

 This work is licensed under a
This work is licensed under a Abstract
Objective
Maternal hypothyroidism during pregnancy can affect the neurodevelopment of their offspring. This study aimed to investigate the effects of maternal subclinical hypothyroidism (SCH) on spatial learning and memory, and its relationship with the apoptotic factors in cerebral cortex of the offspring.
Methods
Female adult Wistar rats were randomly divided into three groups (n = 15 per group): control (CON) group, SCH group and overt hypothyroidism (OH) group. Spatial learning and memory in the offspring were evaluated by long-term potentiation (LTP) and Morris water-maze (MWM) test. The protein expression of the p75 neurotrophin receptor (p75NTR), phospho-c-Jun N-terminal kinase (p-JNK), the pro-apoptotic protein p53 and Bax were detected by Western blotting.
Results
The Pups in the SCH and OH groups showed longer escape latencies in the MWM and decreased field-excitatory post synaptic potentials in LTP tests compared with those in the CON group. p75NTR, p-JNK, p53 and Bax expression levels in the cerebral cortex increased in pups in the SCH and OH groups compared with those in the CON group.
Conclusions
Maternal SCH during pregnancy may impair spatial learning and memory in the offspring and may be associated with the increased apoptosis in the cerebral cortex.
Keywords: subclinical hypothyroidism, long-term potentiation, p75 neurotrophin receptor signaling pathway, cerebral cortex
Introduction
Thyroid hormones (THs) are vital for fetal neurodevelopment. The prevalence of subclinical hypothyroidism (SCH) in the general population is 4–10% and can reach 5% in pregnant women. Haddow et al. investigated the intelligence levels of 62 children aged 7–9 years who were born to mothers with thyroid dysfunction during early pregnancy. They found that even mild and asymptomatic hypothyroidism in pregnant women, including SCH, could affect the development of intelligence in their offspring (1). Several experimental studies also found that maternal TH deficiency during pregnancy could cause irreversible brain damage in the offspring, resulting in varying degrees of mental retardation, cognitive impairment and learning and memory dysfunction (2, 3, 4, 5). We previously found that decreased activation of the cAMP response element-binding protein signaling pathway in pups was related to impaired cognitive function caused by maternal SCH (6). However, the relationship between cognitive impairment due to SCH and the apoptosis in cerebral cortex remains unclear.
The p75 neurotrophin receptor (p75NTR) is an important member of the death receptor family. Although the role of the p75NTR-mediated signal transduction pathway in promoting apoptosis is currently unclear, the activation of the JNK signaling pathway is thought to be an important step in this process (7, 8). Previous studies showed that the expression of p75NTR increased in the cerebral cortex of pups from mothers with perinatal hypothyroidism, and the pro-apoptotic protein such as p53 and Bax increased (9, 10). However, whether neuronal apoptosis induced by the p75NTR signaling pathway exists in the offspring of pregnant SCH rats remains unknown.
This study aimed to determine if the p75NTR signaling pathway was activated in the cerebral cortex in the offspring of rats with SCH during pregnancy and to investigate its role in the apoptosis of cerebral cortex.
Materials and methods
Animals
This experiment used 45 female rats (weighing 190–210 g) that had never been pregnant. All female rats were kept in the Experimental Animal Department at China Medical University under specific pathogen-free conditions, in a constant environment at 23–25°C and 55% relative humidity with a 12-h light/darkness cycle, and provided with normal feed and water. Rats were fed normally for 1 week before the experiment. All animal experiments and procedures were approved by the Animal Care and Use Committee at China Medical University, which complies with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978). The rats were randomly assigned to three groups: control group (CON, n = 15), overt hypothyroidism group (OH, n = 15) and SCH group (n = 15). Rats in the OH and SCH groups were treated by intraperitoneal (i.p.) injection of 3% pentobarbital sodium (0.1 mL/100 g) and underwent thyroidectomy. CON rats underwent sham thyroid surgery. After surgery, rats were kept at 34 ± 2°C under an electric blanket until they awoke. One month after surgery, rats in the SCH group were injected subcutaneously with l-thyroxine (l-T4, Sigma, USA) 1.0 μg/100 g/day on the back or neck. Rats in the CON and OH groups were injected subcutaneously with physiological saline (50 µL/100 g/day) on the back or neck. Calcium lactate (0.1% w/v) was added to the drinking water for all rats after surgery. After 9 days of injections, all rats were mated with normal male rats (male: female = 1: 2). Then, pregnant rats were placed in solitary confinement until delivery. The female rats were observed daily until they gave birth, and the delivery day was recorded as postnatal day 0 (PND0). All pups were weaned and the mothers stopped receiving subcutaneous injections on PND21. Venous blood (approximately 2 mL) was collected from all pregnant rats via the inner canthus vein on the 9th day after initial subcutaneous injection of L-T4 and on PND0, to detect total thyroxine (TT4) and thyroid-stimulating hormone (TSH) levels. Six to eight pups from each group on PND7 were decapitated on ice and the cerebral cortex was removed for Western blotting. The remaining pups were raised to PND39 and subjected to the Morris water-maze (MWM) test (n = 10) and long-term potentiation (LTP) experiments (n = 6), finishing on PND44. The project timeline is described in Fig. 1.
