

Abstract
We previously purified a yeast protein kinase that specifically hyperphosphorylates the carboxylterminal repeat domain (CTD) of RNA polymerase II largest subunit and showed that this CTD kinase consists of three subunits of 58, 38, and 32 kDa. We have now cloned, sequenced, and characterized CTK1, the gene encoding the 58 kDa a subunit. The CTK1 gene product contains a central domain homologous to catalytic subunits of other protein kinases, notably yeast CDC28, suggesting that the 58 kDa subunit is catalytic. Cells that carry a disrupted version of the CTK1 gene lack the characterized CTD kinase activity, grow slowly and are cold-sensitive, demonstrating that the CTK1 gene product is essential for CTD kinase activity and normal growth. While ctk1 mutant cells do contain phosphorylated forms of the RNA polymerase II largest subunit, these forms differ from those found in wild type cells, implicating CTK1 as a component of the physiologically significant CTD phosphorylating machinery. As befitting an enzyme with a nuclear function, the N-terminal region of the CTK1 protein contains a nuclear targeting signal.
The carboxyl-terminal domain (CTD) of the largest subunit (subunit IIa, 210 ± 10 kDa) of eukaryotic RNA polymerase II comprises multiple repeats of a unique heptamer sequence, Tyr-Ser-Pro-Thr-Ser-Pro-Ser (Allison et al, 1985; Corden et al., 1985; Zehring et al., 1988; reviewed in Corden, 1990). Hyperphosphorylation of this domain in vivo and in vitro generates a form of the subunit with reduced mobility in SDS polyacrylamide gels, subunit IIo (Dahmus, 1981; Buhler et al., 1976; Bell et al., 1977; Cadena and Dahmus, 1987; Lee and Greenleaf, 1989; Cisek and Corden, 1989; Guilfoyle, 1989; Corden, 1990). As one approach to investigating the phosphorylation of the CTD, we used CTD-containing fusion proteins as substrates and purified a CTD kinase to near homogeneity from the yeast Saccharomyces cerevisiae; we also detected similar activities in extracts of insect and mammalian cells (Lee and Greenleaf, 1989). The yeast CTD kinase consists of three subunits (α, β, γ) of 58, 38, and 32 kDa, respectively, and it extensively phosphorylates the CTD of the largest subunit of RNA polymerase II to generate a mobility-shifted band in SDS gels. The properties of the yeast CTD kinase enzyme, including substrate specificity, cyclic nucleotide independence, and subunit composition, reveal that the enzyme is distinct from previously described protein kinases. A notable feature of CTD phosphorylation by the yeast kinase is its apparent processivity or cooperativity (Lee and Greenleaf, 1989).
Using as substrate short synthetic peptides containing 4 or 6 heptamer repeats, Cisek and Corden (1989) purified a mouse cell CTD kinase composed of two subunits, the smaller of which (34 kDa) is a murine homologue of the Schizosaccharomyces pombe cell cycle control protein, cdc2, itself the functional homologue of S. cerevisiae CDC28 (see also Zhang and Corden, 1991a). However, results presented below argue that this mouse enzyme is not the mammalian counterpart of the yeast CTD kinase we have characterized. Another mammalian protein kinase activity was partially purified on the basis of its phosphorylation of a synthetic repeat peptide (Stevens and Maupin, 1989); intriguingly, this activity is inhibited by the nucleoside analogue DRB, which affects transcription in vitro and in vivo (reviewed in Sawadogo and Sentenac, 1990). In addition, a repeat peptide phosphorylating activity has been detected in stressed HeLa cells (Legagneux et al., 1990). How these mammalian activities (and a plant CTD kinase activity [Guilfoyle, 1989]) may be related to each other or to the yeast CTD kinase is not yet clear. For example, the activity from mouse does not seem to display the apparent processivity that characterizes the yeast enzyme (Zhang and Corden, 1991b).
While the CTD is essential in vivo (Nonet et al., 1987; Zehring et al., 1988; Bartolomei et al., 1988; Allison et al., 1988), it is not required for accurate transcription at several promoters in vitro (Zehring et al., 1988; Kim and Dahmus, 1989; Thompson et al., 1989; Zehring and Greenleaf, 1990; Buratowski and Sharp, 1990), although it is required for some (Thompson et al., 1989). Neither its specific functions nor the role of its phosphorylation has been determined, though several possibilities have been suggested (Allison et al., 1985; Corden et al., 1985; Sigler, 1988; reviewed in Corden, 1990; also see Discussion). Studies on the in vitro phosphorylation of mammalian polymerase IIA initiating at the adenovirus–2 major late promoter suggest that CTD phosphorylation occurs after polymerase’s interaction with the promoter but before initiation of transcription (Laybourn and Dahmus, 1990). However, since equally accurate and efficient initiation and elongation are carried out by RNA polymerase II entirely lacking the CTD (Kim and Dahmus, 1989; see also Zehring et al., 1988), the functional significance of the observed phosphorylation is not yet clear. Determining the relevance of phosphorylation to in vitro initiation and elongation will require establishing a system in which transcription is dependent on the presence and phosphorylation state of the CTD.
With the purification of the yeast CTD kinase, it became feasible to consider cloning genes encoding the kinase subunits as a foundation for genetic investigations which could help to elucidate the in vivo roles of CTD phosphorylation. Toward this end we have screened an expression library with antibodies raised against the CTD kinase, and we report here the isolation and characterization of CTK1, the gene encoding the α subunit of the enzyme. In addition, we present results of experiments utilizing ctk1 mutant cells which demonstrate that CTKl-containing CTD kinase plays an important role in vivo and which suggest that it is involved in physiologically significant CTD phosphorylation.
Materials and methods
Strains and media
The strains used in this study are S. cerevisiae DBY 1091 (a/α ura3–52/ura3–52 + lhis4 + lcan1 + lade2–101, ATCC #52278), SGY65 (a/α ura3–52/ura3–52 leu2–3,112/leu2–3,112 his3/his3 lys2/lys2 + lade2–101 + ltrp1, from Dr. S. Garrett, Duke University). A yeast strain containing 105/7 repeats of the CTD, CY199 (a HO-LacZ RPB1Δ:: HIS3 pRY2203 :LEU2 lys2– 801 ade2– 101 trp1Δ – 1 ura3– 52 leu2Δ – 1) was a gift from I. Herskowitz (University of California, San Francisco). Plasmid pRY2203 carries the rpbl allele from strain C3 (Nonet et al., 1987). Cells were cultured in YPD (1% yeast extract, 2% peptone, 2% glucose) or minimal media (2% glucose, 0.67% yeast nitrogen base without amino acids) supplemented with required nutrients as described (Sherman et al., 1986).
E. coli XL-1 Blue (Bullock et al., 1987) was obtained from Stratagene. E. coli cells were grown in LB broth (Maniatis et al., 1989).
Antibodies and Western blotting
To raise antibody against the CTD kinase, 0.1 mg of Mono S purified CTD kinase (about 50% pure) was subjected to electrophoresis in a 12% polyacrylamide SDS gel which was stained with 1 M KC1 at 4°C. The three bands of the kinase were cut out and frozen. They were then ground in a mortar in liquid N2, mixed with 0.5 ml complete Freund’s adjuvant (Gibco), and injected subcutaneously into a rabbit. A second injection using incomplete Freund’s adjuvant was performed similarly four weeks later. Serum was prepared two weeks after the second injection. Affinity purification of antibody against each subunit of the kinase was performed using purified CTD kinase bound to nitrocellulose membrane, essentially as described (Kelly et al., 1986). The nitrocellulose membrane with the enzyme bound was used several times, and purified antibodies were pooled. The titer of the purified antibodies was about 20-fold lower than that of the immune serum, as estimated by Western blot.
The 18-residue peptide corresponding to the C-terminus of CDC28 and the affinity-purified antibody against the peptide were kind gifts from Dr. S. Reed (Mendenhall et al., 1987). Where appropriate, the peptide at 1 μg/ml was preincubated with the antibodies for 30 minutes on ice.
Anti-DmE2 antibody was obtained from goat anti-RNA polymerase II serum (Weeks et al., 1982) by affinity purification using a protein that fused exon 2 of the Drosophila RpII215 gene (Jokerst et al., 1989) to β-galactosidase. This construct was made by inserting the exon 2-containing 2.5 kb EcoRI fragment of RpII215 into the EcoRI site of pUR288 (Rüther and Müller-Hill, 1983; construction by E. Wong, Cornell University). The fusion protein was induced by IPTG, and the insoluble fractions after lysis were solubilized using 8 M urea followed by dialysis as described (Rio et al., 1986). The protein was immobilized on Reactigel according to the manufacturer’s protocol (Pierce). Affinity purification was performed as described (Robbins et al., 1984).
Anti-CTD antibody (anti-CTD) was affinity-purified from serum of a rabbit injected with gel-purified RNA polymerase II largest subunit (Weeks et al., 1982) using immobilized β-gal-yeast CTD fusion protein (Y-FP of Lee and Greenleaf, 1989) as for anti-DmE2 antibody. Anti-phosphorylated CTD antiserum (anti-PCTD) was prepared by injection into a rabbit of yeast CTD fusion protein phosphorylated by CTD kinase in vitro. Affinity purification of IgG from the resulting immune serum was then performed as before, though using a column carrying phosphorylated CTD fusion protein.
Western blot analysis was carried out as described (Weeks et al., 1982; modified as Kelly et al., 1986). Bound antibody was detected by either [125I]protein A, or alkaline phosphatase-conjugated secondary antibody followed by reaction with Nitroblue tetrazolium (NBT, 100 μg/ml) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP, 50 μg/ml) in 50 mM Tris-HCl pH 9.6, 50 mM MgCl2 buffer. In some cases the chemiluminescent substrate AMPPD for alkaline phosphatase was used to detect bound antibody following supplied protocols (Western Light, Bios Inc.).
