNA was used to direct protein synthesis in a rifampicin treated cell-free system as described (Goldman, 1982), using E. coli strain BL15 (ma - , rel - ; Studier, 1975). The gene 10 amino terminus was specifically labeled by using N-formyl-[35S]-methionyl-tRNA (Goldman and Lodish, 1972), whereas gene 10 methionine-containing peptides were labeled using [35S]-methionine. Proteins made from the in vitro reaction mixtures were precipitated by 5% TCA, and the pellets were then washed with 90% acetone before being dissolved in sample buffer for SDS-polyacryl-amide gel electrophoresis (performed essentially as described in Ausubel et al., 1987). Following autoradiography of the dried gel, the 10A and 10B bands (apparent molecular weights 38 and 45 kDa; Studier, 1972) were excised from the gel, washed sequentially with 25% isopropanol then 10% methanol, dried, and placed in 1% NH4HCO3. The N-formyl-[35S]-methionyl-tRNA labeled amino terminal peptides were digested/ eluted from the gel slice (Goldman, 1982) at 37°C with 50 μg/ml trypsin (Worthington) overnight, followed by an additional 7 hours of digestion with 50 μg/ml S. aureus V8 protease (Millipore). The [35S]-methionine labeled peptides were digested/eluted with 50 μg/ml S. aureus V8 protease overnight, followed by a 7 hour digestion with 50 μg/ml α-chymotrypsin (Millipore). Following digestion, peptide mapping (Goldman and Hatfield, 1979) was carried out as follows: the reaction supernatant was lyophilized 3 times and subjected for two hours to electrophoresis at pH 3.5 (5% acetic acid, 0.5% pyridine) on Whatman 3mm paper at 4 kV in a Varsol-cooled tank. After autoradiography of the dried electrophoretogram on Kodak X-OMAT XAR-5 x-ray film, individual peptide bands were excised, sewn onto a fresh sheet of Whatman 3mm, and subjected to a second dimension of electrophoresis at pH 1.9 (8% acetic acid, 2% formic acid) for 2 hours at 4 kV, followed by autoradiography.
Labeling of the gene 10 clone and isolation of C-terminal tryptic peptide by peptide mapping
Strain BL21(DE3)/pAR3625, pLysS was used to synthesize 10A and 10B in vivo. Plasmid pAR3625 (ampr), which is slightly smaller than pAR436, lacks a cryptic E. coli promoter that is just upstream of the ϕl0 promoter in pAR436; otherwise it is identical to pAR436. Plasmid pAR3625, which contains a cloned sequence beginning at bp 22,880 of T7 DNA, is more stable than pAR436 because it has a lower level of expression in uninduced cells. BL21(DE3) is a lysogen that has the gene for T7 RNA polymerase under control of the IPTG inducible lac UV5 promoter, and pLysS (cmr) is a plasmid that supplies a low level of T7 lysozyme which reduces the basal activity of T7 RNA polymerase (Studier et al., 1990); this further improves pAR3625 plasmid stability. Cultures of BL21-(DE3)/pAR3625, pLysS were started from single colonies and grown to mid-log phase in M9 medium (Miller, 1972) containing chloramphenicol (25 μg/ml), and ampicillin (20 μg/ml), then induced with 0.4 mM IPTG for 30 minutes, prior to pulse labeling with [14C] amino acids (0.5 M.Ci/ml) for 4 minutes. The following [U-14C]-labeled L-amino acids from Amersham Corp. were used (specific activities in millicuries per millimole are given within parentheses): Leu (342), Ile (342), Val (285), Thr (228), Glu (285), Ala (171), Arg (342), Lys (324). Incorporation of label was monitored by removing a 10 nl aliquot into 25 μl 0.1 M NaOH, incubating at 37°C for 10 minutes, adding 35 μl of 10% TCA and 1 drop of BSA (0.5 mg/ml) as carrier; precipitation was on ice for 30 minutes before filtering through glass fiber filters and counting in a Packard 