

Abstract
Glycopeptide antibiotics (GPAs) are a key weapon in the fight against drug resistant bacteria, with vancomycin still a mainstream therapy against serious Gram-positive infections more than 50 years after it was first introduced. New, more potent semisynthetic derivatives that have entered the clinic, such as dalbavancin and oritavancin, have superior pharmacokinetic and target engagement profiles that enable successful treatment of vancomycin-resistant infections. In the face of resistance development, with multidrug resistant (MDR) S. pneumoniae and methicillin-resistant Staphylococcus aureus (MRSA) together causing 20-fold more infections than all MDR Gram-negative infections combined, further improvements are desirable to ensure the Gram-positive armamentarium is adequately maintained for future generations. A range of modified glycopeptides has been generated in the past decade via total syntheses, semisynthetic modifications of natural products, or biological engineering. Several of these have undergone extensive characterization with demonstrated in vivo efficacy, good PK/PD profiles, and no reported preclinical toxicity; some may be suitable for formal preclinical development. The natural product monobactam, cephalosporin, and β-lactam antibiotics all spawned multiple generations of commercially and clinically successful semisynthetic derivatives. Similarly, next-generation glycopeptides are now technically well positioned to advance to the clinic, if sufficient funding and market support returns to antibiotic development.
Keywords: antibiotics, glycopeptides, antimicrobial resistance, vancomycin
Gram Positive Infections and Current Therapies
The rise of multidrug resistant (MDR) bacteria is of global concern, due to the rapid spread of resistance coupled with a sharp decline in the number of new antibiotics under development.1−3 Most attention is focused on the threat posed by highly resistant Gram-negative pathogens, such as carbapenem-resistant Enterobacteriaceae (CRE), with recent incentives emphasizing the development of new Gram-negative antibiotics (such as CARB-X; www.carb-x.org/). Although it appears that the Gram-positive pipeline is currently well served, especially with the recent introduction of second generation glycopeptide antibiotics (GPAs) oritavancin and dalbavancin, there is no compelling reason to abandon the development of new Gram-positive therapies in the face of inevitable glycopeptide resistance. Despite the economic hurdles faced by new Gram-positive GPAs in the contemporary landscape, similar skepticism did not hinder the introduction of daptomycin (FDA approved in 2003), which achieved annual sales of over $US1b/yr by 2013.4 On the basis of the 2013 US CDC report on antimicrobial resistance,5 it is apparent that the number of infections caused by resistant strains of only two Gram-positive organisms (methicillin-resistant Staphylococcus aureus (MRSA) and multidrug resistant (MDR) Streptococcus pneumoniae) dwarfs the number of resistant Gram-negative infections (including MDR Pseudomonas aeruginosa, MDR Acinetobacter, and extended-spectrum β-lactamase CRE), with >1 000 000 vs <50 000 cases, respectively (Figure 1). This translates into a similar imbalance in the number of deaths, with over 6-fold more deaths caused by Gram-positive organisms (approximately 18 000 vs 3200). A more recent epidemiological review of MRSA rates in the USA found no definitive evidence of a reduction in the incidence of MRSA infections between 2013 and 2015.6 While it is unclear how many of these deaths could be prevented by more effective antibiotic treatment and/or improvements in infection prevention strategies, improved therapies are desirable. Promisingly, of the 43 antibiotics in clinical development at the end of 2015, 39 have Gram-positive activity compared to only 25 with Gram-negative activity.1
Figure 1.
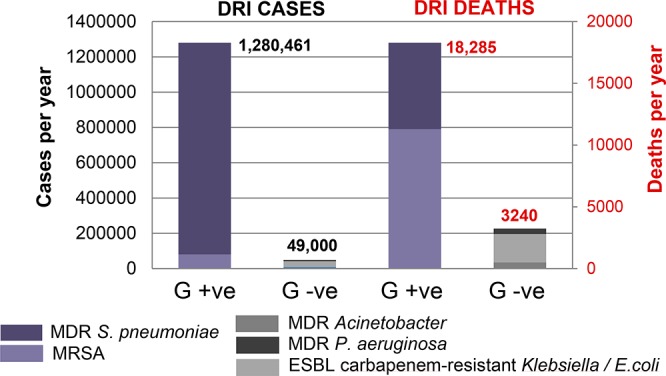
Comparison of annual drug-resistant infection (DRI) cases and deaths in the USA due to Gram-positive (MRSA or MDR S. pneumoniae) or Gram-negative (MDR P. aeruginosa, MDR Acinetobacter, and extended-spectrum β-lactamase CRE) drug-resistant bacteria.5
Current therapies for Gram-positive infections7,8 include the glycopeptides (inhibition of peptidoglycan synthesis via binding to Lipid II: vancomycin 1, teicoplanin 3, and more recently telavancin 4, dalbavancin 5, and oritavancin 6b), the lipopeptide daptomycin (membrane disruption), the oxazolidinones (inhibition of protein synthesis: linezolid and, more recently, tedizolid), cephalosporins (inhibition of peptidoglycan synthesis via binding to penicillin binding proteins: fifth generation ceftaroline and ceftobiprole), and glycylcycline tetracyclines (inhibition of protein synthesis: tigecycline).
Compared to other antibiotics, the development of resistance to vancomycin 1 has taken time to appear (Figure 3);9−12 high-level resistance to vancomycin was first reported in enterococci (VRE) in 1988, some 30 years after its clinical introduction. Although resistance spread rapidly thereafter,9,11 glycopeptide resistance actually predates modern clinical antibiotic use, with the discovery of glycopeptide resistance genes in ancient DNA recovered from 30 000 year old permafrost.13 MRSA with reduced vancomycin susceptibility was not reported until 1997,14 with a moderate reduction in MIC (3–8 μg/mL) leading to its classification as vancomycin intermediate-resistant S. aureus (VISA). Subsequent retrospective studies revealed the prevalence of VISA some 10 years earlier in United States, Europe, and Japan.15 High-level vancomycin resistance in S. aureus (VRSA; MIC ≥ 16 μg/mL), arising from the horizontal gene transfer of the vanA gene cluster from Enterococci, was first reported in the US in 2002.16 Fortunately, the number of cases is still very limited with 13 isolates in the United States at the end of 2013.17
Figure 3.

Timeline of discovery for the clinically used glycopeptide antibiotics vancomycin 1, ristocetin 2, teicoplanin 3, telavancin 4, dalbavancin 5, and oritavancin 6b.
Although vancomycin has been the mainstay of parenteral therapy for MRSA infections for several decades, increasing evidence suggests that it may be losing its clinical efficacy against serious MRSA infections with MICs at the higher end of the susceptibility range. Furthermore, vancomycin is only slowly bactericidal and is characterized by suboptimal properties such as PK (requiring twice daily dosing and serum level monitoring) and complex variable tissue penetration. Despite many decades of successful use, the optimal dosing of vancomycin in critically ill patients remains a contentious issue and highlights the need for improved GPAs.18
Mechanism of Action and Resistance Mechanisms
The glycopeptide antibiotics work by binding to the membrane-bound Lipid II precursor of peptidoglycan, preventing its incorporation into the vital structural cell wall component (Figure 2B). Binding of vancomycin, and other glycopeptides, to Lipid II is enhanced by a cooperative back-to-back dimerization that increases their Lipid II binding affinity (Figure 2A).19−21 A common glycopeptide resistance mutation, especially in enterococci, is a divergent biosynthesis of Lipid II from the d-Ala-d-Ala terminating muropeptide to a d-Ala-d-Lac (vanA, vanB, vanD) or less commonly d-Ala-d-Ser (vanC, vanE, vanG) phenotype. The replacment of d-Ala with d-Lac removes one of the five hydrogen bonds formed with the vancomycin central pocket. More importantly, it introduces a repulsive lone pair–lone pair interaction between the two oxygen atoms that is believed to be mainly responsible for the 1000-fold loss in affinity of vancomycin for Lipid II.22 VISA resistance arises from thickened cell walls due to accumulation of excess amounts of peptidoglycan, while VRSA follows the VRE Lipid II VanA modification.
Figure 2.

(A) Molecular dynamics simulation of vancomycin 1 interacting with membrane-bound Lipid II, demonstrating vancomycin dimerization. (B) Hydrogen bond interactions between vancomycin 1 backbone and d-Ala-d-Ala component of Lipid II.