Figure 1.

Schematic of experimental timeline. E0, gestational day 0; LTP, long-term potentiation; MWM, Morris Water Maze; PND, postnatal day.
Measurements of TT4 and TSH
Blood samples obtained from the rats were immediately centrifuged at 20,913 g for 13 min and stored at −80°C for subsequent chemiluminescence immunoassay (Immulite, Diagnostic Products Corporation, Los Angeles, CA, USA) to measure serum TT4 and TSH. The inter- and intra-assay coefficients of variation (CVs) for TT4 was 3.83–7.09% and 1.58–3.68%, respectively. The inter- and intra-assay (CVs) for TSH were 1.87–5.43% and 2.34–3.47%, respectively. Based on >95% specific binding, the limit of detection for TT4 was 1.0 μg/dL. And the results samples below the level of detection were recorded at 1.0 μg/dL for statistical purposes.
MWM test
The MWM test was used to assess the spatial learning abilities of the pups in all groups. The MWM consists of a black circular swimming pool (100 cm diameter, 60 cm deep) divided into four equal quarters (I, II, III, IV) by two straight lines. A circular platform (10 cm diameter) is located 1 cm below the surface in the middle of quadrant IV. The rats were allowed to swim freely for 120 s on the first day (PND39) to adapt to the water maze and platform, and training was then started on the following day (PND40). Each pup trained four times a day, and rested for 1 min after each training session. The time required for the pup to climb the platform was defined as the escape latency. If the pup had not found the platform after 120 s, the escape latency was recorded as 120 s. All the pups that completed the experiment were lifted out gently, dried with a towel and then returned to their cages. The procedure was repeated for 4 days (PND40, PND41, PND42, PND43). On the sixth day, the platform was removed and the experiments performed in the previous 4 days were repeated. We recorded the number of times each pup reached the platform area and the time spent in the area for 120 s and assigned this as a probe trial. The escape latency for pups in each group was recorded daily. The time spent in the platform quadrant and the number of times they crossed the platform quadrant were measured in the last day’s probe test.
LTP induction in vivo
We measured field-excitatory postsynaptic potentials (f-EPSPs) in the hippocampus of the pups in each group using a MED64 planar microelectrode matrix recording system (Alpha Med Science, Osaka, Japan). On PND40, the pups were decapitated on ice after anesthesia with 3% sodium pentobarbital (30 mg/kg, i.p.). Brain tissue was cut into 300-μm thick slices, removed and placed into oxygenated artificial cerebrospinal fluid containing 7.25 g/L NaCl, 0.22 g/L KCl, 2.18 g/L NaHCO3, 0.12 g/L MgSO4, 0.17 g/L KH2PO4, 1.8 g/L glucose and 0.22 g/L CaCl2 (pH 7.35–7.45). The temperature of the brain was maintained at 34 ± 1°C and the flow rate of the artificial cerebrospinal fluid was 1–1.5 mL/min. The tissues were kept in a mixture of 95% O2 and 5% CO2. Synaptic plasticity in the CA1 area of the hippocampus at 6 weeks can be evaluated by high-frequency stimulation (HFS)-induced increase in f-EPSP after LTP. The baseline level before HFS was measured for 10–15 min and allowed to stabilize. Input/output curves were obtained by increasing the intensity of the stimulus and adjusting it to elicit 70% of the maximal response. The f-EPSP was recorded and the stimulus value corresponding to a 50% amplitude difference was recorded as the HFS. The tissue was stimulated twice with HFS, and LTP was induced 10 s later and recorded for at least 30 min. The percentage f-EPSP (f-EPSP%) increased after HFS was used as an indicator to evaluate LTP.
Western blotting
On PND7, the pups were decapitated on ice. The cerebral cortex was removed immediately and 0.1 g cerebral cortex tissue was added to 200 μL buffer containing protease and phosphatase inhibitors, homogenized by shaking (KeyGenBiotech, Nanjing, China) and centrifuged at 20,913 g for 10 min at 4°C. Protein concentrations were determined by the bicinchoninic acid method (Beyotime, Shanghai, China), and the samples were then stored at −80°C. Tissue lysates were diluted to the same protein concentrations (5 μg/μL), boiled for 8 min and 10 μL (50 g protein) samples from each group were then electrophoresed in 10% SDS-polyacrylamide gels. The marker was separated at constant voltage of 80 V for 30−60 min, and the proteins were then separated at a constant voltage of 120 V for 1 h. The proteins were then transferred to PVDF membranes (Millipore) at a constant voltage of 70 V for 2 h. Non-specific binding was blocked using a mixture of skimmed milk powder with Tris-buffered saline and Tween 20, with bovine serum albumin for p-JNK. The membranes were then incubated for 4 h in the dark with the following antibodies: rabbit anti-p75NTR (1:1000 dilution; Cell Signaling Technologies); rabbit anti-total JNK (1:1000 dilution; Cell Signaling Technologies); rabbit anti-p-JNK (1:1000 dilution; Cell Signaling Technologies); rabbit anti-p53 (1:1000 dilution; Cell Signaling Technologies) and rabbit anti-Bax (1:1000 dilution; Cell Signaling Technologies). The blots were then incubated for 2 h with horseradish peroxidase-conjugated secondary antibody (1:10,000 dilution; Zhongshan Golden Bridge, Beijing, China) and developed by chemiluminescent Western blotting (ALPHAVIEW, version 1.3; ProteinSimple Inc., San Jose, CA, USA). The optimal time to expose the blot to the membrane was determined by standardization experiments.