3255 scintillation counter in 4 g/1 of Omnifluor (New England Nuclear) dissolved in toluene. Protein synthesis was similarly terminated by addition of NaOH to 0.2N and incubation for 10 minutes at 37°C. Cultures were then made 10% TCA, precipitated on ice for 30 minutes, then centrifuged at 10,000 rpm for 10 minutes. The protein pellets were washed twice with 5% TCA, twice with ethanol, once with ether, then lyophilized to dryness. Samples were dissolved in 200 μl of buffer containing 0.01% sodium phosphate (pH 7.2), 1% SDS, 6M urea, 1% β-mercaptoethanol and 0.01% bromophenol blue. It usually required 20–30 minutes of incubation at 37°C and moderate mixing to dissolve the sample pellets. A 3mm thick agarose gel system designed for electrophoretic separation of SDS-denatured proteins (Pro Sieve Gel System, FMC) was utilized. This consisted of a resolving gel of 6% agarose in a Tris-borate buffer (pH 8.5), and a stacking gel solution of 1.5% agarose in Tris-HCl buffer (pH 6.8) and 5 ng/ml phenol red as a tracking dye. Running conditions were 40 V constant voltage for 17 hours, then 80 V for 3 hours until the dye front ran off the gel. The gel was then dried under vacuum without heat and subjected to autoradiography. The two major protein bands, 10A and 10B, were cut out of the dried gel, and each was dissolved in 1 ml of DMSO. After 48 hours at 37°C, the dissolved samples were dialyzed twice for 6 hours each time against 2 liters of 1% NH4HCO3 at 37°C. The samples were then removed from the dialysis tubing and centrifuged (10,000 rpm for 5 minutes) to remove any rehydrated agarose gel fragments. The supernatant was digested with 0.5 mg/ml TPCK-trypsin (Worthington) at 37°C for 4 hours in a 1% solution of NH4HCO3. The agarose gel pellets were resuspended and similarly digested, then centrifuged again to remove the gel fragments. The initial supernatant (∼ ⅔ of cpm recovered) and the second supernatant (from the agarose pellet digestion/extraction, ∼ ⅓ of cpm recovered) were pooled and lyophilized to dryness in a speed vac (Savant). The samples were resuspended in buffer at pH 3.5 (5% acetic acid, 0.5% pyridine) and subjected to electrophoresis on Whatman 3mm paper for 3.5 hours at 4.5kV, followed by autoradiography. Identity of peptides was deduced by the presence or absence of radioactive marker amino acids run in parallel tracks. Candidates for the 10B C-terminal peptide were subjected to two additional dimensions of electrophoresis, first at pH 1.9 (8% acetic acid, 2% formic acid), and next at pH 4.7 (2% acetic acid, 2% pyridine).
Partial sequence analysis of the overlapping “junction” peptide
Strain BL21(DE3)/pAR3625, pLysS, described above, was used to produce the 104 and 10B proteins, labeled with both [35S]-methionine and [3H]-alanine. Following SDS-PAGE, the 10A and 10B bands were located by autoradiography; individual gel slices containing the separated bands were excised and subjected to elution and digestion with a solution containing trypsin, similar to the procedure described above. The resulting peptide mixture was again subjected to SDS-PAGE, this time on a 20% polyacryla-mide gel. Following autoradiography, a large 705-specific methionine-containing peptide band was excised from the gel; the gel slice was rehydrated and crushed to release this peptide band into solution. The peptide was then subjected to multiple rounds of Edman degradations on a Beckman 890C Microsequencer by Dr. C. W. Anderson and J. Wysocki at Brook-haven National Laboratory’s microprotein facility. The positions of elution of 35S and 3H labels were determined by liquid scintillation counting.