History of Glycopeptide Discovery
The first two Actinobacteria-derived glycopetide antibiotics, vancomycin 1 and ristocetin 2 (Figure 3), were discovered by Eli Lilly and Abbott Laboratories from Amycolatopsis orientalis and A. lurida, respectively, in the mid-1950s.15,23 Both were used clinically to treat Gram-positive infections; however, ristocetin 2 was later withdrawn from the market as it led to lowered blood platelet counts (thrombocytopenia) in some patients.24 Vancomycin 1 was approved for use in the clinic in 1958, but its structure was only unambiguously determined in 1982,25 which was 27 years after its first use in humans in 1955. As newer generation antibiotics entered the market, the use of vancomycin 1 declined until the 1980s, when significant resistance to standard β-lactam therapies evolved with the rise of MRSA in hospitals. Importantly, the sparse use of vancomycin had prevented the development of widespread glycopeptide resistance and vancomycin re-emerged in the clinic as the drug of choice until high level vancomycin resistance was reported.
The other naturally occurring marketed glycopeptide antibiotic, teicoplanin 3, is a ristocetin-type lipoglycopeptide complex first reported from Actinoplanes teichomyceticus in 1978.26,27 Teicoplanin 3 was approved in Europe in 1998 and is currently used in many countries. However, it has never been approved for use in the United States.
The uniqueness of vancomycin in terms of its clinical success, lack of cross resistance with other classes of antibacterial agents, and the significant lag time between discovery and the appearance of resistance reignited a focus on the discovery of new natural product GPAs. This renewed interest, facilitated by advances in spectroscopy enabling rapid structural determination, led to an explosion in the number of new glycopeptides identified in the period from 1982 to 1996, thus heralding the development of contemporary glycopeptide therapeutics.28,29 However, since the mid 1990s, the discovery of new glycopeptides has waned, requiring innovative methods to identify and purify relatively uncommon producing strains of actinomycetes.28,30 Despite these improvements, since the 1990s very few new glycopeptides have been identified as natural products, with analogs instead produced semisynthetically or via genetic manipulation. One exception is the discovery of pekiskomycin 38 (Figure 14) in 2013 by Wright and co-workers, applying a novel resistance screening and genetics approach.31 A strategy of screening for novel biosynthetic gene clusters was combined with a glycopeptide resistance prefilter to assist in initial selection of isolates,32 leading to the isolation of 38(31) and demonstrating the utility of this novel approach.
Figure 14.

New glycopeptide scaffolds (removed biaryl linkages highlighted in yellow).
Recently Approved Glycopeptides
Telavancin
Telavancin (Vibativ, TD-6424) 4 (Figure 4) is a second generation lipoglycopeptide introduced by Theravance in 2009. It is derived from the chemical modification of vancomycin 1 to include attachment of a (decylaminoethyl) lipophilic tail on the vancosamine sugar and a hydrophilic [(phosphonomethyl)aminomethyl] group on the 4′-position of aromatic amino acid 7. The former modification improves potency against a range of Gram-positive pathogens, whereas the latter provides favorable ADME properties.33−35 Its spectrum of antibacterial activity is similar to that of vancomycin 1. It is approved for the treatment of Gram-positive associated complicated SSSIs in the US and Canada and hospital-acquired and ventilator-associated bacterial pneumonia caused by S. aureus in the US and Europe (see Table 1 for a comparison of dosing and pharmacokinetic properties of marketed glycopeptides).36−38 It is also being evaluated for efficacy and safety for the treatment of subjects with complicated S. aureus bacteremia and S. aureus right-sided infective endocarditis.39 A recent study40 suggests telavancin 4 may find utility in the treatment of the emerging multidrug-resistant pathogen Corynebacterium striatum, and its effectiveness for eradicating biofilms in vitro and in vivo has been reviewed recently.41 It is highly protein bound (∼93% in human plasma),42 but the presence of albumin does not influence the in vitro antibacterial activity of telavancin to any great extent.43
Figure 4.
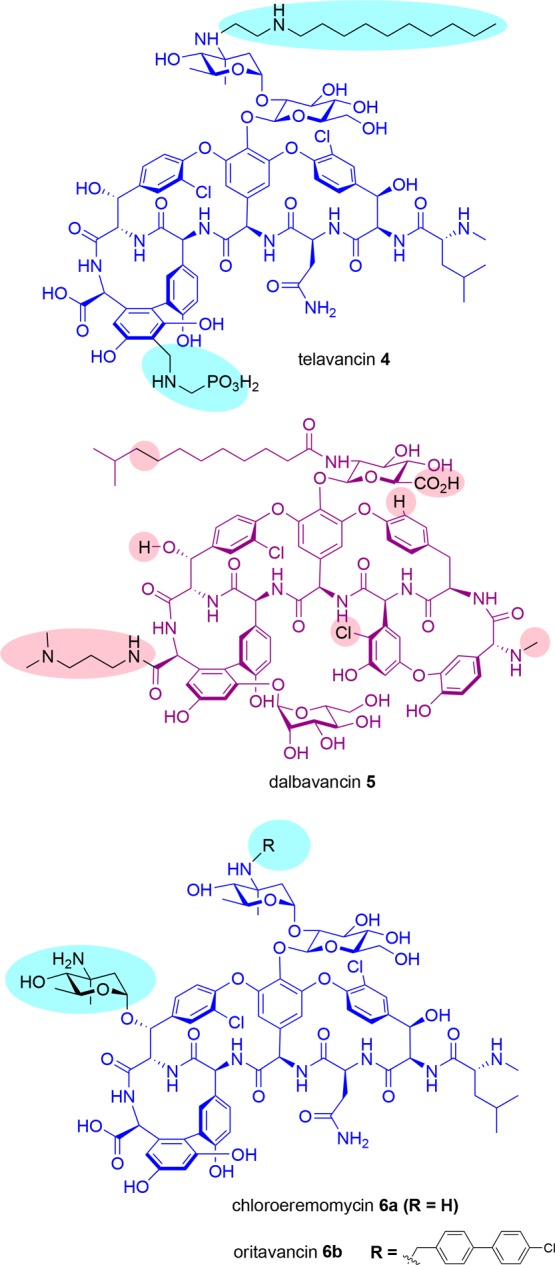
Structures of clinically approved semisynthetic glycopeptides telavancin 4, dalbavancin 5, and oritavancin 6b (derived from chloroeremomycin 6a). Differences from vancomycin are highlighted in blue for 4 and 6 and from teicoplanin, in red for 5.
Table 1. Indications and Pharmacokinetic Properties of Marketed Glycopeptides.
| vancomycin52 | telavancin53 | dalbavancin54 | oritavancin55 | |
|---|---|---|---|---|
| indication | ABSSSI, HAP/VAP,a endocarditis, osteomyelitis, colitisb | ABSSSI, HAP/VAPa | ABSSSIa | ABSSSIa |
| dosage | 25 mg/kg, ivc | 10 mg/kg, iv | 1500 mg, iv | 1200 mg, iv |
| dosage frequency | twice daily | once daily | single dosed | single dose |
| terminal t1/2 (h) | 6–1256 | 8 | 346 | 245 |
| pharmacodynamic predictor | AUC/MIC | AUC/MIC | AUC/MIC | AUC/MIC |
| % protein binding | <5057 | 9342 | 93–9858 | 85–9059 |
| susceptibility breakpoint (S. aureus)60 | ≤2 μg/mL | ≤0.12 μg/mL | ≤0.12 μg/mL | ≤0.12 μg/mL |
Acute bacterial skin and skin structure infections (ABSSSI), hospital-acquired pneumonia (HAP), and ventilator-associated pneumonia (VAP) caused by S. aureus.
Oral dosing: pseudomembranous colitis (C. difficile) and enterocolitis (S. aureus).
Provided as initial loading dose, followed by 1000 mg maintenance dose at 12 h.
Can also be delivered as 1000 mg on day 1 followed 1 week later by 500 mg.