Statistical analysis
Data processing and statistics were performed using SPSS 22.0 software (SPSS). The results were expressed as the mean ± standard error of the mean (s.e.m.). Multiple group comparisons were performed using one-way ANOVA test followed by Dunnett’s T3 test. Independent sample t tests were used for comparisons between two groups. A value of P < 0.05 was considered to be statistically significant.
Results
TH levels in pregnant rats
We measured serum TT4 and TSH levels in pregnant rats to confirm successful establishment of the SCH and OH models. TSH levels were significantly higher in the SCH and OH groups compared with those in the CON group before pregnancy (P < 0.05). TT4 levels were the lowest in the OH group among the three groups (P < 0.05). And TT4 levels in the SCH group were significantly higher compared with those in the OH group (P < 0.05). However, there was no significant difference between the SCH group and the CON group (P = 0.755). On PND1, TSH levels in SCH group were higher than those in CON group, and TT4 levels were similar to those in CON group. These results indicated that the SCH models in rats were established successfully throughout the pregnancy (Fig. 2).
Figure 2.

Thyroid-stimulating hormone (TSH) and serum total thyroxine (TT4) levels in pregnant rats (9 days of injection, n = 10 per group; PND1, n = 6 per group). *P < 0.05 vs same day CON group; #P < 0.05 vs same day SCH group. CON, control; OH, overt hypothyroidism; PND 1, postnatal day 1; SCH, subclinical hypothyroidism; TSH, thyrotropin; TT4, total thyroxine.
Decline in learning and memory in offspring of rats with SCH during pregnancy
We assessed spatial learning and memory in the offspring of rats with and without SCH using the MWM test. Escape latency decreased with increased training in all groups. Escape latency was significantly longer in the SCH group compared with the CON group, but escape latency was the longest in the OH group (P < 0.05; Fig. 3). The time spent in the platform quadrant in the probe trial was 29.5 ± 2.4 s in the CON group, 21.4 ± 1.3 s in the SCH group and 17.6 ± 0.5 s in the OH group (all P < 0.05; Fig. 4). We also calculated the number of times that the pups crossed the platform quadrant, which was 7.98 ± 0.37 times in the CON group, 6.53 ± 0.16 times in the SCH group and 5.30 ± 0.34 times in the OH group (all P < 0.05; Fig. 5). These results showed that spatial learning and memory reduced in the offspring of mothers with SCH during pregnancy.
Figure 3.
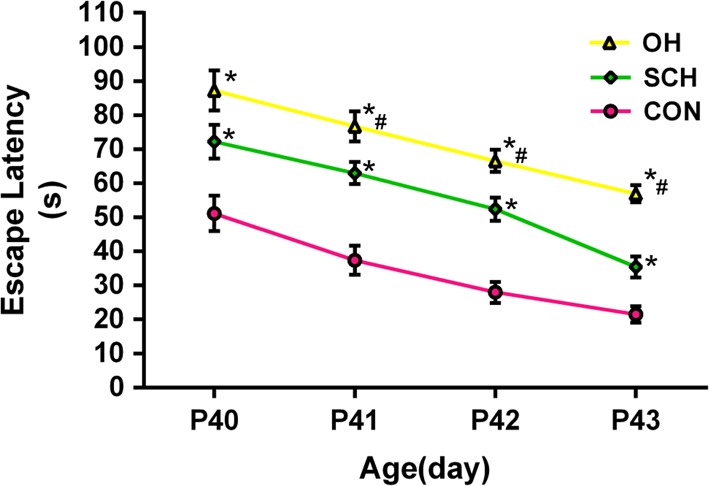
Performance of pups in the MWM test. Data are expressed as the mean ± s.e.m. (n = 10 for each group). Average time to find the hidden platform was longer in the SCH and OH groups compared with the CON group from PND 40–43.
Figure 4.

Probe trial test recorded the time spending by each pup in the platform area. Pups in the SCH and OH groups spent less times than pups in the CON group. *P < 0.05 vs same day CON group; #P < 0.05 vs same day OH group.
Figure 5.

Probe trial test recorded the number of times pups crossed the platform quadrant. Pups in the SCH and OH groups crossed fewer times than pups in the CON group. *P < 0.05 vs same day CON group; #P < 0.05 vs same day OH group.
LTP in offspring of rats with SCH during pregnancy
The LTP results were evaluated by measuring the percentage of baseline f-EPSP (f-EPSP%) after HFS. There was no significant difference in f-EPSP among the three groups before HFS (P > 0.05); however, all three groups showed an increase in f-EPSP after HFS, with significant differences in the f-EPSP% among the three groups. The f-EPSP% reduced in the SCH (175 ± 1%) and OH groups (148 ± 1%) compared with the CON group (209 ± 1%) (n = 6, P < 0.05). However, the amplitude of the increase in the SCH group was significantly greater than that in the OH group (P < 0.05; Fig. 6). These results suggested that SCH during pregnancy could damage LTP in the offspring.