Results
Peptide map comparisons of 10A and 70S demonstrate close similarity of the proteins
Figure 1 depicts the two gene 10 products 10A and 10B and both of the proposed open reading frames (boxed). Protein 10B presumably arises by a −1 frameshift during translation of the gene 10 mRNA at or near a run of 4 U residues in the C-terminal region of 10A spanning codons 340(val)–341(phe). Having established that a coding sequence past the 10A termination codon (nucleotide 24001) is required for 10B synthesis, Dunn and Studier (1983) performed experiments that localized the site of the frameshift (see the expanded portion of the gene 10A C-terminal region of Figure 1). These experiments were based on sequence analysis of 10A which is predicted to contain no tryptophan. Depending on where the reading frame switch to the −1 frame occurs prior to the 10A termination codon, the 10B protein would either contain or not contain tryptophan. Since [3H]-tryptophan labeling of proteins made during T7 infection revealed no incorporation into 10A or 10B, the frameshift must occur after codon 337. A −1 translational frameshift after codon 337 of the gene 10 mRNA predicts that the amino acid sequence of 10B should be identical to 704 except for approximately the last 50 additional amino acids generated by the bypass of the 10A termination codon (24001) to the next in-frame (−1) termination codon (24159). We tested this prediction of common protein sequence between 10A and 10B in a series of peptide mapping experiments. Figure 2 presents a peptide mapping experiment designed to test whether both proteins share the same amino terminus. A T7 gene 10 clone, pAR436 (Dunn and Studier, 1983), was used to direct in vitro transcription by T7 RNA polymerase. The resulting RNA was translated in a rifampicin-treated S-30 extract with N-formyl-[35S]-methionyl-tRNA as the label. Following protein synthesis, the 10A and 10B proteins were separated on an SDS polyacrylamide gel and visualized by autoradiography as presented in Figure 2, panel I. The 10A (apparent molecular weight 38kDa) and 10B bands (apparent molecular weight 45kDa) were subjected to trypsin digestion/elution from the dried gel slice (Goldman, 1982), followed by a second digestion with S. aureus V8 protease (which cleaves after Glu). The 10A and 10B digests were applied side by side on Whatman 3mm paper and subjected to electrophoresis for comparison of peptides. Panel II shows the comigration of three peptide bands (A,B,C) in 10A and 10B. Each of the three peptide bands was cut out, sewn onto a fresh sheet of paper, and subjected to another dimension of electrophoresis, presented in panel III. As predicted, the two proteins share identical amino terminal peptide maps, as each peptide band present in 10A is also present in 10B. The presence of several bands from a single amino terminus is believed to reflect both formylated and deformylated ends, as well as some variability in extent of protease digestions.
Figure 2.
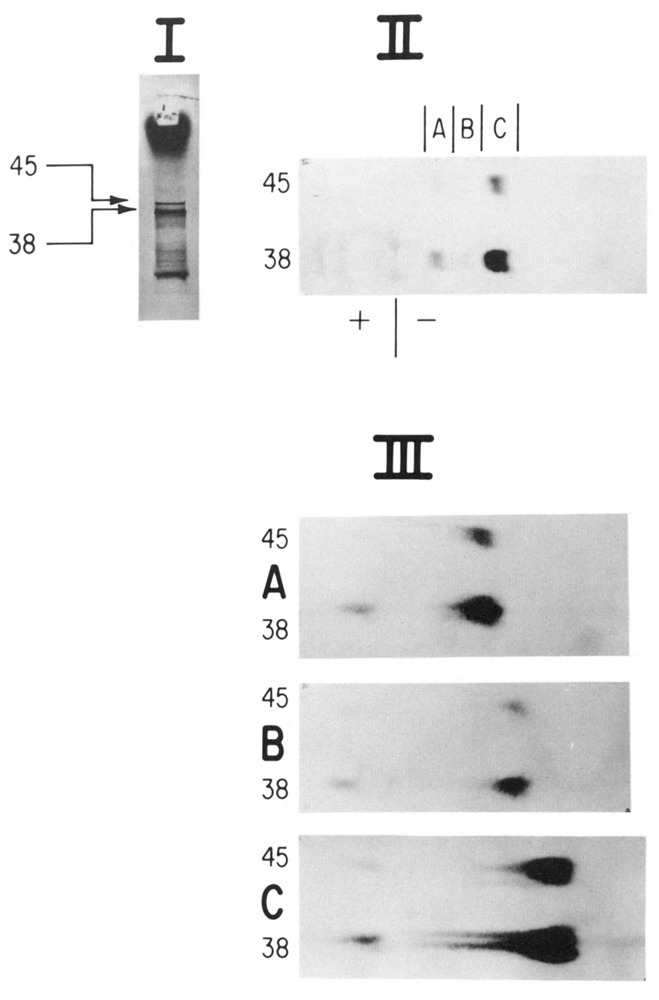
Analysis of amino terminal peptides of 10A and 10B by peptide mapping. In vitro synthesized gene 10 mRNA was used to direct protein synthesis in a rifampicin treated S-30 as described (Goldman, 1982). The gene 10 amino terminus was specifically labeled using N-formyl-[35S]-methionyl-tRNA (Goldman and Lodish, 1972). Proteins made from the in vitro reaction mixtures were subject to SDS-PAGE (panel I). Following autoradiography of the dried gel, the 10A and 10B bands (apparent molecular weights 38 and 45 kDa; Studier, 1972) were excised from the gel and digested first with trypsin (50 μg/ml) and next with S. aureus V8 protease (50 μg/ml). The 10A and 10B peptides were then applied side by side on Whatman 3mm paper and subjected to high voltage paper electrophoresis at pH 3.5 (5% acetic acid, 0.5% pyridine) and exposed to film (panel II). Individual peptide bands (A,B,C) were excised from the pH 3.5 electrophoretogram, sewn onto a fresh sheet of Whatman 3mm, and subjected to another electrophoresis at pH 1.9 (8% acetic acid, 2% formic acid), followed by autoradiography (panel III).