Screening of yeast genomic DNA library
A λgt11 library of yeast genomic DNA (from Dr. A. Sugino, NIEHS) was screened using a mixture of the affinity-purified antibodies against all three subunits at 1:10 dilution essentially as described (Hyunh et al., 1984). As secondary antibody, alkaline phosphatase conjugated goat anti-rabbit IgG antibody (Bio-Rad, 1:2000), was used, and bound antibody was visualized by reaction with NBT and BCIP as described above. Purified recombinant phages were lysogenized into E. coli Y1089 strain using the protocols described by Huynh et al. (1984). From the lysogens, fusion proteins were produced by induction with 5 mM IPTG for 2 hours at 37°C. Affinity purification of antibodies against the fusion proteins from these lysogens using nitrocellulose membrane was carried out as described above. Phage DNAs were purified by standard protocols (Maniatis et al., 1989).
One purified phage (λYJ31) from the screening was determined to contain the gene for the α subunit of CTD kinase by the following criteria: (1) the fusion protein produced by this phage reacted strongly with α subunit-specific affinity-purified antibody, (2) IgG from the anti-kinase serum affinity-purified using the αYJ31 fusion protein in turn strongly reacted with the a subunit of the enzyme, and (3) this reaction was proportional to the amount of kinase activity present in fractions from different stages of the kinase purification.
The 3.8 kb EcoRI insert DNA from this phage DNA was subcloned into pBluescript SK-(Stratagene) to give pBJ31 (Fig. 2).
Figure 2.
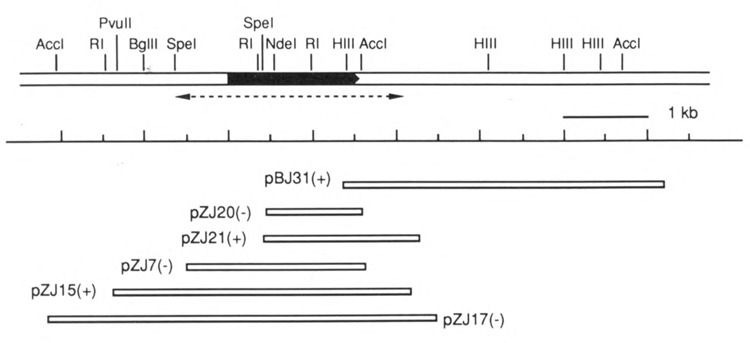
Restriction map of CTK1 gene and surrounding region. The black box represents the open reading frame, pointing in the 5′ to 3′ direction. The sequenced region is indicated as a dotted arrow underneath the map. The sizes of inserts subcloned in recombinant plasmids and their corresponding positions are drawn at the bottom. + / – indicates the orientation of the subcloned insert with respect to the β-galactosidase gene in the vector pSK-. Plasmid pBJ31 contains the insert DNA from phage λYJ31 subcloned into pBluescript SK-. All other plasmids, designated pZJ, were obtained from recombinant λZAP phage by in vivo excision procedures. RI: EcoRI; HIII: HindIII.
A 0.23 kb HindIII/AccI fragment from pBJ31 was tagged with Digoxigenin-dUTP by random-primed DNA labeling using the Genius system (Boehringer/Mannheim) following the protocols of the manufacturer. The λZAP (Short et al., 1988) library of yeast genomic DNA (from Dr. A. Sugino, NEIHS) was screened using the labeled DNA as probe. The hybridization and washing conditions were as described in the protocols (Genius), except that the incubation temperature was 58°C. Hybridized probe DNAs were reacted with alkaline phosphatase-conjugated anti-Digoxigenin antibody, and bound antibody was visualized by reaction with NBT and BCIP in 50 mM Tris-HCl pH9.6, 50 mM MgCl2) 100 mM NaCl buffer.
Phagemids (recombinant plasmids in pBlue-script SK-) were obtained from purified recombinant λZAP phage by an in vivo excision method using the manufacturer’s protocols (Stratagene; and also Short et al., 1988), and restriction enzyme mapped.
DNA sequence analysis
For nucleotide sequencing, a series of nested deletions was constructed from plasmids pZJ15 and pZJ17 DNAs (Fig. 2) using the ExoIII-Mung Bean Nuclease Kit from Stratagene. From the strains harboring the pBluescript SK- plasmids containing the resulting deleted CTK1 genes, single strand DNA was prepared using VCS-M13 helper phage essentially according to the manufacturer’s protocols (Stratagene). Sequencing was performed manually by the dide-oxynucleotide termination method (Sanger et al., 1977) and by the Dupont Genesis 2000 Automatic DNA Sequencer (Prober et al., 1987) using the 16 oligonucleotide M13 reverse primer (5′AACAGCTATGACCATG 3′).
Most computer analyses of DNA and amino acid sequences were performed on a Micro VAXII using the Genetics Computer Group programs (UWGCG, University of Wisconsin). Some analyses were done using the Bionet resource and the McGene Plus II programs. For searching for homologous sequences, both the EMBL and NBRF data bases were used.
DNA isolation and Southern blot
Genomic DNA was prepared from yeast grown in YPD (A600 = 1.0–2.0) following the protocols of Holm et al. (1986). Probe DNA was labeled with Digoxigenin-dUTP as above. Southern blotting was performed as described (Maniatis et al., 1989). Transfer of DNA from gel to Gene Screen nylon membrane (NEN Research Products) was performed using a Vacublot (ABN) by following the manufacturer’s procedures. Hybridized probe DNA was reacted with alkaline phosphatase-conjugated anti-Digoxygenin antibody and visualized by reaction with NBT and BCIP as described above.
The yeast chromosomes were prepared and separated by pulsed-field gel electrophoresis and blotted onto Nytran membrane as described (Rose et al., 1990). This membrane was hybridized with same CTK1 DNA probe described in the legend of Figure 6.
Figure 6.
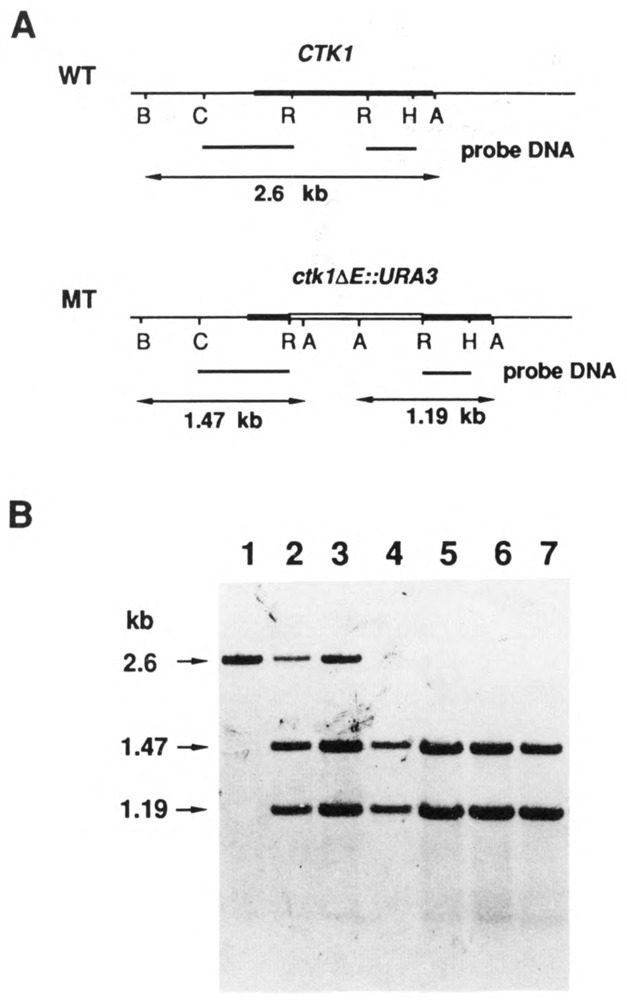
Southern blot analysis of gene disruptions. Total genomic DNAs were digested with restriction enzymes AccI and BglII and analyzed by Southern blotting as described in Materials and Methods (1% agarose gel). A. Schematic diagram of wild type and mutant CTK1 genes. The top line of each is a restriction map. Below that is shown the probe DNA fragments used for the Southern blotting. At the bottom are the expected sizes of DNA bands which will hybridize with the labeled DNA probes. B: BglII; C: ClaI; R: EcoRI; H: HindlII; A: AccI. B. Southern blot of genomic DNA from wild type diploid strain DBY1091 (lane 1), from two independent transformants of DBY 1091 (lanes 2 and 3), and from four haploid mutant ctk1 strains (lanes 4 to 7) derived meiotically from the diploids of lanes 2 and 3. The sizes of hybridized DNA bands are denoted at left. (The sibling wild type CTK1 haploids contained only the 2.6 kb band).
Plasmids and gene disruption
Gene disruption experiments were performed by a one-step gene replacement method (Rothstein, 1983). A mutant CTK1 gene, ctk1ΔE:: URA3, was constructed by substituting the CTK1 internal EcoRI fragment of 0.64 kb size with a 1.2 kb URA3 fragment. The URA3 EcoRI fragment was isolated from plasmid pGB310 (constructed by Dr. Craig Giroux, Wayne State University, and obtained from Dr. A. Sugino, NIEHS), in which the URA3 gene was subcloned into the EcoRI site of the β-galactosidase gene with the polylinker, EcoRI-BamHI-SacI-SmaI-HindIII, on both sides of the URA3 gene. A 2.86 kb BgIII-HindIII fragment of CTK1 was isolated from plasmid pZJ17 (Fig. 2) and subcloned into the BamHI and HindIII sites of the vector pSK-, so that only the two internal EcoRI sites were left. The resulting plasmid pDZ17 was digested with EcoRI and the DNA was isolated with the 0.64 kb internal EcoRI fragment deleted. This DNA was ligated with 1.2 kb EcoRI fragment of URA3, and a plasmid containing the mutant gene, pSZ17, was obtained.
The PSZ17 was digested with ClaI (nucleotides −295 in Figure 3 and one in the polycloning site in the vector), and the 2.37 kb DNA containing the ctk1ΔE ::URA3 construct was isolated and used to transform homozygous ura3 diploid strains DBY1091 and SGY65. Stable Ura+ transformants were selected on plates of synthetic minimal media lacking uracil, and the heterozygosity of the gene disruption of the CTK1 (CTK1/ctk1ΔE::URA3) was confirmed by Southern blot. These cells were sporulated, and the asci were dissected (Sherman et al., 1986) and incubated at 30°C on a YPD plate.