Figure 6.

Maternal SCH caused LTP damage in the hippocampal CA1 region in their pups. LTP was induced by HFS and measured as an increase in f-EPSP slope, expressed as percent of the baseline of f-EPSP slope after HFS in different groups. f-EPSP slopes were reduced in the SCH and OH groups compared with the CON group (P < 0.05).
Expression of the transcription factor p75NTR in offspring of rats with SCH during pregnancy
We measured the expression levels of p75NTR in the cerebral cortex of pups from mother rats in the three groups. p75NTR levels were significantly increased in the SCH and OH groups to 1.56-fold and 2.36-fold higher compared with those in the CON group, respectively (n = 6, both P < 0.05). p75NTR levels in the OH group increased to 1.54-fold higher compared with levels in the SCH group (n = 6, P < 0.05; Fig. 7).
Figure 7.

Protein expression levels of p75 neurotrophin receptor (p75NTR), phospho-c-Jun N-terminal kinase (p-JNK) and pro-apoptotic proteins p53 and Bax in the cortex of pups (n = 6 per group). *P < 0.05 vs CON group, #P < 0.05 vs OH group.
JNK and p-JNK expression in offspring of rats with SCH during pregnancy
p-JNK expression and activation were determined by Western blotting, and p-JNK and total JNK were identified by rabbit polyclonal antibodies. p-JNK levels increased significantly in the pups of SCH (1.47-fold) and OH group (1.93-fold) compared with those in the CON group (n = 6, both P < 0.05; Fig. 6). However, there was no significant difference in total JNK among the three groups.
p53 expression in offspring of rats with SCH during pregnancy
Hypothyroidism promotes N-methyl-d-aspartate receptor activation, which in turn increases p53 expression, probably by increasing DNA damage and triggers apoptosis leading to cell death (11). p53 is an important member of the p75NTR signaling pathway, and it can induce cell apoptosis. We detected p53 and other components of the p75NTR signaling pathway in this study. The expression levels of p53 increased significantly in the cerebral cortex of pups in the SCH (1.64-fold) and OH group (2.16-fold) compared with those in the CON group (n = 6, both P < 0.05), as shown by Western blotting (Fig. 7).
Bax expression in offspring of rats with SCH during pregnancy
p53 can induce apoptosis by raising Bax levels, leading to caspase activation (12). Bax expression increased in pups from the SCH (1.55-fold) and OH group (2.08-fold) compared with those in the CON group (n = 6, P < 0.05; Fig. 7). These results were consistent with a previous report (9).
Discussion
Epidemiological studies suggest that SCH during pregnancy may affect the offspring’s intellectual development (1). Our group has performed a series of experiments to explore the mechanism in it (6). The current study explored the mechanism further from other aspect.
THs play a crucial role in fetal brain development and are involved in the proliferation, differentiation, maturation and migration of neurons in the brain. However, the fetal brain begins to develop before the thyroid, so THs needed for fetal brain development during early pregnancy are therefore derived exclusively from the mother. Moderate or even temporary hypothyroidism in early pregnancy may thus affect the normal migration of cortical neurons and the cellular architecture of the cerebral cortex and hippocampus (13). TH is also involved in apoptosis, which is another important physiological process in cerebral neuron development. Recent research reported that neonatal hypothyroidism could cause extensive apoptosis of granular cells in the cerebellum (14, 15, 16, 17), while maternal hypothyroidism could induce apoptosis in the fetal cortex (9). We therefore investigated if maternal perinatal SCH could also induce apoptosis in their offspring’s cerebral cortex.
Experimental hypothyroidism in neonatal rats was associated with structural defects in the nervous system accompanied by behavioral abnormalities (18). Methimazole and iodine deficiency have traditionally been used to establish models of thyroid deficiency, but these could potentially have other effects on embryogenesis. Surgical removal of the thyroid gland, together with TH supplementation, is thus a preferable method for achieving SCH. We successfully established a surgical model of SCH by this method.
The previous study performed by our group has reported that offspring of rats with either OH or SCH had cognitive impairment (6). Spatial learning and memory, measured by MWM test, were significantly decreased in the SCH group compared with the CON group and were more severely affected in the OH compared with the SCH group. In addition, these differences became more apparent with more training sessions. The probe test also indicated sustained memory impairment in the SCH and OH groups compared with the CON group. These results suggest that the pups born to mothers with either SCH or OH developed irreversible neuronal damage, and the neural network connectivity could not be reconstructed even after prolonged training. This was consistent with previous findings (6, 19, 20).
Hippocampal LTP is considered to be a suitable electrophysiological model for assessing synaptic plasticity in relation to learning and memory. The current results showed that prenatal exposure to OH and SCH led to a smaller increase in amplitude of f-EPSP after HFS, indicating LTP damage, consistent with previous studies (6, 19, 21). This supports the hypothesis that both OH and SCH may cause LTP impairment.