Telavancin 4 has a rapid bactericidal action that is believed to be due to a cooperative effect resulting from binding to the acyl-d-alanyl-d-alanine subunit in nascent peptidoglycan concomitant with membrane insertion of the lipophilic tail, which ultimately disrupts the functional integrity of the bacterial membrane through membrane depolarization and leakage but not cell lysis.34,35
Telavancin 4 is subject to the same resistance mechanisms as vancomycin, namely, VanA modification of the terminal d-Ala-d-Ala to d-Ala-d-Lac, as well as the cell wall thickening characteristic of VISA and heterogeneous VISA (hVISA) phenotypes.44 Telavancin 4 induces expression of the vanA operon in VRE but not in VRE harboring vanB.45 Similarly, telavancin 4 induces VanX activity (reduction in levels of peptidoglycan precursors ending in d-Ala-d-Ala by action of a d,d-dipeptidase) in VanA strains but not in VanB strains.45 High-level telavancin 4 resistance in MRSA, MSSA (methicillin-susceptible S. aureus), or VRE has not been observed during in vitro resistance selection studies;44,46 continued surveillance indicates that telavancin 4 remains potent in vitro against a range of Gram positive pathogens.47−50 Examples of selection of resistance in vivo are not widely reported; in one example, a 3-fold increase in the MIC of telavancin was observed during the treatment of a patient with persistent bacteremia and mediastinitis with MRSA that evolved into hVISA during prior treatment with vancomycin and daptomycin.51
Dalbavancin
Dalbavancin 5 (Dalvance; formerly known as MDL 63,397, BI 397, A-A1, and VER 001) is a second generation teicoplanin-type glycopeptide marketed by Durata Therapeutics/Allergan. It has had a checkered history, with development spanning more than 15 years under four different companies, with final approval for clinical use granted in 2014.61 It is derived from the natural product glycopeptide A4092662 by amidation of the peptide-carboxy group of amino acid 7 with 3-(dimethylamino)-1-propylamine.63 The introduction of this substituent increases potency against staphylococci, particularly coagulase-negative staphylococci (CoNS).64 It is approved for the treatment of Gram-positive-associated acute bacterial skin and skin structure infections (ABSSSI) in adult patients and is currently being evaluated for efficacy and safety in adult subjects with osteomyelitis.65 It was also undergoing a Phase II evaluation in early 2017 in patients with complicated bacteremia or infective endocarditis, but the study was terminated due to “business reasons”.66 A recent study described the unique off-label use of dalbavancin 5 to treat MRSA tricuspid valve endocarditis in a pregnant patient.67 The treatment failed due to reinfection with VISA, perhaps due to inadequate dalbavancin 5 exposure, highlighting the uncertainties associated with off-label use (see Table 1 for a comparison of indications, dosing, and pharmacokinetic properties).68,69 Despite relatively high protein binding (93–98%), dalbavancin 5 is able to exert potent bactericidal activity.58
Like other lipoglycopeptides, dalbavancin 5 targets the C-terminal acyl-d-Ala-d-Ala subunit of peptidoglycan precursors. It is bactericidal and possesses in vitro activity against a wide range of Gram-positive organisms.70 There is no published data implicating the role of the lipid side chain in destabilization of cell membranes. Instead, its interaction with serum proteins likely contributes to its extended half-life.71 Furthermore, the positively charged C-terminal dimethylaminopropyl group may interact with the negative phospholipid head groups of the bacterial membrane.71 Whereas the dimerization of vancomycin-type glycopeptides is cooperative with ligand-binding, dalbavancin 5 dimerizes in an anticooperative manner.72
Dalbavancin 5 is inactive against VanA enterococci but remains active against the VanB phenotype:73in vitro resistance to the latter (>128-fold increase in MIC) was observed during serial passage (20 cycles) of VanB E. faecalis in the presence of dalbavancin 5.46 By virtue of its long half-life, concerns74 have been raised about the potential for resistance to develop during the course of treatment, as there is extended exposure to subtherapeutic levels; the recent emergence of a dalbavancin nonsusceptible VISA strain in a patient with a MRSA central line-associated bloodstream infection supports this hypothesis.75
Oritavancin
Oritavancin 6b (Orbactiv, LY-333328) is a second generation semisynthetic lipoglycopeptide developed by Eli Lilly and subsequently marketed by The Medicines Company, though it has recently (November 2017) been sold to Melinta Therapeutics. It is derived from the chemical modification of the natural product chloroeremomycin 6a to include attachment of an N-alkyl-p-chlorophenylbenzyl substituent onto the epi-vancosamine of the disaccharide attached to the ring 4 amino acid. Lilly discovered chloroeremomycin 6a in 1988, noting that it was uniquely different to vancomycin in terms of the vancosamine sugar moieties, possessing two l-4-epi-vancosamine subunits on the ring 4- and 6-amino acids instead of a single l-vancosamine subunit at position 4 in vancomycin 1. The influence of the additional l-4-epi-vancosamine in chloroeremomycin 6a imparts additional benefits, conferring enhanced antimicrobial activity to 6a against vancomycin-susceptible bacteria compared to vancomycin 1.76 In light of reported vancomycin resistance in E. faecium and E. faecalis in 1988–89, the genesis of oritavancin 6b was inspired by Lilly’s discovery of chloroeremomycin combined with their historical observation that vancomycin derivatives with alkyl (not acyl) side chains substituted on the vancosamine sugar displayed impressive activity against vancomycin-resistant bacteria.76 Oritavancin 6b was clinically approved in 2014 for treatment of Gram-positive associated ABSSSI in adults. Although not approved for other indications, it has shown promise in the treatment of prosthetic joint infections77 and prosthetic valve endocarditis caused by VRE78 (see Table 1 for a comparison of indications, dosing, and pharmacokinetic properties).59,79 Oritavancin, like dalbavancin, is quite highly protein bound, with 85–90% bound to serum proteins.59,80
In contrast to telavancin 4 and dalbavancin 5, oritavancin 6b is the only lipoglycopeptide to retain potent activity against both VRSA and VanA-type VRE due to its ability to act by multiple modes of action.81,82 In addition to inhibiting transglycosylation by binding to acyl-d-Ala-d-Ala termini, it can also disrupt transpeptidation by binding to the pentaglycyl bridging segment of Lipid II, thus maintaining affinity for the modified peptidoglycan peptide termini of vancomycin-resistant organisms.83,84 Second, the interaction with Lipid II is enhanced by oritavancin 6b being anchored to the cell membrane via the hydrophobic N-alkyl-p-chlorophenylbenzyl substituent, leading to increased target avidity promoting the self-association of oritavancin 6b into dimers.21 The interaction of oritavancin 6b with the membrane in this manner also leads to its third mode of action, disruption of the integrity of the bacterial membrane leading to depolarization and increased permeability.82
Although oritavancin 6b nonsusceptible isolates have not been reported in a clinical setting, it is possible to select for resistant enterococcal isolates in vitro; serial passage (20 cycles) of VRE (VanA and VanB) with oritavancin 6b led to 2- to 32-fold increases in MIC.46,85
New Glycopeptide Derivatives
Over the years, there have been many semisynthetic glycopeptide derivatives made, employing a range of derivatization strategies. The glycopeptide scaffold contains several substituents that lend themselves to facile modification, particularly the free C-terminal carboxylic acid group, the vancosamine sugar primary amine (when present), and the N-terminal primary or secondary amine (Figure 5). Comprehensive reviews of structural modifications of glycopeptide antibiotics,86 their total synthesis,87 and their biosynthesis88,89 are available.
Figure 5.
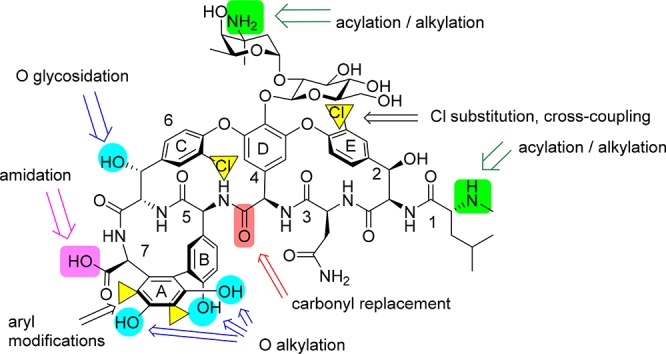
Potential sites for modification of vancomycin 1.
In recent years, several strategies have been published that focus on rational design to improve the potency of glycopeptide antibiotics and overcome resistance, including membrane-targeting approaches, backbone modifications, and hybrid antibiotic conjugates.
Membrane Targeting Approaches
The vancapticins 7 (Figure 6) are a series of vancomycin analogues developed at The University of Queensland that have been modified at the C-terminus with a modular assembly containing a bis-amine linker group, a cationic peptide (the electrostatic effector peptide sequence, EEPS), and a hydrophobic cap (the membrane insertive element, MIE).90−92 These substituents are designed to selectively deliver the vancapticin molecule, containing the Lipid II-binding vancomycin substructure, to bacterial membranes. The MIE provides a general lipid anchoring group, and the positively charged EEPS interacts with the predominantly anionic components of the bacterial membranes. Given that the target of vancomycin, Lipid II, is membrane bound, the membrane-targeting groups enhance the concentration of vancomycin on the membrane surface, not only providing a greater chance of interacting with Lipid II but also promoting the dimerization of vancomycin on the surface of the membrane. This allows for “bidentate” binding to further increase overall potency. Indeed, the combined modifications lead to over 100-fold increases in potency against MRSA, with both MIE and EEPS components required for maximum effect, e.g., 7b MRSA MIC = 0.003 μg/mL versus vancomycin MRSA MIC = 1 μg/mL.92 Additional mechanisms of action beyond Lipid II binding and peptidoglycan inhibition are evident from membrane perturbation studies, which clearly demonstrate that the vancapticins also result in membrane disruption, presumably due to the insertion of the MIE component.92
Figure 6.