Figure 3.

Sequence of CTK1 gene. The nucleotide and amino acid sequences of the CTK1 gene and its open reading frame are shown. The 5′ region upstream of start ATG codon is expressed by - numbers. The total sequenced region covers from −654 to +1991. Every 30 nucleotides and every 10 amino acid residues are indicated by dots. Several restriction enzyme sites are also noted on the sequence. The polyadenylation signal is underlined.
Plasmid pJYC1511 was constructed by sub-cloning a 3.2 kb BgIII-Sa1I CTK1 DNA from pZJ15 (Fig. 3) into BamHI-Sa1I sites in plasmid pRS313 (Sikorski and Hieter, 1989). Domain I-deleted CTK1 gene, pZJ15dl, was constructed from pZJ15 by deletion of amino acid Tyr3 to Ser181 in CTK1 (Fig. 3) by oligonucleotide-directed mutagenesis (Kunkel, 1985) using the oligonucleotide GTAACAATGTCCGTTTACCTAAGG. Note that this deletion contains all of domain I and 17 amino acids in domain II preceding consensus sequences of protein kinase catalytic subunits. The resulting 2.75 kb BgIII-Sa1I fragment from this plasmid was subcloned into pRS313 BamHI-Sa1I sites, yielding pJYC1512. These plasmids were transformed into haploid ctk1 his3 strains obtained from tetrad analysis of SGY65 ctk1/CTK1 heterozygotes.
Yeast manipulations
Transformation of yeast was done using the LiCl/PEG 4000 method (Ito et al., 1983). Sporulation, spore dissection, and tetrad analysis were performed as described (Sherman et al., 1986). Diploid strains were constructed by mating appropriate haploids and selecting for prototrophy. To test viability of ctk1 in cells containing only shortened versions of the CTD, a ctk1 strain, SJY11 (a ctk1ΔE:: URA3 his3 leu2 trp1), obtained from tetrad analysis of transformed SGY65, was crossed with CY199 (Ura − Leu+ His+ CTDΔ105/7; see above), and diploids were selected by prototrophy. These diploids were sporulated and tetrads analyzed. Growth of spore colonies showing Ura+ Leu+ His+ phenotype, which are RPB1Δ ::HIS3 pRY2203:LEU2 (CTDΔ10) ctk1ΔE::URA3, was studied.
Assay of subunit ll0
Yeast cells were grown until A600 = 1.0 – 2.0 in YPD broth, and extracts were prepared by one of two methods. Method 1. Cells from 50 ml culture were centrifuged, washed with ice cold H2O, and resuspended in 1 ml ice cold buffer (25 mM Tris-HCl, pH7.8, 25 mM KCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 20 mM NaF, 0.5 mM NaV04). Cells were disrupted using a Mini-bead beater (Biospec Products) in 1.5 ml screw cap microfuge tubes containing 1 ml washed, cold glass beads (0.5 mm dia.), 3 times for 1 minute with 3-minute intervals on ice for cooling. The supernatant was removed, centrifuged for 5 minutes at 4°C, and used for immunoprecipitations (or Western blotting). Method 2. Cells from 5 ml culture were centrifuged, resuspended in 200 μl ethanol, and immediately placed on dry ice (samples could be stored at – 80°C for later use). Cells were disrupted by vortexing with glass beads (3 times for 1 minute with 1-minute intervals on dry ice). The resulting material was separated from the beads and collected by centrifugation through a small hole punched in the bottom of the tube. The pellet was dried, resuspended in 100 μl SDS sample buffer, and heated. Samples to be compared were run on SDS mini-gels which were stained with Coomassie Blue to estimate relative protein concentrations. We have found that this second method of extract preparation yields Western blot results virtually identical to the first method, except that it is more reproducible and more convenient; it was used for Figure 8A–C.
Figure 8.
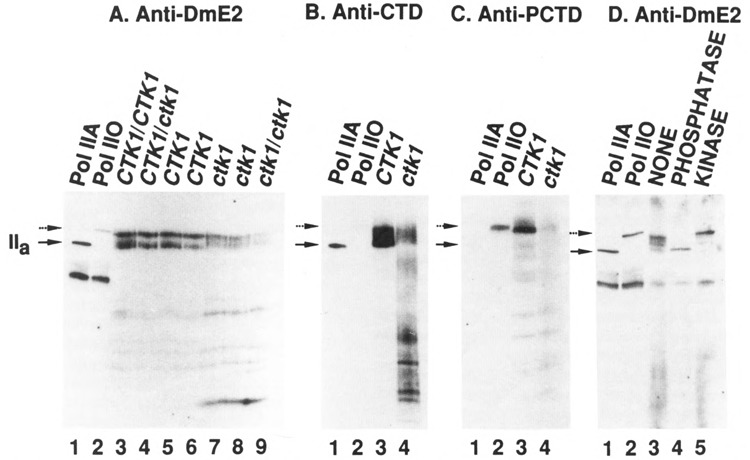
Western blot analysis of subunit IIa/IIo in crude extracts from CTK1 and ctk1 strains using different antibodies. Crude extracts were prepared from different yeast strains and samples containing similar amounts of total protein were analyzed by Western blotting using different largest subunit-specific affinity-purified antibodies (all described in Materials and Methods). Lanes 1 and 2 of each blot contained purified yeast RNA polymerase II (Pol IIA) and the same polymerase II after phosphorylation in vitro by CTD kinase (Pol IIO), respectively. Bound antibody was detected with [125I]protein A for A–C, or with alkaline phosphatase-conjugated goat anti-rabbit antibody followed by reaction with chemiluminescent substrate AMPPD for D. The positions of subunit IIa and IIo are denoted as solid and dotted arrows respectively. A. Blot was reacted with anti-DmE2, directed against determinants outside the CTD (major bands in lanes 1 and 2 represent IIb, the proteolyzed form of IIa). Lane 3: wild type diploid strain (DBY 1091). Lane 4: diploid transformant. Lanes 5 and 6: haploid wild type strains. Lanes 7 and 8: haploid mutant strains. (Strains in lanes 5–8 were siblings from one tetrad derived from the strain of lane 4). Lane 9: a diploid mutant strain constructed by crossing two haploid mutant strains. The low IIo/IIa ratio in this blot (lane 2 vs lane 1) is presumably an artifact (compare blot in D). B. Blot was reacted with anti CTD. Lanes 3 and 4 contained extracts from wild type and mutant strains, respectively (replicate samples of those used in A, lanes 5 and 7). C. Blot was reacted with anti-PCTD, directed against phosphorylated CTD. The lanes contain samples as in B. D. Crude extract prepared from a wild type strain (lane 5 in A) was immuno-precipitated with anti-DmE2 antibody and the immuno precipitate analyzed after no treatment (lane 3), treatment with alkaline phosphatase (lane 4), or treatment with yeast CTD kinase (lane 5). (See Materials and Methods.) Blot was reacted with anti-DmE2 antibody.
Immunoprecipitation of RNA polymerase II was performed by incubating 100 μl of extract (Method 1) with anti-DmE2 antibody followed by protein A-Sepharose, similarly to Kolodziej et al. (1990). Precipitated polymerase was resuspended in 100 μl kinase reaction buffer (Lee and Greenleaf, 1989), and 10 μl aliquots were treated with CTD kinase (20 units) or alkaline phosphatase (0.5 unit, Boeringer/Mannheim) for 30 minutes at room temperature or 37°C, respectively. Phosphorylation of purified yeast RNA polymerase II (from Dr. J. Jaehning, Indiana University) by CTD kinase was done as described previously (Lee and Greenleaf, 1989).
Localization of CTK1/β-gal fusion proteins by indirect immunofluorescence
CTK1-LacZ fusions were constructed by subcloning CTK1 gene fragments into the multicopy (β-galactosidase fusion vector YEp366 (Myers et al., 1986). Plasmids pCTK1(118)-LacZ and pCTK1(194)-LacZ were constructed by ligation of 1.35 kb BgIII-EcoRI(Klenow) and 1.58 kb Bg1II-Ndel(Klenow) CTK1 gene fragments into BamHI-Sa1I(Klenow) site of the vector, respectively. Plasmids pCTK1(463)-LacZ and pCTK1(463, Δ3-181) were constructed by ligation of the wild type 2.4 kb Bg1II-HindIII fragment or the 1.89 kb Bg1II-HindIII domain I-deleted CTK1 fragment (from pZJ15d1) into the BamHI-HindIII site of the vector, respectively. These subclones were transformed into yeast SJY65.
Immunostaining of transformed cells was done as described (Rose et al., 1990) using affinity purified rabbit anti-β-galactosidase antibody (Miles-Yeda, Ltd., 1:500 dilution) and rhodamine-conjugated goat anti-rabbit IgG (Boeringer/Mannheim, 1:1000 dilution). Cells were also stained with DAPI (4′,6-diamidino-2-phenylindole, Sigma) and were observed and photographed using Leitz microscope Laborlux S.
Results
Isolation and sequencing of the gene encoding the largest subunit of yeast CTD kinase
From an antiserum that reacted with all three subunits of the CTD kinase (Fig. 1A), we affinity purified antibodies directed specifically against each subunit (Fig. 1B). The unique specificities of the anti-α and -γ affinity purified antibodies strongly suggest that the CTD kinase subunits are not related in primary structure, presumably because they are products of different genes. The β subunit-specific antibodies, which were recovered in lower yields probably because of poor binding of that subunit to the nitrocellulose membrane (unpublished), also showed unique specificity upon longer exposure. The antibodies thus represent potentially useful reagents for attempting to clone genes for each subunit of the kinase.
Figure 1.