Maternal hypothyroidism can increase neuronal apoptosis during brain development (9), and previous studies showed that hypothyroidism increased apoptosis in the cerebellum (14, 15, 16, 17) and hippocampus (22, 23). Maternal hypothyroidism during pregnancy can also increase apoptosis during cerebral cortex development in the pups, especially in the surface layer of the primary somatosensory cortex (I-III level), which mainly affects neurons (9).
TH modulates levels of nerve growth factor (NGF) in the cerebral cortex, hippocampus and cerebellum in rats (14, 24). Synaptic plasticity, neuronal excitability and cognitive function are all regulated by NGF, which also plays critical roles in regulating hippocampal plasticity and learning in rodents (25, 26, 27, 28). NGF regulates neuronal cell proliferation and apoptosis by binding to two different receptor tyrosine kinase receptors A (TrkA) and p75NTR (29, 30). When NGF levels decreases, the TrkA signal pathway is inhibited and the p75NTR pathway is activated (31), thereby affecting learning- and memory-related protein expression, resulting in neuronal degeneration, apoptosis and mental retardation (32). ProNGF is a precursor protein of NGF and can induce apoptosis in many kinds of cells by combining preferentially with p75NTR (31, 33, 34, 35, 36). p75NTR can also transmit signals independently to promote the apoptosis of a variety of cells, including developing neurons (34, 35). The current results showed that p75NTR expression increased in the cerebral cortex of pups of hypothyroid rats, even in the case of SCH, suggesting that elevated levels of maternal TSH may increase apoptosis in the cerebral cortex in their offspring.
Several models of p75NTR-dependent cell death have demonstrated the involvement JNK activity (29, 35, 37, 38, 39, 40, 41). Increased p75NTR has been reported to activate the JNK pathway, suggesting a link between JNK activation and cell death. Overexpression of the p75NTR adaptor protein NRAGE was shown to induce apoptosis in a JNK-dependent manner (37). JNK activation is also considered to be a molecular switch in stress signal transduction (42). Some stimuli, including cytokines (such as tumor necrosis factor and interleukin-1) and reactive oxygen species, can activate the JNK pathway and play an important role in regulating cell fate, including cell proliferation, gene expression and apoptosis (43). Sherrin et al. also reported a role for JNK in the hippocampus in terms of memory and synaptic plasticity (44). JNK expression in the hippocampus and cortex may thus influence learning and memory.
p75NTR can transduce a dichotomous signal with both pro-apoptotic and anti-apoptotic functions. The tumor suppressor p53 is a nuclear phospho-protein that can promote arrest or apoptosis, and it is an essential factor in JNK-mediated neuronal apoptosis. When DNA is damaged, p53 induces the expression of the cyclin-dependent kinase inhibitor p21, leading to cell cycle stagnation (45). Previous studies found that increased JNK activation was consistent with increased expression of the pro-apoptotic proteins p53 and Bax in hypothyroidism (10) (Fig. 8).
Figure 8.
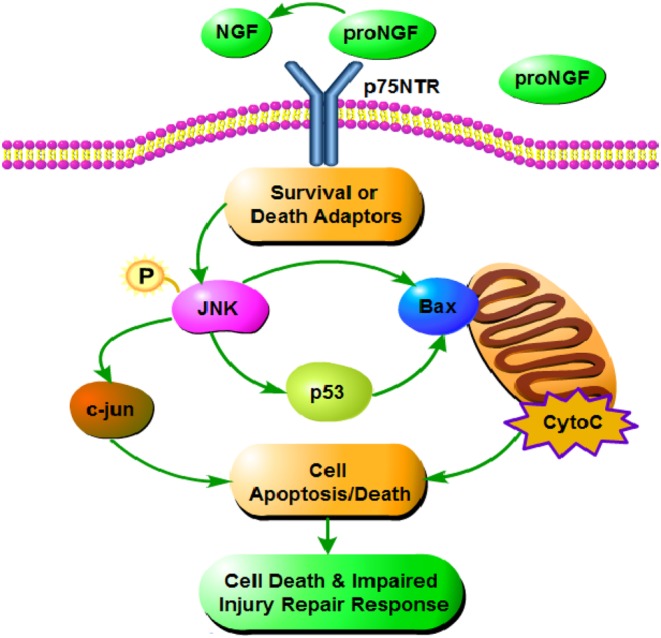
Effects of SCH and OH on p75NTR signaling pathway. ProNGF, as a precursor protein of NGF, can combine with p75NTR preferentially. p75NTR activates the JNK pathway to increase the p53 signaling pathway. p53 and Bax play essential roles in JNK-mediated neuronal apoptosis. In the present study, maternal SCH and OH increased p75NTR expression in the offspring, thus increasing activation of p-JNK, Bax and p53 signaling pathways and thereby increasing neuronal apoptosis.