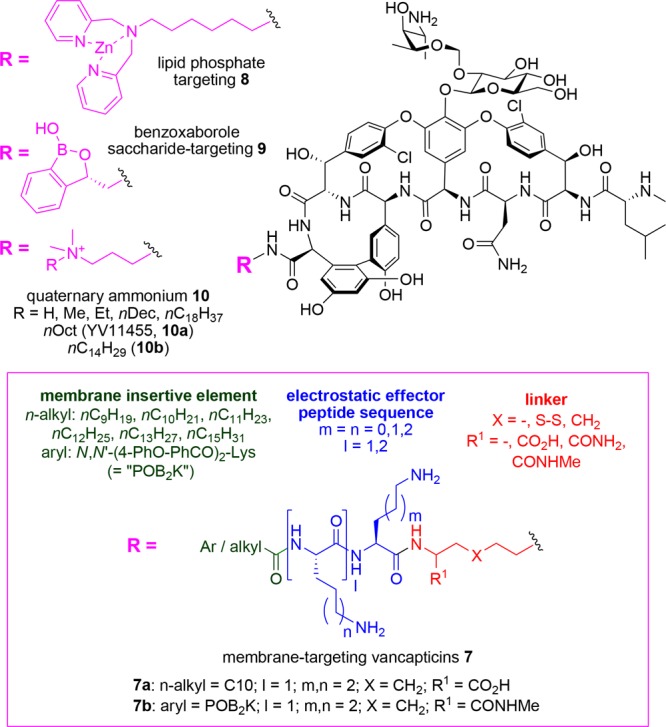
Membrane-targeting strategies to increase vancomycin potency.
The synthesis of the vancapticins initially relied on a disulfide ligation to attach the MIE/EEPS to a thiol-substituted vancomycin core, but these derivatives were biologically unstable. Replacement of the disulfide with a carbon-based linker provided a series of compounds (e.g., 7a, 7b) with excellent plasma stability and half-lives substantially greater than that of vancomycin in mice (e.g., t1/2 = 1.1 to 7.0 h vs 0.8 h for vancomycin). The carbon-linked compounds also demonstrated excellent efficacy in multiple mouse infection models, including S. aureus thigh infection, S. aureus lung infection, S. pneumoniae lung infection, and S. aureus peritonitis studies. For example, a single 25 mg/kg dose of compound 7a was equivalent to a single 200 mg/kg dose of vancomycin 1, with a 6-log reduction compared to saline control at 24 h in a neutropenic mouse thigh infection model against MRSA.92
The Haldar group at the Jawaharlal Nehru Centre for Advanced Scientific Research in India attached a zinc-binding dipicolyl moiety to the C-terminus of vancomycin to give 8 (Figure 6). Their hypothesis was that the Zn-chelated complex would bind to the pyrophosphates of cell-wall lipids, providing enhanced activity. While activity against vancomycin-sensitive MRSA was similar (0.5 μM), under standard testing conditions, the adduct was 10-fold more potent than vancomycin against VISA and >150-fold higher against VRE. In the presence of added Zn2+ ions, activity was further enhanced by a factor of 2 to 3. No induction of resistance was observed after 25 passages, and at 12 mg/kg in a VRE renal murine infection model, the compound reduced the cfu by 5 logs, compared to 2 logs for vancomycin. No hemolysis was seen at 1000 μM, and single iv dosing at 100 mg/kg was tolerated.93
Researchers at the Gause Institute for New Antibiotics in Russia and Anacor Pharmaceuticals have tethered a benzoxaborole group to the C-terminus of vancomycin, eremomycin, and teicoplanin aglycon, as well as to the amino group of the vancomycin vancosamine sugar, or to the N-terminus of vancomycin or teicoplanin aglycon (Figure 6). Their rationale was that the benzoaborazole group would form additional interactions with the 1,2- and 1,3-diols of saccharides coating the cell surface. The vancosamine and N-terminal substitutions on vancomycin generally reduced activity, but the C-terminal 3-substituted oxaborole derivative 9 was 4–16-fold more potent than vancomycin against S. epidermidis, VISA, and enterococci.94
A number of other glycopeptide derivatization strategies have introduced lipophilic groups, or positively charged lipophilic groups, with the goal of increasing membrane interactions. The Haldar group attached a quaternary ammonium propylamine substituent to the C-terminus of vancomycin to give 10 (Figure 6).95 They found the optimum chain length for the quaternary substitution varied with bacterial species: MSSA, MRSA, and VISA were best inhibited with a decyl chain, but vancomycin-susceptible enterococci (VSE) and VRE, with a more lipophilic C14H29 substituent. VRE potency of 0.7 μM was achieved, equating to an over 100-fold improvement compared to vancomycin (750 μM). VISA activity was also improved (0.36 μM vs 13 μM), but the same derivative had identical MRSA activity as vancomycin, 0.6 μM. The compounds increased the permeability of bacterial membranes but showed no mammalian cell cytotoxicity at 100 μM. The analog with R = nOct (YV1145, 10a) was not hemolytic at 1000 μM. Importantly, it was also shown to be efficacious in a neutropenic mouse thigh infection study, with greater efficacy than vancomycin when both were dosed at 12 mg/kg. 10a underwent additional in vivo testing. The 50% effective dose in the MRSA thigh infection model was found to be 3.3 mg/kg, and dose fractionation studies showed that a single dose was preferred, with a half-life of 1.6 h. Acute toxicity studies gave an LD50 of 78 mg/kg (more toxic than vancomycin but similar to other lipoglycopeptides; e.g., vancomycin: median lethal dose in rats, 319 mg/kg;96 telavancin: minimum lethal dose in mice, 100 mg/kg;97 dalbavancin: LD50 for mice and rats, 200 mg/kg;98 oritavancin: median lethal dose in mice and rats, 63–98 mg/kg99) with no abnormalities observed in major body organs after a single iv dose of 12 mg/kg.10010a also demonstrated intracellular activity, killing MRSA contained within macrophages to a much greater extent than vancomycin or linezolid.101 These quaternary ammonium derivatives were also tested against a range of Gram-negative bacteria,102 against which vancomycin is inactive, and found to possess significant activity (C8, C10, and C14 analogs). The C14 derivative 10b possessed Gram-negative activity vs Escherichia coli (1–5 μM), A. baumannii (3–5 μM), K. pneumoniae (9 μM), and P. aeruginosa (6 μM), presumably due to disruption of the integrity of the outer membrane allowing the glycopeptide to reach the peptidoglycan layer. 10b at 15 mg/kg showed potency equivalent to 5 mg/kg colistin in an A. baumannii murine thigh infection model, and a resistance induction experiment showed no development of resistance after 20 passages, compared to a 32-fold increase for colistin.102
Yoganathan and Miller from Yale University lipidated the three different hydroxyl groups on vancomycin in a selective fashion by employing different peptide catalysts, producing 11a–g (Figure 7).103 The tri- and penta-peptide catalysts were identified by screening a peptide library containing a catalytic π(methyl)-histidine residue, which enables the selective transfer of acyl groups. The introduction of a decanoyl group had similar effects at all three positions, leading to an approximately 4-fold improvement in potency compared to vancomycin against MSSA, MRSA, and E. faecalis and even larger improvements against VanA and VanB E. faecalis.
Figure 7.

Additional membrane-targeting strategies to increase vancomycin potency.