We sought to establish further common sequence identity between the two proteins in a second peptide mapping experiment. Figure 3 compares [35S]-methionine-labeled gene 10 proteins which were isolated and treated in a manner similar to that in Figure 2; however S. aureus V8 protease was used in the first digestion/elution from the gel slice, followed by a second digestion with chymotrypsin. Figure 3, panel II reveals the isolation of 14 shared peptide regions between 10A and 10B (sections A through N) after paper electrophoresis. The next dimension of electrophoresis in panel III (6 of these regions are presented) shows virtually identical peptide maps between the two proteins, indicating similar sequence. From DNA sequence analysis, the 10B protein is predicted to contain one additional methionine (encoded by double underscored triplet in Figure 1B) containing peptide; however, this peptide was not resolved under these conditions of separation.
Figure 3.
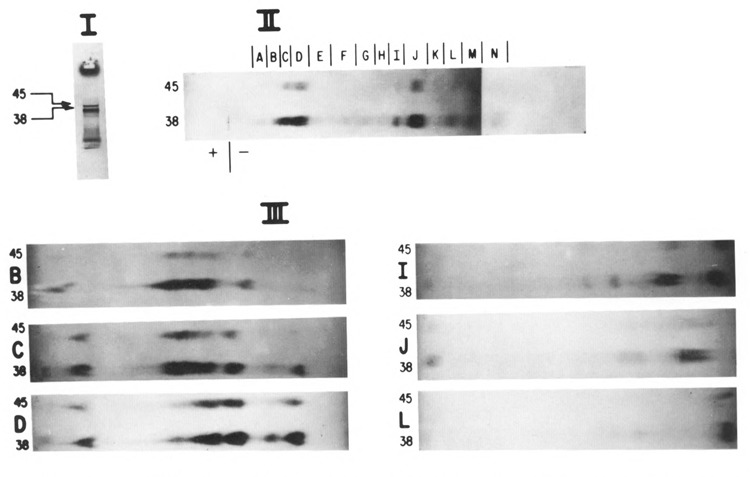
Analysis of [35S]-methionine labeled peptides of 10A and 10B by peptide mapping. The same procedures were utilized as in Figure 2; however, the proteins were labeled with [35S]-methionine, separated by SDS-PAGE (panel I), then subjected to digestion initially with S. aureus V8 protease (50 μg/ml) followed by chymotrypsin (50 μg/ml). Individual peptide regions (panel II) were identified as A through N; they were cut out and subjected to electrophoresis at pH 1.9 and exposed to film (6 of these regions are shown in panel III).
Isolation of unique carboxyl-terminal peptide from 10B
Having established common sequence identity between 10A and 10B, an additional peptide mapping experiment was performed to demonstrate that 10B is different from 10A in the C-terminal region after the frameshift (predicted codons 342–397). We set out to identify specifically the predicted tryptic peptide fragment of 10B containing the 10B C-terminus. [14C] amino acids were used to label proteins produced in a host strain bearing a plasmid with the gene 10 clone under control of a T7 RNA polymerase promoter. Selection of the appropriate [l4C] amino acid markers (leu, ile, val, thr, glu, ala, arg, lys) in parallel cultures was expected to allow us to differentiate the predicted 11 amino acid C-terminal tryptic peptide (glu-ala-glu-leu-ala-ala-ala-thr-ala-glu-gln)from all other peptides in 10B or 10A.