Activation of p75NTR, JNK and p53 signaling have also been associated with increased neuronal apoptosis in Alzheimer’s disease (46). Our results suggest that similar changes in terms of increased apoptosis occurred in the cerebral cortex of pups exposed to perinatal SCH. This may explain the mechanism involved in the damage to the neurodevelopment of the offspring from SCH mothers partly. However, some other potential mechanisms may exist. Zhang et al. found decreased activation of the CREB signaling pathway in pups was related to impairments of cognitive function caused by maternal SCH (6). Lu et al. reported maternal SCH affects cytoarchitecture and cell migration in the developing brain of the progeny (47). A recent study of patients with Alzheimer’s disease and SCH showed that regional cerebral blood flow was significantly reduced, suggesting that SCH may affect the perfusion of brain regions associated with memory function (48). This may provide a new direction for research into SCH-induced memory deficits.
Limitations
Several limitations exist in our study. We did not perform Western blotting or other molecular biological experiments in the pups following MWM experiments, because we considered that the training may have affected the neurons in the cerebral cortex and thus affected spatial learning and memory. Furthermore, some rats died after thyroid surgery and some fetuses were aborted, which limited the availability of the cortical tissue for PCR, immunofluorescence, immunohistochemistry and other molecular experiments.
Conclusions
In summary, this study showed that perinatal SCH could increase the expression of p-JNK and the pro-apoptotic proteins p53 and Bax and increase neuronal apoptosis in the cerebral cortex of pups exposed to perinatal SCH, by activating the p75NTR signaling pathway. These results help to explain the cellular mechanisms whereby maternal SCH during pregnancy may adversely affect the development of the offspring’s cerebral cortex.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This study was supported by the Chinese National Natural Science Foundation (grant: 81500605) and the National Science and Technology Support Program (grant: 2014BAbib6B02). The funder had no role in study design, data collection or analysis or in the presentation or publication of the results.
References
- 1.Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, O’Heir CE, Mitchell ML, Hermos RJ, Waisbren SE, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. New England Journal of Medicine 1999. 341 549–555. ( 10.1056/NEJM199908193410801) [DOI] [PubMed] [Google Scholar]
- 2.Anderson GW, Schoonover CM, Jones SA. Control of thyroid hormone action in the developing rat brain. Thyroid 2003. 13 1039–1056. ( 10.1089/105072503770867219) [DOI] [PubMed] [Google Scholar]
- 3.Bernal J, Guadano-Ferraz A, Morte B. Perspectives in the study of thyroid hormone action on brain development and function. Thyroid 2003. 13 1005–1012. ( 10.1089/105072503770867174) [DOI] [PubMed] [Google Scholar]
- 4.Mastorakos G, Karoutsou EI, Mizamtsidi M, Creatsas G. The menace of endocrine disruptors on thyroid hormone physiology and their impact on intrauterine development. Endocrine 2007. 31 219–237. ( 10.1007/s12020-007-0030-y) [DOI] [PubMed] [Google Scholar]
- 5.Zoeller RT, Rovet J. Timing of thyroid hormone action in the developing brain: clinical observations and experimental findings. Journal of Neuroendocrinology 2004. 16 809–818. ( 10.1111/j.1365-2826.2004.01243.x) [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Fan Y, Yu X, Wang X, Bao S, Li J, Fan C, Shan Z, Teng W. Maternal subclinical hypothyroidism impairs neurodevelopment in rat offspring by inhibiting the CREB signaling pathway. Molecular Neurobiology 2015. 52 432–441. ( 10.1007/s12035-014-8855-x) [DOI] [PubMed] [Google Scholar]
- 7.Hempstead BL. The many faces of p75NTR. Current Opinion in Neurobiology 2002. 12 260–267. ( 10.1016/S0959-4388(02)00321-5) [DOI] [PubMed] [Google Scholar]
- 8.Roux PP, Barker PA. Neurotrophin signaling through the p75 neurotrophin receptor. Progress in Neurobiology 2002. 67 203–233. ( 10.1016/S0301-0082(02)00016-3) [DOI] [PubMed] [Google Scholar]
- 9.Kumar A, Sinha RA, Tiwari M, Pal L, Shrivastava A, Singh R, Kumar K, Kumar Gupta S, Godbole MM. Increased pro-nerve growth factor and p75 neurotrophin receptor levels in developing hypothyroid rat cerebral cortex are associated with enhanced apoptosis. Endocrinology 2006. 147 4893–4903. ( 10.1210/en.2006-0027) [DOI] [PubMed] [Google Scholar]
- 10.Sinha RA, Pathak A, Kumar A, Tiwari M, Shrivastava A, Godbole MM. Enhanced neuronal loss under perinatal hypothyroidism involves impaired neurotrophic signaling and increased proteolysis of p75(NTR). Molecular and Cellular Neurosciences 2009. 40 354–364. ( 10.1016/j.mcn.2008.12.001) [DOI] [PubMed] [Google Scholar]