The Haldar group that prepared 8 has also developed another vancomycin derivative, YV54465 12a, (Figure 7) in which the vancosamine amine is alkylated with a decanyl group and the carboxylic acid is amidated with a cyclic-acyclic polyhydroxylated sugar moiety.104,105 Nominally, this sugar was designed to produce additional hydrogen bonding interactions with the Lipid II target. 12a was 2-fold more potent than vancomycin against MRSA but 35- and >100-fold more active against VISA and VRE, with an MIC of 2 μg/mL vs both VanA and VanB VRE. Pharmacokinetic studies in mice demonstrated a half-life of 2.76 h, while efficacy in a VISA neutropenic thigh infection model at a single dose of 12 mg/kg gave a >5 log reduction in cfu (compared to approximately 1 log reductions for 2 × 12 mg/kg vancomycin or linezolid). A 100 mg/kg single dose toxicity study in mice was also conducted, with no mortality up to 14 days following administration.105 The vancosamine has also been alkylated with a positively charged pyridinium group containing a range of alkyl substituents (12b–f).106 The combination of a lactobiono sugar and positively charged lipophilic moiety significantly improved activity compared to either component alone. An nOct pyridinium substituent (12c) was optimal, with 0.1 μM MIC against most strains (MRSA, VISA, vancomycin sensitive S. epidermidis VSSE, and VanA VRE) and 1.3 μM MIC against VanB VRE. Membrane disruption in VRE was shown. 12c disrupted MRSA biofilms, did not induce resistance after 25 passages (vs 15-fold increase in MIC for vancomycin), and showed efficacy in a VRE renal infection model at 12 mg/kg per day, with 6 log cfu reduction after 72 h compared to 4 log for linezolid and 2 log for vancomycin; no toxicity was seen with a single 100 mg/kg bolus iv injection.106
The vancosamine amino group has also been alkylated with a set of O-substituted glyceric acid derivatives 13 (Figure 7), in an attempt to mimic the transglycosylation inhibition properties of moenomycin and introduce lipophilic substituents while maintaining solubility. With the exception of a highly lipophilic chlorobiphenyl substituent (13a), which was 4- to >8-fold more potent than vancomycin against Newman S. aureus (MIC < 0.125 vs 2 μg/mL) and Mu50 S. aureus (MIC 2 vs 8 μg/mL), the remaining analogs were equipotent or worse than vancomycin.107
Glycopeptide Core Modifications
In 2006, the Boger group from the Scripps Research Institute published the first of a series of papers describing a novel approach to overcome vancomycin resistance, by replacing a key vancomycin carbonyl group involved in binding interactions with the terminal amide NH in the Lipid II Lys-d-Ala-d-Ala tripeptide (Figure 8). As described earlier, high levels of resistance are induced when this tripeptide is modified to Lys-d-Ala-d-Lac, with the ester oxygen of the lactate unable to form a hydrogen bond with the vancomycin carbonyl, substantially reducing binding. In 2006, the group reported the total synthesis of vancomycin aglycon analog 14b in which the carbonyl of residue 4 in vancomycin aglycon 14a was replaced with a methylene group.108 Affinity for an Ac-Lys(Ac)-d-Ala-d-Ala tripeptide ligand (representative of native Lipid II) was reduced by 35-fold compared to binding of the native aglycon, but the corresponding lactate ligand had a 40-fold increase in affinity. Subsequently, a total synthesis campaign in 2011 delivered an aglycon where the carbonyl was replaced with a thioamide (14c), an intermediate that was then elaborated into an amidine (14d).109−111 While the thioamide 14c lost all binding activity, the amidine 14d only lost 2-fold activity against the tripeptide ligand and maintained similar activity against the lactate derivative, representing a 600-fold increase in binding relative to vancomycin aglycon 14a. More importantly, this ligand binding activity translated into potent activity against VRE, with an MIC of 0.31 μg/mL for VanA E. faecalis.
Figure 8.

Backbone modifications to overcome vancomycin VRE/VRSA resistance.
In 2014, the fully glycosylated version of vancomycin was prepared with the amidine modification (15a), along with an analog that introduced the vancosamine chlorobiphenyl substituent of oritavancin (15b).112,113 While 15a had similar activity to the aglycon, the additional substituent of 15b improved potency by over 100-fold against VRE, with MIC of 0.005 μg/mL for VanA E. faecalis or E. faecium and 0.06 μg/mL for VanB E. faecalis. Excellent potency was also observed against MRSA (0.03–0.06 μg/mL for 15b; not determined for 15a). Finally, in 2017, the carboxylic acid group was also modified,114 by amidation with quaternary aminoalkylamine substituents somewhat similar to the dimethylaminopropylamine group found on dalbavancin or the quaternary ammonium groups in 10. This final modification enabled additional mechanisms of action (i.e., membrane disruption) but was only constructed with the methylene-modified vancomycin. Presumably, similar modification of the amidine derivative would lead to substantially more potent activity. The preferred analog 16, with a trimethylaminopropylamine group, had impressive VanA VRE activity of 0.01–0.005 μg/mL. Negligible induced resistance (4-fold) was observed after 50 serial passages in the presence of 0.5 MIC levels of compound. Despite the widespread publicity this publication received, there are a number of limitations. Surprisingly, no activity against strains other than VRE was reported, including MRSA. More importantly, evidence of in vivo activity for any of these analogs has yet to be described, possibly due to limited access to larger quantities of material; indeed, the total synthesis of 16 required over 30 steps, and it is difficult to envision how this could be commercially viable, unless a biosynthetic strategy could be developed.
Aryl Ring Functionalization
In the past decade, several groups have reported novel modifications to the glycopeptide aryl rings (Figure 9). The Boger group examined the influence of E-ring substitution vancomycin aglycon activity, by selective functionalization of the E-ring aryl chloride via Pd-catalyzed conversion to a boronic acid, followed by substitution with a range of functional groups (see 17d–z).115 Replacement of the E-ring chloride with hydrogen or polar groups reduced activity, while nonpolar group derivatives were similar to the parent chloro moiety. Permethylation of the phenolic hydroxyls and carboxylic acid of the same E-ring substituted aglycon series gave derivatives with up to an 8-fold improvement in activity against VanB E. faecalis compared to vancomycin aglycon, whereas potency was generally lost across the series (∼2-fold) against vancomycin-sensitive S. aureus (ATCC 25923). In a separate study by the same group, the influence of the C- and E-ring chlorides was also examined.116 In vancomycin-sensitive S. aureus (ATCC 25923), removal of the C-ring chloride while retaining the E-ring chloride in the aglycon reduced activity 8-fold relative to vancomycin or its aglycon, while the opposite positional isomer was 4-fold less active; removal of both caused a 16-fold loss of potency (17a–c).116 The E-ring chloride of vancomycin itself can be selectively cross-coupled under Suzuki-Miyaura conditions with a range of boronic acids, with the adducts 18a–i possessing similar or slightly improved activity compared to vancomycin 1, indicating that this position is not critical for activity.117 More forcing conditions produced the bis-alkenylated products 18j–l, with a 32-fold loss in activity. The Miller group at Yale selectively dechlorinated the E-ring of vancomycin, and then used the less reactive remaining C-ring chloride for cross-coupling reactions with relatively bulky lipophilic groups. All analogs 18m–s suffered from loss of activity against MSSA, MRSA, and VRE (VanA, VanB), with MICs > 64 μg/mL.118
Figure 9.

Aryl ring modifications.
N-Bromophthalimide regioselectively brominates the A/B aromatic rings of residues 5 and 7 of vancomycin 1 in the presence of a peptide scaffold catalyst, producing mono-, di-, and tribromovancomycins. The catalyst, developed by the Miller group, incorporates the d-Ala-d-Ala motif of the natural Lipid II peptide binding partner in addition to an N-terminal N,N-dimethylamide group designed to accelerate the bromination reaction.119 The catalysts had a profound effect on the intrinsic product distribution, enabling the preferential formation of regioisomers depending on the reaction conditions. The brominated compounds lost approximately 4-fold potency compared to vancomycin against MRSA (ATCC 43300) and E. faecalis (VanA, VanB). When the same strategy was applied to teicoplanin 3, bromination could be directed to the aryl rings of either residue 7 or residue 3, with the adducts retaining similar activity to the parent.120 Within the same series, Pd-catalyzed cross-coupling introduced a series of aryl or alkenyl substituents onto the aryl rings of residue 2 and/or 3; however, no significant improvements in activity were observed.
In 2018, a group from Shanghai reported on vancomycin analogs 19 (Figure 9) with an additional sugar residue attached to the aryl ring of residue 7 via an aminomethyl modification akin to that used in telavancin 4, in combination with vancosamine alkylations as in oritavancin 6b, (Figure 9).121 Their rationale was that while the lipophilic substitutions on vancomycin enhance bacterial cell wall interactions, they also lead to long elimination half-life and accumulative toxicity, which might be alleviated by the addition of hydrophilic sugar units. They tested combinations of 12 different lipophilic substituents and 9 sugar residues, generating 24 analogs (including 3 that also included a C-terminal dimethylaminopropyl amide group). The best analogs, including 19a and 19b, incorporated the rigid lipophilic biphenyl moiety of oritavancin 6b along with linkages to either glucosamine or galactose carbohydrates. Improvements in activity of 128- to 1024-fold were observed against MSSA ATCC 5904 and VISA Mu50 (19a, 19b MIC = 0.03–0.25 μg/mL), and several vanA, vanM (19a, 19b MIC = 1–8 μg/mL), and vanB (19a, 19b MIC = < 0.06–0.25 μg/mL) Enterococci strains. Both were equivalent to telavancin 4 at 7 mg/kg ip injection in a liver abscess VISA (Mu50) mouse infection mode, with approximately 2 log reduction in cfu compared to saline or vancomycin 1 at 7 mg/kg. They also demonstrated 88–93% survival in a MRSA mouse lethal challenge model (again at 7 mg/kg ip), compared to 7% survival for vancomycin 1 and 93% survival for telavancin 4. The compound’s pharmacokinetic properties showed substantially enhanced half-lives (t1/2 = 3.8, 2.9 h for 19a, 19b) compared to vancomycin 1 (t1/2 = 0.6 h) or telavancin 4 (t1/2 = 1.1 h) but substantially less than a related analog with a lipophilic vancosamine substituent but no extra sugar (t1/2 = 6.0 h), demonstrating that a balance of properties could regulate half-life and clearance. The compounds showed similar or reduced cytotoxicity against two human cell lines, compared to vancomycin or televancin 4. NMR studies of 19 bound to a Lipid II Ac2-Lys-d-Ala-d-Ala tripeptide showed indications of interactions with the new carbohydrate, supported by an H-bond seen in molecular modeling.121
Hydroxyl Modifications
The Miller group also reported a site selective phosphorylation strategy using similar peptide catalysts as described earlier for vancomycin hydroxyl acylation and aryl bromination. The catalysts employed a π(methyl)-histidine residue, with some containing the Lys-d-Ala Lipid II motif, leading to the selective phosphorylation of any one of the three hydroxymethyl sugar substituents on teicoplanin 3 (20, Figure 10).122 The resulting analogs were 8-16-fold less potent than teicoplanin 3 against MRSA/MSSA (MIC 4−8 μg/mL), but did show somewhat improved activity against VanA E. faecalis. The Boger group has selectively methylated one or more of the four phenolic groups of vancomycin aglycon 14a:123 analogs 21 possessing either four free phenols or four methyl ethers gave similar activity (MIC 1.25 μg/mL) against vancomycin-sensitive S. aureus (ATCC 25923), while partially methylated versions were 4- to 8-fold less active, demonstrating that the phenolic groups are not critical for activity (Figure 10). Against E. faecalis (VanA, VanB), the trends were similar, though the tetramethyl ether analog was 8- to 16-fold more active (MIC 2.5 μg/mL) than 14a (MIC 40 μg/mL).123
Figure 10.