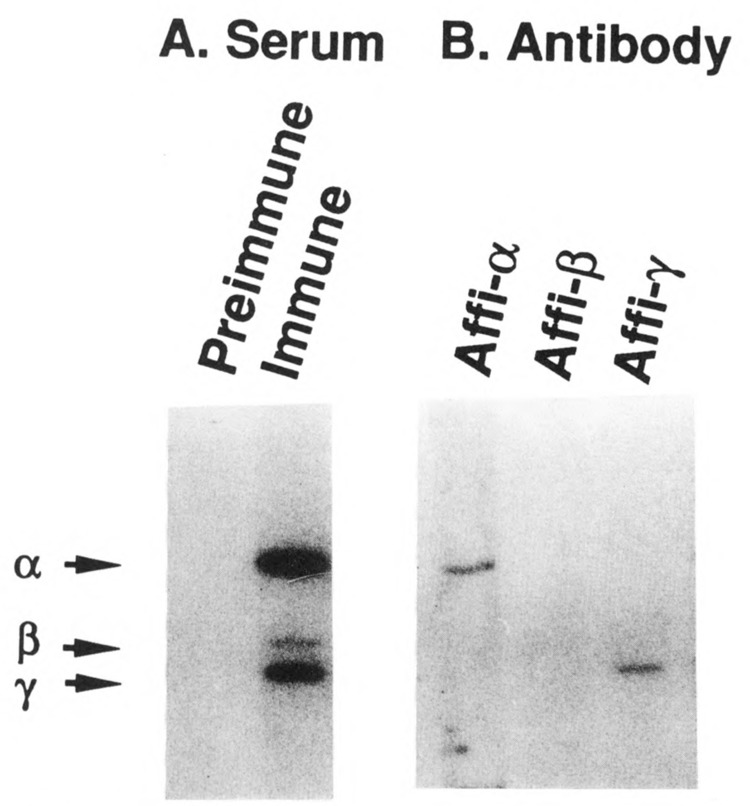
Immunoblots of CTD kinase. Mono S purified CTD kinase was run in a 12% polyacrylamide SDS gel and analyzed by Western blotting as described in Materials and Methods (2 μl [37 ng protein] per lane; see Lee and Greenleaf, 1989). [125I]Protein A was used to reveal the bound antibody. A. Lanes were reacted with preimmune serum (lane 1) or immune serum (lane 2) at 1:100 dilution. B. Each lane was reacted with antibodies affinity purified against a single subunit of CTD kinase (1:20 dilution). Lane 1, anti-α subunit (58 kDa); lane 2, anti-β subunit (38 kDa); lane 3, anti-γ subunit (32 kDa). The positions of the three subunits of the CTD kinase are denoted at left.
We screened a λgt11 expression library of yeast genomic DNA with the affinity purified antibodies and succeeded in isolating a phage containing part of the gene for the large subunit (see Materials and Methods). Subsequently we used a fragment from this phage as a DNA hybridization probe to isolate clones spanning the whole gene, which we call CTK1 (CTK1 for the protein). The restriction map of the CTK1 region is shown in Figure 2.
Sequencing 2645 nucleotides of CTK1 DNA (Fig. 3) revealed one long open reading frame of 1582 nucleotides which predicts a 528 amino acid protein with a molecular weight of 60.4 kDa, in good agreement with the subunit size estimate of 58 kDa derived from SDS polyacrylamide gel electrophoresis. The sequence surrounding the predicted N-terminus of CTK1 is consistent with the yeast translation start consensus sequence (Cigan and Donahue, 1987, Hamilton et al., 1987). At the DNA level, we noticed several T and A clusters in the region upstream of the open reading frame, a characteristic of constitutively-expressed yeast genes (Struhl, 1985); whether the CTK1 gene is expressed constitutively or inducibly has yet to be determined.
We analyzed codon usage in the CTK1 open reading frame, calculating a codon bias value (Bennetzen and Hall, 1982) of – 0.2, which indicates that codon usage in this gene is non-randomly biased toward nonpreferred triplets and suggests that the gene product should exist at a very low level. This is in agreement with the purification data described earlier (Lee and Greenleaf, 1989).
To determine on which chromosome CTK1 is located, we hybridized a labeled CTK1 DNA probe to a blot of separated yeast chromosomes; it hybridized to chromosome XI.
Homologies to protein kinases
We searched the NBRF and EMBL data banks using the total amino acid sequence of CTK1 and found that all of the proteins with significant homology to CTK1 are protein kinases or catalytic subunits of protein kinases. The relevant portion of CTK1 possesses all of the nine invariant amino acids found in catalytic subunits of protein kinases and shows strong homology to most of the other conserved sequences (Hanks et al., 1988). These data strongly suggest that the gene product of CTK1, the α subunit of the CTD kinase, is a catalytic subunit.
The proteins most homologous to CTK1 are CDC28 of Saccharomyces cerevisiae and its homologues, including human CDC2 and cdc2 of the yeast Schizosaccharomyces pombe. Amino acid sequences of CTK1, CDC28 and cdc2 are compared in Figure 4. The CTK1 protein kinase-homologous region comprises an internal segment of the protein, from amino acid 183 to 471, which displays homologies spanning virtually the entire length of CDC28/cdc2. While the overall amino acid identity between CDC28 and the homologous region of CTK1 is 40.3%, some stretches are nearly identical (e.g., eleven of twelve amino acids from Val190 to Ala201 [CTK1 numbering]).
Figure 4.

Sequence homologies among cdc2 of S. pombe, CDC28 of S. cerevisiae, and CTK1. Identical sequences are shown as white letters in the dark background; amino acid numbers are denoted at left.
The CDC28/cdc2 kinases are 60% identical overall and share several highly conserved regions, such as an invariant stretch of 16 amino acids between Glu49 and Glu64, EGVPSTAIREISLLKE (CDC28 numbering; Lee and Nurse, 1988). This “PSTAIR” region is thought to play a common critical role in the CDC28/cdc2 enzymes, such as influencing substrate recognition. Significantly, the CTK1 sequence in this region differs in 6 of 16 amino acids from the CDC28/cdc2 proteins. In addition, of four perfectly conserved tryptophan residues in all CDC28/cdc2 kinases (Krek and Nigg, 1989), only three are present in CTK1. These differences hint that the substrate specificity of CTK1 should be different from CDC28/cdc2, which is consistent with results described below.
Many protein kinases have been found to be phosphorylated and/or autophosphorylated, and in the CDC28/cdc2 kinases phosphorylation and dephosphorylation are functionally important (Simanis and Nurse, 1986; Draetta et al., 1988; Dunphy and Newport, 1989; Morla et al., 1989; Pondavin et al., 1990; Felix et al., 1990; Jessus et al., 1990). Consensus sequences surrounding a potential phosphorylation site in CDC28/cdc2 proteins (Nurse, 1985; Russel and Nurse, 1987) are also found in CTK1 (residues 304 to 373; Fig. 4). Providing possible significance to the presence of these sequences in CTK1 is our observation that the largest subunit of CTD kinase is autophosphorylated in vitro (as is the smallest subunit; J. M. L., unpublished data).
Analysis of CTK1 structure
Hydrophobicity values (Kyte and Doolittle, 1982) for CTK1, displayed in Figure 5, reveal a striking structural feature of this protein. The N-terminal and C-terminal regions of the protein comprise completely hydrophilic domains, while the central section is composed of a more common combination of alternating hydrophilic and hydrophobic segments. The long hydrophilic segment at the N-terminus (“domain I”, residues 1–165) is predicted (Gamier et al., 1978) to contain only one segment of α-helix, the remainder most likely being random coils or turns (not shown). Thus we expect this region to have an extended, solvent-exposed structure, reminiscent of the CTD itself. The shorter hydrophilic segment at the C-terminus (“domain III”, residues 490–528) probably manifests a similar extended structure, part of which may be α-helical. On the other hand, the middle region (“domain II”, residues 166–489) is predicted to contain all types of secondary structures, suggesting that this region is probably globular. Domain II contains the sequences homologous to CDC28/cdc2 which are conserved in catalytic subunits of the protein kinases. These facts suggest that overall the CTK1 protein probably contains a globular CDC28-like central catalytic domain with solvent-exposed N-terminal and C-terminal extensions.
Figure 5.
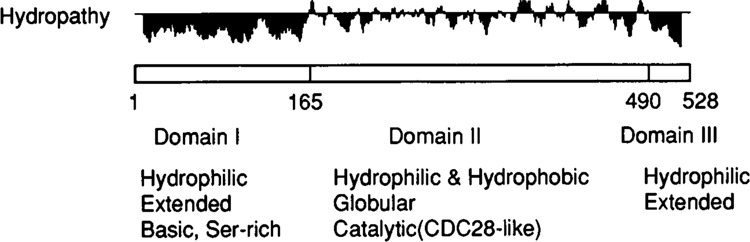
Structural features of CTK1 protein. Hydropathy values (Kyte and Doolittle, 1982) were calculated and plotted using MacGene II Plus computer program. The three tentatively assigned domains, as described in text, are indicated at the bottom. For hydropathy the + values (above the horizontal lines) indicate hydrophobicity, and the - values (below the line) the hydrophilicity. The numbers underneath the box indicate the amino acid number.
Gene disruptions and partial deletions
Southern blots of genomic DNA digested with restriction enzymes which cut only once within the CTK1 coding region clearly indicated that the gene was single copy (data not shown). We asked whether the CTK1 gene was essential or not by a one-step gene disruption experiment (Rothstein, 1983), followed by tetrad analysis. The 0.64 kb internal EcoRI fragment of the wild type CTK1 gene (Fig. 6) was excised and replaced by a 1.2 kb URA3 DNA fragment to create ctk1ΔE::URA3 (Materials and Methods); deleting the EcoRI fragment removes a region containing putative ATP binding and essential catalytic sequences, and the mutant gene should therefore be completely devoid of kinase activity. Diploid transformants of DBY1091 carrying one copy of the disrupted gene were isolated (Fig. 6), one of these was sporulated, and tetrads were dissected. All four spores germinated and grew (data not shown).
Among four colonies from each tetrad, two were large (Ura−) and two were small (Ura+), suggesting that disrupting the CTK1 gene leads to a reduced growth rate. That the CTK1 gene was disrupted in the Ura+ cells was confirmed by Southern blot analysis (Fig. 6). The data in Figure 6 also confirmed that the CTK1 gene is a single copy gene, because no wild type gene was detected in the mutant cells. These results demonstrate that the CTK1 gene is not absolutely essential in vivo at normal growth temperatures, but indicate that disruption of the gene is detrimental to the cells.
We compared the growth rates of the wild type and mutant cells and found that doubling time of the mutant cells in YPD broth at 30°C was about 2-fold longer than wild type. At lower temperatures the mutant cells became progressively more debilitated relative to wild type, until at a temperature of 12°C they failed to grow at all.