- 11.Alva-Sanchez C, Rodriguez A, Villanueva I, Anguiano B, Aceves C, Pacheco-Rosado J. The NMDA receptor antagonist MK-801 abolishes the increase in both p53 and Bax/Bcl2 index induced by adult-onset hypothyroidism in rat. Acta Neurobiologiae Experimentalis 2014. 74 111–117. [DOI] [PubMed] [Google Scholar]
- 12.Miller FD, Pozniak CD, Walsh GS. Neuronal life and death: an essential role for the p53 family. Cell Death and Differentiation 2000. 7 880–888. ( 10.1038/sj.cdd.4400736) [DOI] [PubMed] [Google Scholar]
- 13.Auso E, Lavado-Autric R, Cuevas E, Del Rey FE, Morreale De Escobar G, Berbel P. A moderate and transient deficiency of maternal thyroid function at the beginning of fetal neocorticogenesis alters neuronal migration. Endocrinology 2004. 145 4037–4047. ( 10.1210/en.2004-0274) [DOI] [PubMed] [Google Scholar]
- 14.Neveu I, Arenas E. Neurotrophins promote the survival and development of neurons in the cerebellum of hypothyroid rats in vivo. Journal of Cell Biology 1996. 133 631–646. ( 10.1083/jcb.133.3.631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh R, Upadhyay G, Godbole MM. Hypothyroidism alters mitochondrial morphology and induces release of apoptogenic proteins during rat cerebellar development. Journal of Endocrinology 2003. 176 321–329. ( 10.1677/joe.0.1760321) [DOI] [PubMed] [Google Scholar]
- 16.Singh R, Upadhyay G, Kumar S, Kapoor A, Kumar A, Tiwari M, Godbole MM. Hypothyroidism alters the expression of Bcl-2 family genes to induce enhanced apoptosis in the developing cerebellum. Journal of Endocrinology 2003. 176 39–46. [DOI] [PubMed] [Google Scholar]
- 17.Xiao Q, Nikodem VM. Apoptosis in the developing cerebellum of the thyroid hormone deficient rat. Frontiers in Bioscience 1998. 3 A52–A57. ( 10.2741/A252) [DOI] [PubMed] [Google Scholar]
- 18.Cragg BG. Synapses and membranous bodies in experimental hypothyroidism. Brain Research 1970. 18 297–307. ( 10.1016/0006-8993(70)90330-6) [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Wei W, Wang Y, Dong J, Song B, Min H, Teng W, Chen J. Neurotoxicity of developmental hypothyroxinemia and hypothyroidism in rats: impairments of long-term potentiation are mediated by phosphatidylinositol 3-kinase signaling pathway. Toxicology and Applied Pharmacology 2013. 271 257–265. ( 10.1016/j.taap.2013.04.034) [DOI] [PubMed] [Google Scholar]
- 20.Dong J, Liu W, Wang Y, Hou Y, Xu H, Gong J, Xi Q, Chen J. Developmental iodine deficiency and hypothyroidism impair spatial memory in adolescent rat hippocampus: involvement of CaMKII, calmodulin and calcineurin. Neurotoxicity Research 2011. 19 81–93. ( 10.1007/s12640-009-9142-x) [DOI] [PubMed] [Google Scholar]
- 21.Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science 2001. 294 1030–1038. ( 10.1126/science.1067020) [DOI] [PubMed] [Google Scholar]
- 22.Huang XW, Zhao ZY, Ji C. Effects of hypothyroidism on apoptosis and the expression of Bcl-2 and Bax gene in the neonatal rat hippocampus neurons. Zhonghua Er Ke Za Zhi 2005. 43 48–52. [PubMed] [Google Scholar]
- 23.Huang XW, Yang RL, Zhao ZY, Ji C, Yang RW. Mechanism for apoptosis of hippocampus neuron induced by hypothyroidism in perinatal rats. Zhejiang Da Xue Xue Bao Yi Xue Ban 2005. 34 298–303. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez-Dolado M, Iglesias T, Rodriguez-Pena A, Bernal J, Munoz A. Expression of neurotrophins and the trk family of neurotrophin receptors in normal and hypothyroid rat brain. Brain Research Molecular Brain Research 1994. 27 249–257. ( 10.1016/0169-328X(94)90007-8) [DOI] [PubMed] [Google Scholar]
- 25.Sola E, Capsoni S, Rosato-Siri M, Cattaneo A, Cherubini E. Failure of nicotine-dependent enhancement of synaptic efficacy at Schaffer-collateral CA1 synapses of AD11 anti-nerve growth factor transgenic mice. European Journal of Neuroscience 2006. 24 1252–1264. ( 10.1111/j.1460-9568.2006.04996.x) [DOI] [PubMed] [Google Scholar]
- 26.Rosato-Siri M, Cattaneo A, Cherubini E. Nicotine-induced enhancement of synaptic plasticity at CA3-CA1 synapses requires GABAergic interneurons in adult anti-NGF mice. Journal of Physiology 2006. 576 361–377. ( 10.1113/jphysiol.2006.114587) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagostena L, Rosato-Siri M, D’Onofrio M, Brandi R, Arisi I, Capsoni S, Franzot J, Cattaneo A, Cherubini E. In the adult hippocampus, chronic nerve growth factor deprivation shifts GABAergic signaling from the hyperpolarizing to the depolarizing direction. Journal of Neuroscience 2010. 30 885–893. ( 10.1523/JNEUROSCI.3326-09.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conner JM, Franks KM, Titterness AK, Russell K, Merrill DA, Christie BR, Sejnowski TJ, Tuszynski MH. NGF is essential for hippocampal plasticity and learning. Journal of Neuroscience 2009. 29 10883–10889. ( 10.1523/JNEUROSCI.2594-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. Journal of Neuroscience 1998. 18 3273–3281. ( 10.1523/JNEUROSCI.18-09-03273.1998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nykjaer A, Willnow TE, Petersen CM. p75NTR--live or let die. Current Opinion in Neurobiology 2005. 15 49–57. ( 10.1016/j.conb.2005.01.004) [DOI] [PubMed] [Google Scholar]