Hydroxy and N-terminal modifications.
N-Terminal Modifications
Vancomycin has an N-terminal N-methyl Leu residue. In China, a des-methyl vancomycin analog with similar activity to vancomycin has been used clinically since 1967.124 Desmethyl vancomycin was selectively alkylated on the vancosamine amino group with a range of aliphatic and aromatic lipophilic substituents (22, desmethyl group highlighted in yellow, Figure 10); at best, a 2- to 4-fold improvement (MIC 0.78 μg/mL) was noted for selected analogs against different strains of MRSA and S. pneumoniae compared to des-methyl vancomycin (MIC 1.56–3.13 μg/mL for both strains) with a phenylhexyl derivative 22 also showing >4-fold improvement against VRE (E. faecalis, MIC 12.5 μg/mL).124,125 The N-methyl Leu residue has been removed from vancomycin aglycon 14a by Edman degradation to provide the desleucylaglucovancomycin, which was then N-acylated with ten other N-methyl amino acids. In general, activity was lost against MRSA but some improvements in VRE activity were seen.126 Similarly, the primary N-terminal amine of teicoplanin pseudoaglycon (missing the sugar on residue 4) has also been modified. Some of these N-terminal derivatives 23 (e.g., triazole substituted with phenol or naphthol) showed modest improvements in activity against VRE strains, though toxicity against mammalian cells also increased.127,128
Glycopeptide Dimers
Vancomycin and other glycopeptides undergo a cooperative back-to-back dimerization that increases their Lipid II binding affinity.19 Over the years, a number of groups have attempted to take advantage of this characteristic by covalently linking two or more vancomycin moieties together.
In 1996, the C-termini of two vancomycin 1 moieties were bridged with alkyl, disulfide, or bisamine linkers (24a–d, Figure 11). Generally, this led to a reduction in S. aureus activity but >100-fold improvement in VRE potency.129 Several years later, the vancosamine amino groups were bridged with disulfide- or methylene-based linkers, producing dimers 25a–h with a range of potency. The best (e.g., 25e, n = 2) was >10-fold more active than vancomycin against MRSA, 3-fold more active against a VISA strain, and 100-fold more active against VRE.130,131 Other multimeric constructs were described at the same time, including a trivalent system bridging three C-termini with a 1,3,5-tris(4-aminomethylanilinide)benzene core.132 This showed exceptionally high avidity to a trimeric d-Ala-d-Ala ligand. However, neither this compound132 nor a rigid p-xylylenediamine C-terminal bridged dimer 24e(133) was tested for antimicrobial activity. The corresponding vancomycin dimer 24f, along with 24a and other dimers derived from 1,6-diaminohexane and des-leucyl vancomycin, were prepared in 2003 by another group.134 Large increases in VRE MIC potency (800-fold) were reported, but only VanA VRE was tested.
Figure 11.

Glycopeptide dimers.
A “head-to-tail” dimer, with 3-aminopropanoic acid or 6-aminohexanoic acid joining a vancomycin N-terminus to a vancomycin C-terminus, was prepared in 1998 but biological activity was not reported.135 Glutaric acid and subaric acid bridged vancosamine group dimers 25i–j were only tested for SPR binding to Lys-d-Ala-d-Ala, with MIC activity not assessed.136 In 2003, Theravance conducted a systematic evaluation comparing 40 different dimers, with attachments via the C-terminus (C), N-terminus (N), vancosamine amino group (V), or an amine installed on the aromatic ring of residue 7 (R) (the site of modification leading to telavancin’s 3).137 Ten unique pairwise combinations (C–C, N–N, V–V, R–R, C–N, C–V, C–R, N–V, N–R, and V–R) and four different linker lengths (11, 19, 27, and 43 total atoms) were investigated. Effects on MIC varied depending on the test organism. For VanB VRE, V–V bridging with short linkers was most effective (400-fold more potent than vancomycin), but other linkages were more promising against VanA VRE. Only one of the dimers, with a V–V bridge, was more active against MRSA.137
More recently, a rigid link between vancosamine groups was formed using oxidative coupling of the adduct generated by reductive alkylation of the amine with 2-benzyloxy-3-nitrobenzaldehyde. The dimers 25k–m had 16- to 32-fold reductions in MRSA/VRSA activity but retained similar activity against Enterococci, gained potency against S. pneumoniae, and had large 8- to 32-fold improvements in VRE activity.138 They were active in an S. pneumoniae mouse infection model. In 2015, the carboxylic acid groups of two vancomycin aglycons were connected by 1,8-diaminooctane, N,N-bis(3-aminopropyl) octylamine, or linkers containing a quaternary amine (24g–i) (Figure 11). All three dimers were slightly more effective than vancomycin aglycon against MSSA and MRSA (0.2–0.8 μg/mL vs 1.0 μg/mL) but were substantially more potent against VISA (0.1–0.6 vs 4 μg/mL) and VRE (2.5–48 vs >100 μg/mL).139
In 2017, the Sharpless group employed the Cu-catalyzed azide–alkyne cycloaddition (CuAAC, “click”) reaction to form a triazole bridge between two vancomycin units substituted with a variety of C-terminal functionalized alkynes and azides (26A, Figure 11).140 MIC against MRSA varied from 0.6 to 5.0 μg/mL for the 30 different dimers (compared to 2.5 μg/mL for vancomycin 1), with substantial improvements in activity against VRE (0.8–1.6 μg/mL vs 25 μg/mL). In addition, heterodimers were prepared linking azide-functionalized C-terminal vancomycin derivatives with alkyne-substituted vancosamine amine vancomycin derivatives (26B) (Figure 11). Activity was reduced against MRSA (5–30 μg/mL) but was slightly improved against VRE (3.1–6.3 μg/mL).140
Glycopeptide Conjugates
Vancomycin has been conjugated with a range of functional moieties, including other antibiotics, siderophores, fluorophores, and specific targeting constructs, in order to create improved therapeutics or useful tool compounds.
Cefilavancin (RD-1792, TD-1792) 27 is a conjugate of vancomycin and a cephalosporin, leading to a dual targeting action against peptidoglycan synthesis (Figure 12).141−145 Originally developed by Theravance Biopharma, Inc. (South San Francisco, CA, USA), it was partnered with R-Pharm (Moscow, Russia) in October 2012 and in March 2015 reported to enter a Phase III trial as a treatment of Gram-positive complicated skin and skin structure infections.146 The conjugate is formed via a linker from the vancomycin C-terminal carboxyl group through an oxime linkage to the cephalosporin lactam amine substituent. A closely related analog, TD-1607 28, uses the same components but a different linker strategy, employing an aminomethylated residue 7 aromatic ring on the vancomycin core to attach the cephalosporin via a pyridinyl substituent off the cephalosporin bicyclic ring system.14728 entered two Phase I trials in 2013 (NCT01791049, NCT01949103). No further development has been reported. It was listed in the 2015 Theravance Biopharma annual report as a “midstage” candidate for MRSA148 but is not mentioned in the 2016 report149 and has been removed from their pipeline chart.
Figure 12.

Hybrid antibiotics.