To confirm the generality of the slow growth and cold sensitivity phenotype of ctk1 mutants, we disrupted the gene in another diploid (SGY65) and in several different haploid strains. All transformants with the CTK1 gene disrupted displayed the same phenotype. Furthermore the slow growth, cold sensitivity phenotype of the ctk1 mutant strains was rescued by introduction of the wild type CTK1 gene on a CEN plasmid (pJYC1511), providing additional evidence that this phenotype is caused by disruption of the CTK1 gene (not shown).
As an initial test of the functional importance of the N-terminal hydrophilic domain I described above, we constructed a plasmid carrying a CTK1 gene lacking this domain and tested its ability to restore wild type growth properties to ctk1 strains. This truncated gene (plasmid pJYC1512) failed to rescue the growth defects, indicating that domain I performs a critical function in vivo.
ctk1 mutant cells lack CTD kinase activity
Because sequence analyses strongly suggest that the CTK1 gene product is a catalytic subunit, the mutant ctk1 cells generated in the gene disruption experiments should lack CTD kinase activity if in fact CTK1 encodes the a subunit of the enzyme. To test this prediction directly we checked for CTD kinase activity in extracts of CTK1 and ctk1 sibling strains.
As mentioned previously (Lee and Greenleaf, 1989), two kinds of phosphorylating activity were observed in unfractionated yeast extracts when a CTD-containing fusion protein was used as substrate. One activity, the CTD kinase we purified, causes a marked mobility shift of the substrate in SDS gels. The other activity does not cause the mobility shift (non-shifting kinase). Since these two activities can be cleanly separated by phosphocellulose column chromatography, we applied crude extracts from wild type and ctk1 mutant strains to parallel phosphocellulose columns and assayed the resulting fractions for kinase activities. As seen in Figure 7A, wild type cells contain both activities as expected (with the CTD kinase peaking in fraction 33). In contrast, ctk1 cells do not contian detectable CTD kinase activity, while they still contain the non-shifting kinase (peaking in fraction 24, Fig. 7B). These results support the conclusion that indeed CTK1 encodes the α subunit of the CTD kinase.
Figure 7.
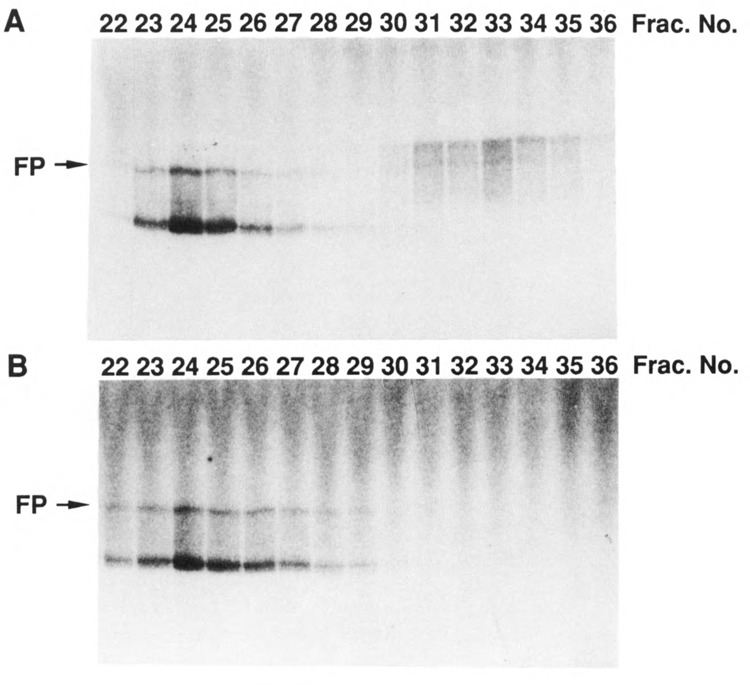
Phosphocellulose column profiles of CTD kinase activity in extracts prepared from CTK1 and ctk1 strains. Crude extracts from wild type CTK1 and mutant ctk1 haploid strains (30 ml culture at A600 = 1.0) were loaded directly onto two identical columns of 3 ml phosphocellulose (P11) at 0.2 M KCl in buffer H (Lee and Greenleaf, 1989) and eluted with a 15 ml gradient of 0.2–0.8 M KCl in buffer H. Fractions of 0.5 ml were collected and dialyzed against 25 mM KCl in buffer H, then 1 μl of each fraction was assayed for CTD kinase activity under standard conditions with yeast CTD fusion protein as substrate (Lee and Greenleaf, 1989). Reaction products were analyzed by 6% SDS polyacrylamide gel electrophoresis, and an autoradiogram was taken from dried gel. The position of the intact fusion protein is indicated. A. Extract of wild type CTK1 strain. B. Extract of mutant ctk1 strain.
These experiments also indicated that the non-shifting kinase was distinct from CTD kinase. Consistent with this, other studies showed that unlike the CTD kinase, the non-shifting kinase was not inhibited by anti-CTD kinase antibody (data not shown). In addition, note that the segment of subunit IIa carried by the fusion protein used here as substrate contains 48 amino acid residues N-terminal to the start of the CTD (“upstream” residues), and that it is approximately 90% proteolyzed (Lee and Greenleaf, 1989). The non-shifting kinase activity preferentially labels the proteolyzed species which probably contain the upstream residues but little if any CTD, whereas the CTD kinase does not label these molecules. We have also prepared a fusion protein substrate lacking these upstream residues and found that it was an extremely poor substrate for the non-shifting kinase, while it remained an excellent substrate for the CTD kinase (unpublished).
Subunit IIo in ctk1 mutant cells
Subunit form IIo has been characterized best in HeLa cells, where in rapidly prepared extracts it is the exclusive form of the large subunit (Kim and Dahmus, 1986), indicating that in vivo essentially all of the HeLa RNA polymerase II is hyperphosphorylated. However, this situation may not be expected to obtain in other types of cells or in other organisms, since even in other mammalian cultured cell extracts, substantial levels of IIa were detected (Kim and Dahmus, 1986). The in vivo phosphorylation state of yeast large subunit has been studied less thoroughly. Earlier studies detected a phosphorylated form we would now call IIo (Buhler et al., 1976; Bell et al., 1977), and a recent investigation found that ca. 50% of immune-precipitated RNA polymerase II large subunit was highly phosphorylated and migrated with reduced mobility in SDS gels, features that define form IIo (Kolodziej et al., 1990).
To investigate the presence of subunit IIo in both wild type and ctk1 cells, we prepared extracts rapidly and assayed polymerase II largest subunit status by immunoblotting with a subunit-specific affinity purified antibody (see Materials and Methods). In wild type extracts we detected both subunit IIa and slower migrating species, as shown in lanes 3–6 of Figure 8A. After immune precipitation and treatment with phosphatase, the upper band(s) disappeared while the amount of IIa increased, indicating that the slow migration of the upper band(s) was due to phosphorylation (Fig. 8D, lane 4). As is most noticeable in the immune precipitate (Fig. 8D, lane 3), more than one slower migrating form of the large subunit is sometimes observed in these analyses, and most of the forms migrate slightly faster than the “IIo” generated by CTD kinase in vitro (compare Fig. 8D, lanes 3 and 2), suggesting that they are not maximally phosphorylated. This suggestion was verified by treating the antibody precipitate with CTD kinase before electrophoresis; as shown in lane 5 of Figure 8D, the upper band now migrated at the same position as the in vitro phosphorylated form. From these results we conclude that the upper band(s) in extracts from wild type cells represents a phosphorylated form of subunit IIa with reduced electrophoretic mobility, that is, IIa.
We next checked for the presence of IIo in extracts from ctk1 mutant cells. These extracts also contained slower migrating forms of the largest subunit (Fig. 8A, lanes 7–9), which, upon phosphatase treatment or CTD kinase treatment, behaved similarly to those from wild type cells (not shown). Thus ctk1 cells, which do not contain the characterized CTD kinase, nevertheless contain an activity capable of phosphorylating the CTD. Careful inspection of lanes 7–9 in Figure 8A reveals that the forms of the largest polymerase II subunit in mutant cells are not precisely the same as those in wild type (lanes 3–6). In particular, form “IIa” in the mutant cells migrates slightly slower than in wild type cells; this observation is reproducible, but we do not yet understand the underlying basis for it.
To compare further the forms of the largest RNA polymerase II subunit in wild type and mutant cells, we prepared two new affinity-purified antisera differentially reactive toward the CTD (Materials and Methods). “Anti-CTD” reacts well with unphosphorylated CTD and very poorly with in vitro phosphorylated CTD (compare lanes 1 and 2 of Fig. 8B), while “anti-PCTD” reacts principally with the in vitro phosphorylated CTD (compare lanes 1 and 2 of Fig. 8C). When reacted with parallel immunoblots of the same sets of extracts, the anti-CTD antibody displays different intensity and pattern of reaction with large subunit forms in wild type vs. mutant extracts (Fig. 8B, lanes 3 and 4). Note that the lower mobility forms that this antibody detects in both extracts are presumably less (or differently) phosphorylated than the in vitro generated IIo, with which it reacts poorly (Fig. 8B, lanes 2). The most noticeable difference was observed using the anti-PCTD antibody, which reveals predominantly a slow-migrating form in wild type extracts but very little of this form in mutant extracts (Fig. 8C, lanes 3 and 4). Taken together, these results provide strong evidence that the in vivo phosphorylation state of pol II largest subunit is altered when cells lack functional CTKl-containing CTD kinase, supporting the idea that this kinase plays a role in CTD phosphorylation in vivo. Similar preliminary analyses of RNA polymerase II largest subunit forms in ctk1 mutants constructed in other strains also consistently reveal differences between the mutant and corresponding wild type (unpublished); however, even among wild type strains the actual IIo/IIa ratio appears to vary, presumably due to effects of different genetic backgrounds.
Other properties of ctk1 mutants
We tested cells lacking CTD kinase for other physiological defects. We found that two haploid ctk1 strains of opposite mating type were capable of mating and producing viable homozygous diploids; thus the complex pathways involved in mating behavior remain functional in the absence of CTD kinase. In addition, the diploid strains thus produced displayed the same slow growth/cold sensitivity phenotype as the haploid mutant strains.