- 31.Fortress AM, Buhusi M, Helke KL, Granholm AC. Cholinergic degeneration and alterations in the TrkA and p75NTR balance as a result of Pro-NGF injection into aged rats. Journal of Aging Research 2011. 2011 460543 ( 10.4061/2011/460543) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capsoni S, Brandi R, Arisi I, D’Onofrio M, Cattaneo A. A dual mechanism linking NGF/proNGF imbalance and early inflammation to Alzheimer’s disease neurodegeneration in the AD11 anti-NGF mouse model. CNS and Neurological Disorders Drug Targets 2011. 10 635–647. [DOI] [PubMed] [Google Scholar]
- 33.Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low-affinity NGF receptor. Science 1993. 261 345–348. ( 10.1126/science.8332899) [DOI] [PubMed] [Google Scholar]
- 34.Frade JM, Rodriguez-Tebar A, Barde YA. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 1996. 383 166–168. ( 10.1038/383166a0) [DOI] [PubMed] [Google Scholar]
- 35.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 1996. 383 716–719. ( 10.1038/383716a0) [DOI] [PubMed] [Google Scholar]
- 36.Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. PNAS 1994. 91 6501–6505. ( 10.1073/pnas.91.14.6501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salehi AH, Xanthoudakis S, Barker PA. NRAGE, a p75 neurotrophin receptor-interacting protein, induces caspase activation and cell death through a JNK-dependent mitochondrial pathway. Journal of Biological Chemistry 2002. 277 48043–48050. ( 10.1074/jbc.M205324200) [DOI] [PubMed] [Google Scholar]
- 38.Harrington AW, Kim JY, Yoon SO. Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. Journal of Neuroscience 2002. 22 156–166. ( 10.1523/JNEUROSCI.22-01-00156.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman WJ. Neurotrophins induce death of hippocampal neurons via the p75 receptor. Journal of Neuroscience 2000. 20 6340–6346. ( 10.1523/JNEUROSCI.20-17-06340.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brann AB, Tcherpakov M, Williams IM, Futerman AH, Fainzilber M. Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. Journal of Biological Chemistry 2002. 277 9812–9818. ( 10.1074/jbc.M109862200) [DOI] [PubMed] [Google Scholar]
- 41.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. Journal of Cell Biology 1998. 143 1691–1703. ( 10.1083/jcb.143.6.1691) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cattani D, Goulart PB, Cavalli VL, Winkelmann-Duarte E, Dos Santos AQ, Pierozan P, de Souza DF, Woehl VM, Fernandes MC, Silva FR, et al. Congenital hypothyroidism alters the oxidative status, enzyme activities and morphological parameters in the hippocampus of developing rats. Molecular and Cellular Endocrinology 2013. 375 14–26. ( 10.1016/j.mce.2013.05.001) [DOI] [PubMed] [Google Scholar]
- 43.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012. 143 307–320. ( 10.1053/j.gastro.2012.06.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sherrin T, Blank T, Todorovic C. c-Jun N-terminal kinases in memory and synaptic plasticity. Reviews in the Neurosciences 2011. 22 403–410. ( 10.1515/RNS.2011.032) [DOI] [PubMed] [Google Scholar]
- 45.Golias CH, Charalabopoulos A, Charalabopoulos K. Cell proliferation and cell cycle control: a mini review. International Journal of Clinical Practice 2004. 58 1134–1141. ( 10.1111/j.1742-1241.2004.00284.x) [DOI] [PubMed] [Google Scholar]
- 46.Costantini C, Rossi F, Formaggio E, Bernardoni R, Cecconi D, Della-Bianca V. Characterization of the signaling pathway downstream p75 neurotrophin receptor involved in beta-amyloid peptide-dependent cell death. Journal of Molecular Neuroscience 2005. 25 141–156. ( 10.1385/JMN:25:2:141) [DOI] [PubMed] [Google Scholar]
- 47.Lu L, Yu X, Teng W, Shan Z. Treatment with levothyroxine in pregnant rats with subclinical hypothyroidism improves cell migration in the developing brain of the progeny. Journal of Endocrinological Investigation 2012. 35 490–496. ( 10.3275/7967) [DOI] [PubMed] [Google Scholar]
- 48.Haji M, Kimura N, Hanaoka T, Aso Y, Takemaru M, Hirano T, Matsubara E. Evaluation of regional cerebral blood flow in Alzheimer’s disease patients with subclinical hypothyroidism. Dementia and Geriatric Cognitive Disorders 2015. 39 360–367. ( 10.1159/000375298) [DOI] [PubMed] [Google Scholar]