In an earlier effort, vancomycin was conjugated with nisin, a peptide antibiotic that binds to the pyrophosphate component of Lipid II (Figure 12). The C-terminus of vancomycin was amidated with various terminal alkyne constructs for the dipolar cycloaddition reaction with an azide-substituted nisin(1–12) derivative.150 The adducts were less potent than the individual components against VSE but around 40-fold more active against VRE, with a C-terminal PEG linker (n = 3) in derivative 29 found to be the most effective. Another antimicrobial peptide, tridecaptin (a Gram-negative membrane disrupting peptide), was prepared with an azide-(PEG)3 substituent on a Lys side chain. CuAAC reaction with a vancomycin derivative amidated on the C-terminus with propargylamine produced the conjugate, which retained some Gram-negative activity in vitro but was substantially less active than coadministering a 1:1 mixture of the antibiotics.151
Catechol and catechol/hydroxamate siderophore ligands have been attached by acylation of the primary vancosamine amino group in an attempt to hijack the bacteria’s need for iron and use a “Trojan horse” approach to get vancomycin inside Gram-negative bacteria (Figure 13). The adducts 30a–b displayed reduced activity against Gram-positive bacteria (MIC = 8–32 μg/mL) compared to vancomycin (MIC = 1–4 μg/mL) and were generally inactive against Gram-negative bacteria under iron sufficient conditions. Under iron-depleted conditions, 30a (X = CH2) displayed moderate activity (MIC = 32 μg/mL) against a P. aeruginosa strain otherwise insensitive to vancomycin (MIC > 128 μg/mL) under the same conditions.152 As discussed earlier, a zinc-binding dipicolyl moiety was attached to the C-terminus of vancomycin in 8, to bind to the pyrophosphates of cell-wall lipids.93 Similarly, a silver-complexing pyridinyl ligand has also been ligated (31), for use in combination with surface-attached pyridinyl groups to create a vancomycin-silver surface coating that had antimicrobial activity.153 The C-terminus of vancomycin has also been functionalized with a bone-targeting methoxyphenylamide moiety that binds strongly to hydroxyapatite, attached via a PEG linker, to develop a more effective therapeutic for bone infections.154 The adduct 32 retained similar activity as vancomycin against 30 MRSA clinical isolates (MIC = 1–2 μg/mL), and bound strongly to hydroxyapatite (Figure 13). It was statistically more effective than vancomycin in a rat osteomyelitis model, with 1.3 log reduction in cfu vs 0.5 log reduction, and demonstrated enhanced bone accumulation in bone compared to vancomycin. However, histology showed significant kidney damage.155
Figure 13.

Glycopeptide conjugates.
A vancomycin prodrug was unexpectedly generated by attaching branched PEG groups to the vancosamine amine through an amide linkage; the resulting amide bond was susceptible to hydrolysis, releasing vancomycin in vivo in the rat.156 The same study derivatized the vancosamine amine with PEG acrylates of various chain lengths, and converted either the vancosamine amine or N-terminal methylamine to an acrylamide, with the alkene then being used for polymerizations through a surface-mediated reaction, creating bactericidal surface coatings.156 Surface modification of titanium alloy or bone cement with the glycopeptide adduct resulted in reduced S. epidermidis biofilm formation.157
Fluorescent probes able to visualize sites of bacterial infection would be useful clinical tools to diagnose infections and monitor treatment efficacy. The vancosamine amine of vancomycin 1 was used to attach a near-IR fluorophore, IRDye 800CW.158 The resulting adduct 33a was successfully used to image S. aureus myositis (intramuscular infection) in mice. Neither an E. coli infection nor induced inflammation resulted in a signal, demonstrating its specificity for Gram-positive infections. It was also able to visualize a S. epidermidis biofilm in a human post-mortem implant 8 mm under the skin.158 Other fluorescent vancomycin derivatives have been reported and used as in vitro mechanistic probes, such as vancomycin labeled with fluorescein 33b,159 BODIPY 33c,159 or Oregon Green 33d(160) on the vancosamine amine or on the exposed N-terminal amine of desleucyl vancomycin (Figure 13).159
Another mode of action study created a vancomycin photoaffinity probe by attaching a biotin moiety (for antibody capture) to the C-terminal carboxylic acid and a benzophenone moiety to the vancosamine amine, for covalent labeling of the protein–probe complex. It was used to identify a vancomycin receptor in Streptomyces coelicolor.161
Biosynthesis
Given that all almost all glycopeptide antibiotics are derived from natural biosynthetic pathways, either directly or following semisynthetic modifications, an alternative approach to creating new glycopeptides is to artificially manipulate their biosynthesis.88 Complestatin 34 (Figure 14) is a type V glycopeptide aglycone that is bicyclic and incorporates a Trp residue instead of a Phe. The biosynthetic gene cluster has been cloned and expressed in Streptomyces lividans, allowing for manipulation of the genes to produce new analogs. Deletion of CYP450 monooxygenase genes produced a monocyclic 35 and linear 36 derivative. Activity was reduced, but the methodology can be applied to other glycopeptides and used for alterations that are difficult to achieve chemically.162 A minimal teicoplanin scaffold (no glycosylation or acylation) has been expressed in Streptomyces coelicolor, testing 13 scaffold-modifying enzymes from seven glycopeptide antibiotic biosynthetic gene clusters from different producer organisms. Combinations of one and two gene integrations into the expression platform were used to identify what modifications are possible and tolerated.163 Methylated, glycosylated, and sulfated analogs were produced, though activity was generally similar across the derivatives. UK-68,597 37 is a teicoplanin-like glycopeptide sulfated on residue 3 that contains the same unusual N-terminal α-keto acid found in complestatin 34. Attempts to reisolate the antibiotic from the original producing strain were unsuccessful, so the biosynthetic gene cluster was identified with the eventual aim of expressing the compound in a better host.164 Several of the tailoring enzymes identified in the cluster were expressed and tested for their ability to modify other glycopeptides. This led to several novel derivatives with different sulfation or glycosyl substituents. Further studies on how to utilize key steps of the glycopeptide biosynthetic machinery165−168 will potentially lead to improvements in the biomimetic synthesis of new glycopeptides.
As mentioned earlier, a strategy of screening for novel biosynthetic gene clusters combined with a glycopeptide resistance prefilter led to the isolation of pekiskomycin 38,31 a sulfated vancomycin-like scaffold containing a Glu residue instead of Asn, an N,N-dimethyl terminal amine, and a different sugar moiety.
Conclusions
There is a continuing need for new and more potent antibiotics with a good safety profile that can treat resistant Gram-positive infections. Currently, a key obstacle to the development of additional glycopeptides to fill this need is the lack of investor and market interest. Sales of the three recently approved semisynthetic glycopeptides do not inspire much confidence. Telavancin 4, which has not been helped by its black box label warning, is languishing at <$3.5 million USA sales per quarter (2Q 2017),169 while oritavancin 6b has had only $16 million in sales in 2016.170 Dalbavancin 5 is more promising, with revenues for the four quarters from 3Q 2016 to 2Q 2017 of $48 million and growing ($15.2 million in 2Q 2017).171−174 The exceedingly long half-lives of dalbavancin 5 and oritavancin 6b, which beneficially allow for once-weekly or single injection treatment (compared to twice daily for vancomycin), may also lead to extended exposure to subtherapeutic drug levels. Inadequate drug exposure may encourage the selection of resistant subpopulations,175 providing treatment failure, as was recently reported for dalbavancin 5.75 The physicochemical properties leading to improved half-lives compared to vancomycin also leads to different tissue penetration properties, which may result in reduced efficacy in some types of infections, as potentially seen with the failure of dalbavancin in an off-label endocarditis case study. The need for more potent antibiotics that can treat resistant Gram-positive infections, yet maintain a good safety profile, means that new glycopeptide antibiotics will continue to play an important role in the ongoing fight against drug resistant infections into the future. As outlined above, many innovative modified glycopeptides have been developed, which exploit different approaches to overcome resistance, and stand ready to be cultivated into new therapies.
Acknowledgments
M.A.C. is supported by a NHMRC Principal Research Fellowship (APP1059354). M.A.T.B., M.S.B., and K.A.H. are supported in part by Wellcome Trust Strategic Grant WT141107 with glycopeptide research supported by a Wellcome Trust Seeding Drug Discovery Award 094977/Z/10/Z, NHMRC Project Grants APP631632, APP1026922, and APP1063214, and NHMRC Development Grant APP1113719.
Author Contributions
M.A.T.B. structured the manuscript. M.A.T.B., K.A.H., M.S.B., and M.A.C. wrote the paper with input from all authors, and Z.J. and A.E.M. conducted the molecular modeling to generate the vancomycin–Lipid II membrane binding figures.
The authors declare the following competing financial interest(s): M.A.C. and M.A.T.B. are inventors on WO 2015/117196 A, describing new glycopeptide derivatives discussed in this Review and subject to commercialization activities.