The availability of homozygous ctk1 mutants allowed us to test their ability to undergo meiosis and sporulation. We found that several diploids derived from different haploid pairs were all defective in sporulation under all conditions tested. While we have not attempted to determine the specific stage at which the process was blocked, we did not observe cells with multiple nuclei after DAPI staining; this suggests that the process was blocked at an early stage. The block was specific to the ctk1 defect, since introducing the wild type CTK1 gene into the diploids restored their ability to sporulate (not shown).
Microscopic examination revealed that the morphology of ctk1 mutant strains differed from that of wild type. The mutant cell population was very heterogeneous, displaying sizes and shapes ranging from almost wild type to large, round cells roughly twice wild type size. Perhaps these morphologies are a consequence of disturbed patterns of transcription resulting from improper CTD phosphorylation.
In yeast and other organisms tested the CTD can be shortened substantially without loss of viability, but about 50% is needed for normal growth (Nonet et al., 1987; Bartolomei et al., 1988; Zehring et al., 1988; Allison et al., 1988). In S. cerevisiae 13 repeats suffice for normal growth rates, fewer than 10 repeats result in lethality, while 10 to 12 repeats yield slow growth rates and cold sensitivity. We tested the possibility that a combination of ctk1 disruption and CTD truncation to 10 repeats would result in either exacerbating or ameliorating the slow growth/cold sensitivity phenotypes of each mutation. To generate the double mutants, we crossed strains possessing a CTD with 105/7 repeats with ctk1 mutants and analyzed tetrads (Materials and Methods). The resulting double mutant haploids were slow growing and cold sensitive, but we observed no other noticeable changes of phenotypes (unpublished); this may suggest that the partially truncated CTD was being phosphorylated in these doubly mutant strains as was the intact CTD in the ctk1 mutant strains described above.
Nuclear targeting of CTK1 protein
If CTD kinase functions in the nucleus to phosphorylate RNA polymerase II, we anticipate that it should be a nuclear enzyme. As one approach to test this idea, we prepared several fusion constructs linking different portions of CTK1 to E. coli lacZ, expressed them in yeast, and located the resultant fusion proteins with antibodies to β-galactosidase (see Materials and Methods). As shown in Figure 9B, a fusion protein carrying the 118 N-terminal amino acids of CTK1 (plasmid pCTKl(118)-LacZ) was localized in the nucleus. Other fusion proteins containing longer N-terminal CTK1 segments were also nuclear (not shown). In contrast, a fusion protein containing CTK1 residues 182–463 (pCTKl (463, Δ3 – 181)-LacZ) was found in the cytoplasm (Fig. 9D). Clearly a nuclear localization signal resides in the first 118 residues of CTKL Inspection of the sequence reveals a motif similar to previously described nuclear localization signals (PPKRIRTD beginning at residue 37; e.g., Estruch and Carlson, 1990).
Figure 9.
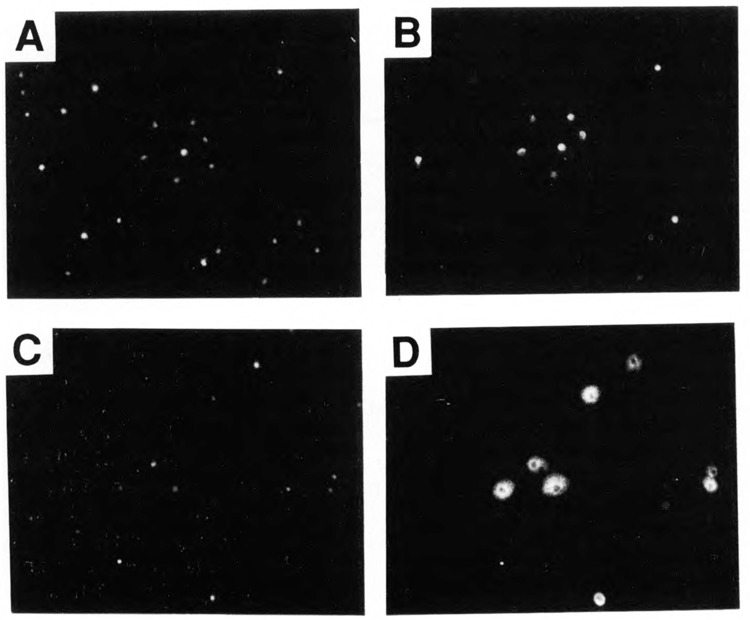
Intracellular localization of CTKl- β-galactosidase fusion proteins. Cells were stained with 4′, 6-diamidino-2-phenylindole (DAPI) (panels A, C) and rabbit anti- β-galactosidase antibody followed by rhodamine-labeled second antibody (panels B, D). The strain containing plasmid pCTKl(118)-LacZ is shown in panels A and B. The strain containing pCTKl (463, Δ3 – 181)LacZ is shown in panels C and D.
No CDC28 protein in CTD kinase
S. cerevisiae CDC28 (ca. 34 kDa) is a major cell-cycle regulatory protein kinase (Reed et al., 1985) whose activity is required for the start of the cell cycle. Functionally and structurally homologous proteins have been found in the fission yeast Schizosaccharomyces pombe (cdc2) and mammals (p34; for review, see Pines and Hunter, 1990). This protein is a component of MPF (maturation promoting factor) isolated from Xenopus (Gautier et al., 1988, Dunphy et al., 1988) and also of M phase-specific histone H1 kinase of starfish (Labbe et al., 1988, Arion et al., 1988). Recently, a CTD kinase was purified from mouse cells which consists of 58 and 34 kDa subunits (Cisek and Corden, 1989). Sequencing the cloned gene encoding the 34 kDa subunit showed it to be a murine homologue of S. cerevisiae CDC28. This observation raised the possibility that the similarly-sized subunit of yeast CTD kinase (γ) might actually be CDC28.
We tested this possibility by immunoblotting with a CDC28-specific antibody, which was raised against a peptide of 18 amino acids corresponding to the C-terminus of CDC28 (see Materials and Methods). Figure 10 shows that the CDC28-specific antibody strongly reacted with a ‘34 kDa’ protein in the crude yeast extract, but not with the CTD kinase (Fig. 10B, lanes 2 and 1). Note that the level of the purified kinase in lane 1 is much higher than in the crude extracts in lane 2. In the control experiments (Fig. 10A) anti-CTD kinase antibody easily detected purified kinase subunits (lane 1), but barely detected kinase subunits (if at all) in the crude extract (lane 2), presumably because of the low abundance of the CTD kinase in yeast (Lee and Greenleaf, 1989). Furthermore, the 18 amino acid peptide blocked the binding of the CDC28 antibody to the CDC28 protein (Fig. 10B, lane 4) but did not block the CTD kinase antibody (Fig. 10A, lanes 3 and 4). These data clearly demonstrate that the CTKl-containing yeast CTD kinase and the cdc2/CDC28-containing mammalian enzyme are distinct.
Figure 10.

Immunoblot of CTD kinase with anti-CTD kinase and anti-CDC28 antibodies. Mono-S purified CTD kinase (lanes 1 and 3, 0.1 μl) and crude extract (lanes 2 and 4, 10 μl) were run in a 12% polyacrylamide SDS gel and blotted onto nitrocellulose membrane. A. Blots were reacted with a mixture of all three affinity-purified antibodies against the subunits of CTD kinase (lanes 1 and 2), or with these antibodies preincubated with an 18 amino acid oligopeptide representing the C-terminus of CDC28 (lanes 3 and 4). B. Blots were reacted with affinity purified anti-CDC28 antibody (lanes 1 and 2), or this antibody preincubated with the 18 amino acid oligopeptide (lanes 3 and 4) (see Materials and Methods). Bound antibodies were detected with alkaline phosphatase conjugated goat anti-rabbit IgG followed by reaction with NBT and BCIP. Positions of the CTD kinase subunits are indicated at left.
Discussion
The results reported here indicate that the αsubunit of yeast CTD kinase contains an internal domain that is highly homologous to catalytic subunits of other protein kinases and in particular contains the residues which are invariant in those subunits. These data strongly suggest that α, the 58 kDa product of the CTK1 gene, is a catalytic subunit. While α (the CTK1 protein) shows most homology to the CDC28/cdc2 protein kinases, it differs from them considerably. For example, it is nearly twice as large as canonical CDC28, containing unusual N- and C-terminal domains. In addition, sequence motifs precisely conserved among CDC28/cdc2 kinases are significantly different in CTK1, suggesting distinct functions.
Besides α, purified CTD kinase contains β and γ subunits of apparent sizes 38 and 32 kDa respectively, for which the genes have yet to be cloned. In view of the report that a mouse CTD kinase activity contains a 34 kDa subunit which in fact is murine p34cdc2 (Cisek and Corden, 1989), we checked the possibility that the small yeast CTD kinase subunit might actually be CDC28, the S. cerevisiae counterpart of p34cdc2; our results establish that it is not. Thus it seems clear that the enzyme we purified using a CTD-containing fusion protein as substrate is different from the enzyme purified using short synthetic repeat peptides as substrate (Cisek and Corden, 1989). On the other hand, since amino acid sequence analysis reveals that CTK1 and p34cdc2/CDC28 belong to the same family of Ser/Thr protein kinase catalytic subunits, we might expect some similarities between the CTKl-containing yeast CTD kinase and the cdc2/CDC28-containing mouse enzyme. In fact, CDC28-containing protein kinases phosphorylate histone H1, pp60src and other substrates at sites with a consensus sequence that can be represented by S/T-P-X-Z, where X is a polar amino acid and Z is generally a basic amino acid (Moreno and Nurse, 1990; see also Shenoy et al., 1989; Belenguer et al., 1990; Lewin, 1990). The Ser/Thr-Pro motif in this consensus is obviously well represented in the CTD repeats. One possibility that derives from these facts is that the residual CTD phosphorylating activity present in yeast ctk1 mutant cells could be due to CDC28. On the other hand, only nine of fifty-two CTD repeats in mammals, and one of twenty-six repeats in yeast, contain basic residues. Interestingly, the p34cdc2,CDC28-containing CTD kinase from mouse cells appears to prefer the CTD repeats that contain basic residues (Zhang and Corden, 1991a). In apparent contrast, the CTK1 containing yeast CTD kinase is not active toward histone H1 (Lee and Green-leaf, 1989), which is a good CDC28 substrate. Conversely, while a CDC28-containing kinase from yeast actively phosphorylates histone H1, as expected, it does not phosphorylate the β-Gal-CTD fusion protein (Y-FP) we used as substrate to purify yeast CTD kinase (S. Reed, personal communication).