In Figure 4, labeled gene 10 proteins were first separated, then removed from an SDS denaturing 6% agarose protein gel, digested with trypsin, and subjected to three sequential dimensions of separation by paper electrophoresis. Panel I displays the peptide map obtained with the first paper electrophoretogram. 10B peptides were spotted on the left 9 lanes of the origin, whereas 10A peptides were spotted in parallel on the right 9 lanes of the origin. Trypsin digestion will always yield peptides with either lysine or arginine at the end, with the sole exception of the actual carboxy terminal peptide of the entire protein, which will not contain either lysine or arginine (unless by chance the protein normally ends with one of these residues). Therefore, our first criterion for identifying potential regions containing the carboxy terminal peptide of 10B was to find a portion of the electrophoretogram which was not labeled by either lysine or arginine, but which was labeled by other amino acids predicted to be in this peptide (glu, leu, ala, and thr). Further, a potential carboxy terminal peptide of 10B had to also fulfill the criterion of being absent from the digests of 10A. A region in the first electrophoretogram was observed (“C-T” in panel I) which appeared to meet these criteria. This region was excised, sewn onto a fresh sheet of Whatmann 3mm paper, and subjected to a second dimension of electrophoresis at pH 1.9 (not shown). The peptide identified in this electrophoresis had the predicted pattern of labeling for the 10B C-terminal peptide, with the exception of comigrating valine label (valine is predicted to be absent from this peptide). Therefore, the peptide was again excised and subjected to a third dimension of electrophoresis, at pH 4.7. Figure 4, panel II presents this last dimension (pH 4.7) electrophoretogram showing the isolation of a peptide unique to 10B containing the appropriate amino acid markers for this C-terminal tryptic peptide, but with a faint band in the (10B) valine position. Analysis of the valine label stock revealed a slight radioactive leucine contamination; leucine is predicted to be present in this peptide and would thus appear to explain this faint band.
Figure 4.
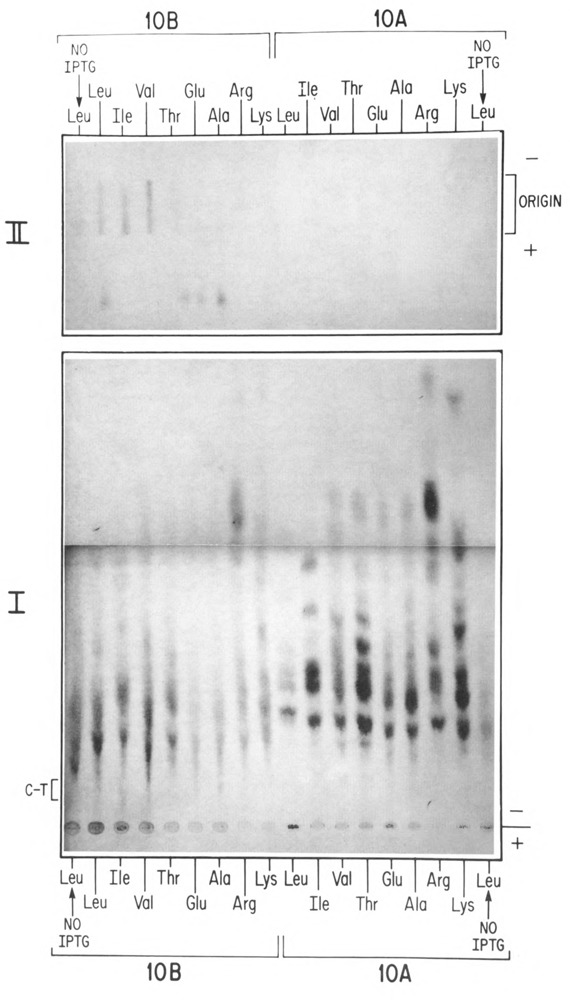
Peptide mapping of the 10B C-terminal tryptic peptide. Induced cultures of BL21(DE3)/pAR3625, pLysS were labeled with amino acids predicted to be in the 10B C-terminal tryptic peptide (glu-ala-glu-leu-ala-ala-ala-thr-ala-glu-gln). As described in Materials and Methods, selection of the appropriate [14C] amino acid markers (leu, ile, val, thr, glu, ala, arg, lys) in parallel cultures was expected to allow us to specifically identify the C-terminal peptide. Following labeling, the 10A and 10B proteins were separated on an SDS denaturing agarose protein gel, removed from the gel, and digested with trypsin (0.5 mg/ml). The peptides were then spotted on Whatman 3mm for electrophoresis at pH 3.5. 10B peptides were applied on the left portion of the origin, whereas 10A peptides were applied in parallel on the right portion of the origin (panel I). Candidates for the 10B C-terminal peptide (C-T), were subjected to two additional dimensions of electrophoresis and autoradiography, first at pH 1.9 (not shown), and next at pH 4.7 (2% acetic acid, 2% pyridine) (panel II). The outer leucine-labeled lanes (No IPTG) are from uninduced cells, which were included for analysis of background incorporation.