In addition to the enzymes purified from yeast and mouse cells, CTD phosphorylating activities have been partially purified from HeLa and plant cells (see Introduction); the complete purification of these activities is needed to determine their molecular identities, and additional characterizations of all these kinases will be required to determine the possible relationships among them. Meanwhile, in view of the inhibition by DRB of a partially purified HeLa CTD kinase activity (Stevens and Maupin, 1989), we tested this nucleotide analogue for effects on the yeast enzyme; it neither inhibited nor stimulated yeast CTD kinase. In addition, we have tested a short synthetic repeat peptide (4 heptamers) as substrate for our yeast CTD kinase and find that while this short peptide is phosphorylated by the enzyme, the Km is ca. 100-fold higher than for the fusion protein containing 26 repeats (unpublished results). While additional studies are needed, these results suggest that the yeast CTD kinase favors long repeat units (the observed difference is more than the 7-fold difference in the number of repeats present in the short and long substrates). It will be instructive to obtain similar information for the CTD phosphorylating activities identified in or purified from other organisms.
The predicted secondary structure of CTK1 suggests a catalytic central domain (II) with two very hydrophilic N- and C-terminal extensions (domains I and III). Genetic studies reported here provide evidence that domain I plays an important role in vivo, because versions of CTK1 with this domain removed failed to rescue phenotypes caused by CTK1 gene disruption. At least one role for domain I is probably to localize the CTD kinase in the nucleus, since we have shown directly that it contains a nuclear targeting signal. Other possible roles are suggested by its structure and sequence. The amino terminal domain I is rich in serine residues in a region with numerous positive charges, indicating that it might contain phosphorylation sites and play a regulatory role influencing CTD kinase activity. We suggest that the many positively charged residues in this domain might interact with phosphate groups being added to the CTD substrate, allowing the enzyme to act processively. This suggestion seems plausible in that the CTD substrate is itself probably an extended structure and when partially phosphorylated might interact effectively with an extended, positively charged domain I. The actual structures and functions of domains I and III remain to be elucidated by further biochemical and genetic investigations.
Gene disruption experiments revealed that cells lacking CTD kinase remained viable at the normal growth temperature of 30°C, although they grew slowly. However, unlike wild type yeast, the ctk1 mutant cells were inviable at lower temperatures, such as 12°. Thus the CTK1 gene product, and by extension the CTD kinase, is required for normal growth. Western blot experiments revealed that the ctk1 mutant cells contained slower migrating, phosphorylated forms of the largest RNA polymerase II subunit, indicating that another protein kinase capable of phosphorylating the CTD is present in yeast cells. However, our results indicate that the extent and/or pattern of CTD phosphorylation differ between wild type and ctk1 mutant strains, arguing that CTKl-containing CTD kinase normally plays a role in phosphorylating the CTD in vivo. Furthermore the slow-growth, cold sensitivity phenotype of ctk1 mutant strains suggests that the activity of the additional kinase is not sufficient by itself to support wild type growth. Available data do not allow us to decide whether or not the additional activity normally participates in CTD phosphorylation in vivo.
We should emphasize that much additional analytical work, including development of improved assays, is required to describe in detail CTD phosphorylation patterns in both wild type and mutant cells. The immunoblot assays used here have several limitations. For example, the reduced mobility of subunit “IIo” in SDS gels indicates addition of multiple phosphate groups, but it does not provide a detailed picture of either the extent or pattern of the phosphorylation (e.g., Corden, 1990). In addition, we find that while we reproducibly detect subunit form IIo, its relative amount and the extent of its mobility shift can differ depending on several variables, including the method and rapidity of extract preparation, the freezing history of the cells, the presence of phosphatase inhibitors, and the genetic background. Once the analytical methods are improved, it will be of some interest to investigate in more detail the physiology of RNA polymerase IIO.
Understanding the in vivo role of CTD phosphorylation and clarifying the relationship between the characterized CTD kinase and the additional CTD phosphorylating activity would be facilitated by having available mutant versions of genes encoding the additional enzyme. The phenotypes of ctk1 mutant cells provide several approaches for identifying mutations in and subsequently cloning of such genes. For example, mutations that inactivate the additional kinase activity in a ctk1 background might result in a synthetic lethal phenotype. Serendipity has already provided candidates for such mutations, because among the meiotic products of diploid strain SGY65 transformed with the disrupted CTK1 gene were haploid ctk1 strains that not only grew slowly and were cold sensitive, but were also temperature sensitive (J. M. L., unpublished). Introducing the wild type CTK1 gene into these strains rescued both the cs/slow-growth and ts phenotypes. One possible basis for this phenotype would be a ts mutation in the additional CTD phosphorylating activity. Attempts to clone the gene carrying this ts synthetic lethal mutation are under way.
Yeast genetics provides additional approaches to identifying activities that affect CTD phosphorylation or interact with CTD kinase in vivo. For example, extragenic suppressors of ctk1 mutations could potentially identify genes for phospho-protein phosphatase activities. Indeed, genetic data consistent with in vivo interactions between the gene product of SIT4, a putative phospho-protein phosphatase, and pol II have been reported (Arndt et al., 1989). Whether or not the SIT4 gene product interacts directly with pol II and whether or not it is involved in de-phosphorylating the CTD are currently unanswered questions.
Models for the involvement of CTD phosphorylation in the process of initiation have been proposed based on the observation that phosphorylation of the CTD accompanies initiation at the adenovirus major late promoter in vitro (e.g., Laybourn and Dahmus, 1990; Payne et al., 1989). However, because pol II lacking the CTD operates as efficiently at this promoter in vitro as intact pol II, it is difficult currently to evaluate the physiological significance of the observed phosphorylation. In order to assess the functional significance of these intriguing observations, a transcription system which responds to the presence and phosphorylation state of the CTD will be required.
The in vitro transcription results comparing pol II either carrying or lacking the CTD are consistent with the idea that at several promoters the CTD does not interact with basal transcription factors. In addition, two recent reports suggest that at this type of promoter some regulatory factors can exert their influence in vitro on polymerase II lacking the CTD (Zehring and Greenleaf, 1990; Buratowski and Sharp, 1990). On the other hand, several in vivo experiments suggest interactions between the CTD and certain regulatory factors. For example, transcription by polymerases with CTDs of different lengths can be affected differently by GAL4 derivatives with activation domains of different strengths (Allison and Ingles, 1989). It has also been observed that partially truncating the CTD reduces the stimulation of transcription mediated by INO1 and GAL10 UAS elements, but not by the HIS4 UAS (Scafe et al., 1990). In this case the response to the UAS signals decreased as the length of the CTD decreased, although the sensitivities of INOl and GAL10 differed. In a follow-up study, decreased response of CTD-shortened Pol II to acidic activators has been observed in nuclear extracts (R. Young, personal communication). Finally, a recent genetic study reveals that consequences of CTD shortening can be counteracted by inactivation of the SIN1 gene, which encodes a negative regulator of transcription, indicating formal genetic interaction between SIN1 and the CTD (Peterson et al., 1991). Together these studies suggest that the CTD may interact with certain transcription factors to mediate or modulate their activities. In fact, a genetic approach to identifying such factors has been described (Nonet and Young, 1989). Whether the interactions suggested by the in vivo studies are direct or indirect is not yet known. In the context of this paper it is also important to point out that we do not know if these interactions involve the phosphorylated or unphosphorylated form of the CTD. Resolving these unknowns and determining actual mechanisms will require developing novel biochemical approaches in addition to proceeding with more genetic studies.
The results presented here demonstrate that the CTK1 gene encodes the α subunit of the CTD kinase we previously characterized, that this kinase is essential for normal growth of S. cerevisiae, and that in the absence of this kinase phosphorylation of the CTD in vivo is abnormal. Considered together with the specificity and processivity displayed by the purified enzyme, these findings support the idea that the CTKl-containing CTD kinase normally plays a role in phosphorylating the CTD in vivo. We can now hope to exploit available genetic and biochemical tools to examine how this CTD kinase and other activities determine the phosphorylation state of the CTD and how that in turn influences gene expression by modulating the properties of RNA polymerase II.
Acknowledgments
This work was supported by NIH grant GM40505.
We thank John Weeks for extensive help with computer analyses; Steven Reed for CDC28 peptide, antibody, and results; Stephen Garrett for much useful advice, help, and materials; Akio Sugino for libraries and blots; Kerstin Leuther for help with tetrads; Rick Young for sharing results, ideas, and suggestions; Steven Hardin and john Weeks for useful comments; C. Peterson and I. Herskowitz for sharing strains and results; and J. Jaehning for RNA polymerase.
The sequence data reported in this paper have been submitted to GenBank and have been assigned accession number M69024.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Allison L. A., Moyle M., Shales M., and Ingles C. J. (1985), Cell 42, 599–610. [DOI] [PubMed] [Google Scholar]
- Allison L. A., Wong J. K., Fitzpatrick V. D., Moyle M., and Ingles C. J. (1988), Mol Cell Biol 8, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison L. A. and Ingles C. J. (1989), Proc Natl Acad Sci USA 86, 2794–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion D., Meijer L., Brizuela L., and Beach D. (1988), Cell 55, 371–378. [DOI] [PubMed] [Google Scholar]
- Arndt K. T., Styles C. A., and Fink G. R. (1989), Cell 56, 527–537. [DOI] [PubMed] [Google Scholar]
- Bartolomei M. S., Halden N. F., Cullen C. R., and Corden J. L. (1988), Mol Cell Biol 8, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenguer P., Caizergues-Ferrer M., Labbe J.-C., Doree M., and Amalric F. (1990), Mol Cell Biol 10, 3607–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G. I., Valenzuela P., and Rutter W. J. (1977), J Biol Chem 252, 3082–3091. [PubMed] [Google Scholar]
- Bennetzen J. L. and Hall B. D. (1982), J Biol Chem 257, 3026–3031. [PubMed] [Google Scholar]
- Buhler J. M., Iborra F., Sentenac A., and Fromageot P. (1976), Febs Lett 72, 37–41. [DOI] [PubMed] [Google Scholar]
- Bullock W. O., Fernandez J. M., and Short J. M. (1987), Biotechniques 4, 376–379. [Google Scholar]
- Buratowski S. and Sharp P. A. (1990), Mol Cell Biol 10, 5562–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena D. L. and Dahmus M. E. (1987), J Biol Chem 262, 12468–12474. [PubMed] [Google Scholar]
- Cigan A. M. and Donahue T. F. (1987), Gene 59, 1–18. [DOI] [PubMed] [Google Scholar]
- Cisek L. J. and Corden J. L. (1989), Nature 339, 679–684. [DOI] [PubMed] [Google Scholar]
- Corden J. L. (1990), Trends Biol Sci 15, 383–387. [DOI] [PubMed] [Google Scholar]
- Corden J. L., Cadena D. L., Ahearn J. M. J., and Dahmus M. E. (1985), Proc Natl Acad Sci USA 82, 7934–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmus M. E. (1981), J Biol Chem 256, 3332–3339. [PubMed] [Google Scholar]
- Draetta G., Piwnica-Worms H., Morrison D., Druker B., Roberts T., and Beach D. (1988), Nature 336, 738–744. [DOI] [PubMed] [Google Scholar]
- Dunphy W. G., Brizuella L., Beach D., and Newport J. W. (1988), Cell 54, 423–431. [DOI] [PubMed] [Google Scholar]
- Dunphy W. G. and Newport J. W. (1989), Cell 58, 181–191. [DOI] [PubMed] [Google Scholar]
- Estruch F. and Carlson M. (1990), Mol Cell Biol 10, 2544–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers R., Hammer A., Köck J., Jess W., Borst P., Memet S., and Cornelissen A. W. C. A. (1989), Cell 56, 585–597. [DOI] [PubMed] [Google Scholar]
- Felix M.-A., Cohen P., and Karsenti E. (1990), EMBO J 9, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamier J., Osguthorpe D. J., and Robson B. (1978), J Mol Biol 120, 97–120. [DOI] [PubMed] [Google Scholar]
- Gautier J., Norbury C., Lohka M., Nurse P., and Maller J. (1988), Cell 54, 433–439. [DOI] [PubMed] [Google Scholar]
- Guilfoyle T. J. (1989), Plant Cell 1, 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton R., Watanabe C. K., and de Boer H. A. (1987), Nucl Acids Res 15, 3581–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks S. K., Quinn A. M., and Hunter T. (1988), Science 241, 42–52. [DOI] [PubMed] [Google Scholar]
- Holm C., Meeks-Wagner D. W., Fangman W. L., and Botstein D. (1986), Gene 42, 169–173. [DOI] [PubMed] [Google Scholar]
- Hyunh T., Young R. A., and Davis R. W. (1984), in DNA Cloning Techniques: A Practical Approach (Glover D., ed.), IRL Press, Oxford, pp. 49–78. [Google Scholar]
- Ito H., Jukuda Y., Murata K., and Kimura A. (1983), J Bacteriol 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokerst R. S., Weeks J. R., Zehring W. A., and Greenleaf A. L. (1989), Mol Gen Genet 215, 266–275. [DOI] [PubMed] [Google Scholar]
- Jessus C., Ducommun B., and Beach D. (1990), FEBS Lett 266, 4–8. [DOI] [PubMed] [Google Scholar]
- Kelly J. L., Greenleaf A. L., and Lehman I. R. (1986), J Biol Chem 261, 10348–10351. [PubMed] [Google Scholar]
- Kim W. Y. and Dahmus M. E. (1986), J Biol Chem 261, 14219–14225. [PubMed] [Google Scholar]
- Kim W. Y. and Dahmus M. E. (1989), J Biol Chem 264, 3169–3176. [PubMed] [Google Scholar]
- Kolodziej P. A., Woychik N., Liao S.-M., and Young R. A. (1990), Mol Cell Biol 10, 1915–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek W. and Nigg E. A. (1989), EMBO J 8, 3071–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T. A. (1985), Proc Natl Acad Sci USA 82, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J. and Doolittle R. F. (1982), J Mol Biol 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Labbe J. C., Lee M., Nurse P., Picard A., and Doree M. (1988), Nature 335, 251–254. [DOI] [PubMed] [Google Scholar]
- Laybourn P. J. and Dahmus M. E. (1990), J Biol Chem 265, 13165–13173. [PubMed] [Google Scholar]
- Lee J. M. and Greenleaf A. L. (1989), Proc Natl Acad Sci USA 86, 3624–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. and Nurse P. (1988), Trends Genet 4, 287–290. [DOI] [PubMed] [Google Scholar]
- Legagneux V., Morange M., and Bensaude O. (1990), Eur J Biochem 193, 121–126. [DOI] [PubMed] [Google Scholar]
- Lewin B. (1990), Cell 61, 743–752. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E. F., and Sambrook J. (1989), Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Mendenhall M. D., Jones C. A., and Reed S. I. (1987), Cell 50, 927–935. [DOI] [PubMed] [Google Scholar]
- Morla A. O., Draetta G., Beach D., and Wang J. Y. J. (1989), Cell 58, 193–203. [DOI] [PubMed] [Google Scholar]
- Myers A. M., Tzagoloff A., Kinney D. M., and Lusty C. J. (1986), Gene 45, 299–310. [DOI] [PubMed] [Google Scholar]
- Nonet M., Sweetser D., and Young R. A. (1987), Cell 50, 909–915. [DOI] [PubMed] [Google Scholar]
- Nonet M. L. and Young R. A. (1989), Genetics 123, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P. (1985), Trends Genet 1, 51–55. [Google Scholar]
- Payne J. M., Laybourn P. J., and Dahmus M. E. (1989), J Biol Chem 264, 19621–19629. [PubMed] [Google Scholar]
- Peterson C. L., Kruger W., and Herskowitz I. (1991), Cell 64, 1135–1143. [DOI] [PubMed] [Google Scholar]
- Pines J. and Hunter T. (1990), New Biol 2, 389–401. [PubMed] [Google Scholar]
- Pondaven P., Meijer L., and Beach D. (1990), Genes Dev 4, 9–17. [DOI] [PubMed] [Google Scholar]
- Prober J. M., Trainer G. L., Dam R. J., Hobbs F. W., Robertson C. W., Zagursky R. J., Cocuzza A. J., Jensen M. A., and Baumeister K. (1987), Science 238, 336–341. [DOI] [PubMed] [Google Scholar]
- Reed S. I., Hadwiger J. A., and Lörincz A. T. (1985), Proc Natl Acad Sci USA 82, 4055–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio D. C., Laski F. A., and Rubin G. M. (1986), Cell 44, 21–32. [DOI] [PubMed] [Google Scholar]
- Robbins A., Dynan W. S., Greenleaf A. L., and Tjian R. (1984), J Mol Appl Genet 2, 343–353. [PubMed] [Google Scholar]
- Rogers S., Wells R., and Rechsteiner M. (1986), Science 234, 364–369. [DOI] [PubMed] [Google Scholar]
- Rose M. D., Winston F., and Hieter P. (1990), Methods in Yeast Genetics: A Laboratory Course Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Rothstein R. J. (1983), Methods Enzymol 101, 202–211. [DOI] [PubMed] [Google Scholar]
- Russel P. and Nurse P. (1987), Cell 49, 559–567. [DOI] [PubMed] [Google Scholar]
- Rüther U. and Müller-Hill B. (1983), EMBO J 2, 1791–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., and Coulson A. R. (1977), Proc Natl Acad Sci USA 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawadogo M. and Sentenac A. (1990), Ann Rev Biochem 59, 711–754. [DOI] [PubMed] [Google Scholar]
- Scafe C., Chao D., Lopes J., Hirsch J. P., Henry S., and Young R. A. (1990), Nature 347, 491–494. [DOI] [PubMed] [Google Scholar]
- Shenoy S., Choi J.-K., Bagrodia S., Copeland T. D., Mailer J. L., and Shalloway D. (1989), Cell 57, 763–774. [DOI] [PubMed] [Google Scholar]
- Sherman F., Fink G. R., and Hicks J. B. (1986), Laboratory Course Manual for Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Short J. M., Fernandez J. M., Sorge J. A., and Huse A. (1988), Nucl Acids Res 16, 7583–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigler P. B. (1988), Nature 333, 210–212. [DOI] [PubMed] [Google Scholar]
- Sikorski R. and Hieter P. (1989), Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanis V. and Nurse P. (1986), Cell 45, 261–268. [DOI] [PubMed] [Google Scholar]
- Smith J. L., Levin J. R., Ingles C. J., and Agabian N. (1989), Cell 56, 815–827. [DOI] [PubMed] [Google Scholar]
- Stevens A. and Maupin M. K. (1989), Biochem Biophys Res Commun 159, 508–515. [DOI] [PubMed] [Google Scholar]
- Struhl K. (1985), Proc Natl Acad Sci USA 82, 8419–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson N. E., Steinberg T. H., Aronson D. B., and Burgess R. R. (1989), J Biol Chem 264, 11511–11520. [PubMed] [Google Scholar]
- Weeks J. R., Coulter D. E., and Greenleaf A. L. (1982), J Biol Chem 257, 5884–5892. [PubMed] [Google Scholar]
- Zehring W. A. and Greenleaf A. L. (1990), J Biol Chem 265, 8351–8353. [PubMed] [Google Scholar]
- Zehring W. A., Lee J. M., Weeks J. R., Jokerst R. S., and Greenleaf A. L. (1988), Proc Natl Acad Sci USA 85, 3698–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. and Corden J. L. (1991a), J Biol Chem 266, 2290–2296. [PubMed] [Google Scholar]
- Zhang J. and Corden J. L. (1991b), J Biol Chem 266, 2297–2302. [PubMed] [Google Scholar]