Partial sequence analysis of the junction peptide
An experiment aimed at confirming directly the frameshift at the amino acid sequence level is presented in Figure 5A–C. In the strain bearing the plasmid with the gene 10 clone, proteins were labeled with [35S]-methionine and [3H]-alanine and separated on an SDS-polyacrylamide gel (Fig. 5A). The 10A and 10B bands were each excised and subjected to digestion with trypsin; the resulting peptides were separated on another SDS-polyacrylamide gel (with 20% polyacrylamide). If a −1 frameshift in fact occurred near the end of the coding region for 10A, the predicted “junction” tryptic peptide, overlapping 10A into the 10B reading frame, was expected to be rather large (at least 54 residues) and would contain 11 alanine and 2 methionine residues.
Figure 5.
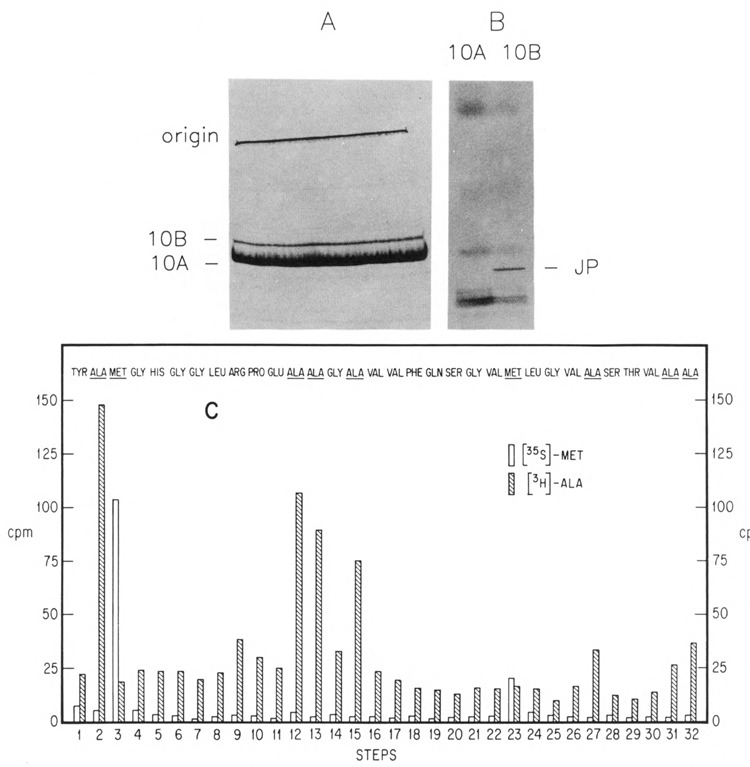
Partial sequence analysis of the overlapping “junction” peptide. A,B. Isolation of the junction peptide. Induced cultures of BL21(DE3)/pAR436 were labeled with [MS]-methionine and PH]-alanine and then subjected to preparative SDS-PAGE on a 10% gel followed by autoradiography (A). The positions of the labeled 10A and 10B proteins are indicated. The bands were excised and digested with trypsin, as described above. The resulting peptide mixture was again subjected to SDS-PAGE, on a 20% gel (B). Shown is an autoradiogram of the gel, with the putative junction peptide (“JP”) indicated. C. Thirty-two cycles of Edman degradation of the junction peptide. Released counts were monitored for each step: 35S [left (open) bar at each step] and 3H [right (hatched) bar at each step]. The predicted amino acid sequence for the first 32 residues of the junction peptide is also shown, aligned to each step. Note that trypsin is not expected to cleave the Arg-Pro bond at positions 9–10 of the peptide.
A large, sharp, methionine-containing peptide band was obtained from 10B (Fig. 5B) and considered a likely candidate for this junction peptide, since it was missing from the 10A digest. The peptide was removed from the gel slice, subjected to repeated cycles of Edman degradation, and monitored for release of the [35S]-methionine and [3H]-alanine labels. The pattern of 35S and 3H obtained matched precisely the predicted pattern for the putative junction peptide. Release of methionine and alanine counts corresponded to the steps where these residues are predicted to be found if Phe-tRNA (which reads both UUC and UUU) slipped back one base at amino acid position 18 in the junction peptide. Thus, the locations of the methionine and alanine residues in this peptide indicate that the frameshift occurs after codon 337, to the −1 reading frame predicted sequence. The frameshift site thus narrows to a position between codon 338 and the 10A termination codon (345), with the run of 4 U’s at codons 340–341 the most likely site of the shift. Complete amino acid sequencing across the junction has confirmed this to be the site of the shift (B. Condron, J. Atkins, and R. Gesteland, submitted to J Bacteriol).
Discussion
In this report we have provided further biochemical evidence for the site and nature of the bacteriophage T7 gene 10 frameshift. The data support the location of the shift as originally proposed (Dunn and Studier, 1983). Peptide mapping experiments show that both 10A and 10B have the same amino terminus, and that both proteins have virtually identical methionine-labeled S. aureus V8/chymotryptic peptide maps. Only one additional methionine-labeled peptide would be expected in 10B; however, this peptide was not resolved in our mapping experiments.
Another peptide mapping experiment was intended to isolate and identify the predicted unique carboxyl-terminal tryptic peptide from 10B. The strategy of this experiment was to label the peptide with a series of radioactive markers in parallel incorporations. A unique combination of markers which were both included and excluded from separated peptides was the criterion for identifying candidates for the C-terminal peptide. A labeling pattern was obtained which fit very well the predicted sequence of the 10B C-terminal tryptic peptide, further substantiating the nature of the frameshift.
Finally, a large 10B-specific tryptic peptide was isolated on a polyacrylamide gel; repeated cycles of Edman degradations indicated this to be the junction peptide overlapping 10A and the frameshift into the 10B reading frame. The alanine and methionine residues found in this junction peptide matched precisely their anticipated locations from the DNA sequence of the two reading frames. Complete amino acid sequencing across the junction between the two reading frames has been accomplished by another group (B. Condron, J. Atkins, and R. Gesteland, submitted to J Bacteriol); this has confirmed that the site of the −1 frameshift is at the run of 4 U residues spanning codons 340–341 of 10A mRNA, as originally proposed by Dunn and Studier (1983).
Although we believe that the 10B protein arises as a consequence of a translational frame-shift by ribosomes during reading of the 10A message, an alternate explanation for the origin of the 10B protein is that T7 RNA poly-merase somehow slips and adds a base during transcription, giving rise to a subpopulation of frameshifted messages from this template, which would then be translated to yield the 10B protein. Arguing against this alternate explanation is the observation that a messenger RNA preparation from this template was shown to yield only the 10A protein in a mammalian in vitro translation system, while yielding both 10A and 10B proteins in an E. coli in vitro translation system (J. Atkins and J. Dunn, unpublished experiments). Although there may be several reasons why the mammalian extract failed to synthesize 10B, the result nevertheless does suggest that the frameshift is a property of the E. coli translation apparatus, and is not a consequence of a subpopulation of messenger RNAs.
Acknowledgments
This work was supported by National Institutes of Health grant GM27711 and the Office of Health and Environmental Research of the United States Department of Energy. E. G. was the recipient of a Research Career Development Award from the National Cancer Institute (CA00890).
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
Diane Stassi is currently at Abbott Laboratories, Abbott Park, IL 60064.
References
- Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., and Struhl K., eds. (1987), in Current Protocols in Molecular Biology, Analysis of Proteins, vol. 2 John Wiley & Sons, New York, pp. 10.2.1–10.2.9. [Google Scholar]
- Dunn J. J. and Studier F. W. (1983), J Mol Biol 166, 477–535. [DOI] [PubMed] [Google Scholar]
- Goldman E. (1982), J Mol Biol l58, 619–636. [DOI] [PubMed] [Google Scholar]
- Goldman E. and Hatfield G. W. (1979), Methods Enzymol 59, 292–309. [DOI] [PubMed] [Google Scholar]
- Goldman E. and Lodish H. F. (1972), J Mol Biol 67, 35–47. [DOI] [PubMed] [Google Scholar]
- Miller J. H. (1972), in Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 431–432. [Google Scholar]
- Studier F. W. (1972), Science 176, 367–376. [DOI] [PubMed] [Google Scholar]
- Studier F. W. (1975), J Bacteriol 124, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W., Rosenberg A. H., Dunn J. J., and Dubendorff J. W. (1990), Methods Enzymol 185, 60–89. [DOI] [PubMed] [Google Scholar]