References
- Butler M. S.; Blaskovich M. A. T.; Cooper M. A. (2017) Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 70 (1), 3–24. 10.1038/ja.2016.72. [DOI] [PubMed] [Google Scholar]
- Fernandes P.; Martens E. (2017) Antibiotics in late clinical development. Biochem. Pharmacol. 133, 152–163. 10.1016/j.bcp.2016.09.025. [DOI] [PubMed] [Google Scholar]
- World Health Organization . (2017) Antibacterial agents in clinical development: An analysis of the antibacterial clinical development pipeline, including tuberculosis, WHO/EMP/IAU/2017.11, WHO, Geneva, http://apps.who.int/iris/bitstream/10665/258965/1/WHO-EMP-IAU-2017.11-eng.pdf?ua=1 (accessed 22 November 2017).
- Grayeff Y. (2014) Cubist Q4 revenues + 22%; Cubicin annual sales top $1B, https://seekingalpha.com/news/1503931-cubist-q4-revenues-plus-22-percent-cubicin-annual-sales-top-1b (accessed 22 November 2017).
- CDC . (2013) Antibiotic Resistance Threats in the United States, 2013, U.S. Department of Health and Human Services (Centers for Disease Control and Prevention), Atlanta, http://www.cdc.gov/drugresistance/threat-report-2013 (accessed 22 November 2017).
- Kavanagh K. T.; Abusalem S.; Calderon L. E. (2017) The incidence of MRSA infections in the United States: is a more comprehensive tracking system needed?. Antimicrob. Resist. In. 6, 34. 10.1186/s13756-017-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Guillamet C.; Kollef M. H. (2014) Treatment of gram - positive infections in critically ill patients. BMC Infect. Dis. 14 (1), 92. 10.1186/1471-2334-14-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eades C.; Hughes S.; Heard K.; Moore L. S. (2017) Antimicrobial therapies for Gram-positive infections. Clinical Pharmacist 9 (9), 1–19. 10.1211/CP.2017.20203363. [DOI] [Google Scholar]
- Cetinkaya Y.; Falk P.; Mayhall C. G. (2000) Vancomycin-resistant enterococci. Clin. Microbiol. Rev. 13 (4), 686–707. 10.1128/CMR.13.4.686-707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardete S.; Tomasz A. (2014) Mechanisms of vancomycin resistance in Staphylococcus aureus. J. Clin. Invest. 124 (7), 2836–2840. 10.1172/JCI68834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M. O.; Baptiste K. E. (2017) Vancomycin-Resistant Enterococci: A Review of Antimicrobial Resistance Mechanisms and Perspectives of Human and Animal Health. Microb. Drug Resist. 10.1089/mdr.2017.0147. [DOI] [PubMed] [Google Scholar]
- McGuinness W. A.; Malachowa N.; DeLeo F. R. (2017) Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 90 (2), 269–281. [PMC free article] [PubMed] [Google Scholar]
- D’Costa V. M.; King C. E.; Kalan L.; Morar M.; Sung W. W. L.; Schwarz C.; Froese D.; Zazula G.; Calmels F.; Debruyne R.; Golding G. B.; Poinar H. N.; Wright G. D. (2011) Antibiotic resistance is ancient. Nature 477 (7365), 457–461. 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- Hiramatsu K.; Hanaki H.; Ino T.; Yabuta K.; Oguri T.; Tenover F. C. (1997) Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 40 (1), 135–136. 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]
- Butler M. S.; Hansford K. A.; Blaskovich M. A.; Halai R.; Cooper M. A. (2014) Glycopeptide antibiotics: Back to the future. J. Antibiot. 67 (9), 631–644. 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- Sievert D. M.; Rudrik J. T.; Patel J. B.; McDonald L. C.; Wilkins M. J.; Hageman J. C. (2008) Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin. Infect. Dis. 46 (5), 668–674. 10.1086/527392. [DOI] [PubMed] [Google Scholar]
- Limbago B. M.; Kallen A. J.; Zhu W.; Eggers P.; McDougal L. K.; Albrecht V. S. (2014) Report of the 13th vancomycin-resistant Staphylococcus aureus isolate from the United States. J. Clin. Microbiol. 52 (3), 998–1002. 10.1128/JCM.02187-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez R.; López Cortés L. E.; Molina J.; Cisneros J. M.; Pachón J. (2016) Optimizing the Clinical Use of Vancomycin. Antimicrob. Agents Chemother. 60 (5), 2601–2609. 10.1128/AAC.03147-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauregard D. A.; Williams D. H.; Gwynn M. N.; Knowles D. J. C. (1995) Dimerization and Membrane Anchors in Extracellular Targeting of Vancomycin Group Antibiotics. Antimicrob. Agents Chemother. 39 (3), 781–785. 10.1128/AAC.39.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z.; O’Mara M. L.; Zuegg J.; Cooper M. A.; Mark A. E. (2013) Vancomycin: ligand recognition, dimerization and super-complex formation. FEBS J. 280 (5), 1294–1307. 10.1111/febs.12121. [DOI] [PubMed] [Google Scholar]
- Cooper M. A.; Williams D. H. (1999) Binding of glycopeptide antibiotics to a model of a vancomycin-resistant bacterium. Chem. Biol. 6 (12), 891–899. 10.1016/S1074-5521(00)80008-3. [DOI] [PubMed] [Google Scholar]
- James R. C.; Pierce J. G.; Okano A.; Xie J.; Boger D. L. (2012) Redesign of Glycopeptide Antibiotics: Back to the Future. ACS Chem. Biol. 7 (5), 797–804. 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine D. P. (2006) Vancomycin: a history. Clin. Infect. Dis. 42 (Suppl 1), S5–S12. 10.1086/491709. [DOI] [PubMed] [Google Scholar]
- Jenkins C. S. P.; Meyer D.; Dreyfus M. D.; Larreu M. J. (1974) Willebrand factor and ristocetin I. Mechanism of ristocetin-induced platelet aggregation. Br. J. Haematol. 28 (4), 561–578. 10.1111/j.1365-2141.1974.tb06675.x. [DOI] [PubMed] [Google Scholar]
- Harris C. M.; Harris T. M. (1982) Structure of the glycopeptide antibiotic vancomycin. Evidence for an asparagine residue in the peptide. J. Am. Chem. Soc. 104 (15), 4293–4295. 10.1021/ja00379a062. [DOI] [Google Scholar]
- Goldstein B. P., Rosina R., and Parenti F. (1994) Teicoplanin in Glycopeptide Antibiotics, (Nagarajan R., Ed.), Vol. Drugs and the Pharmaceutical Sciences, pp 273–307, CRC Press, New York. [Google Scholar]
- Barna J. C. J.; Williams D. H.; Stone D. J. M.; Leung T. W. C.; Doddrell D. M. (1984) Structure elucidation of the teicoplanin antibiotics. J. Am. Chem. Soc. 106 (17), 4895–4902. 10.1021/ja00329a044. [DOI] [Google Scholar]
- Yao R. C., and Crandall L. W. (1994) Glycopeptides. Classification, Occurrence, and Discovery in Glycopeptide Antibiotics (Nagarajan R., Ed.), Vol. Drugs and the Pharmaceutical Sciences, pp 1–27, CRC Press, New York. [Google Scholar]
- Nicolaou K. C.; Boddy C. N. C.; Bräse S.; Winssinger N. (1999) Chemistry, Biology, and Medicine of the Glycopeptide Antibiotics. Angew. Chem., Int. Ed. 38 (15), 2096–2152. 10.1002/(SICI)1521-3773(19990802)38:15<2096::AID-ANIE2096>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Rake J.; Gerber R.; Mehta R.; Newman D.; Oh Y.; Phelen C.; Shearer M.; Sitrin R.; Nisbet L. (1986) Glycopeptide antibiotics: a mechanism-based screen employing a bacterial cell wall receptor mimetic. J. Antibiot. 39, 58–67. 10.7164/antibiotics.39.58. [DOI] [PubMed] [Google Scholar]
- Thaker M. N.; Wang W. L.; Spanogiannopoulos P.; Waglechner N.; King A. M.; Medina R.; Wright G. D. (2013) Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nat. Biotechnol. 31 (10), 922–927. 10.1038/nbt.2685. [DOI] [PubMed] [Google Scholar]
- Thaker M. N.; Waglechner N.; Wright G. D. (2014) Antibiotic resistance-mediated isolation of scaffold-specific natural product producers. Nat. Protoc. 9 (6), 1469–1479. 10.1038/nprot.2014.093. [DOI] [PubMed] [Google Scholar]
- Leadbetter M. R.; Adams S. M.; Bazzini B.; Fatheree P. R.; Karr D. E.; Krause K. M.; Lam B. M.; Linsell M. S.; Nodwell M. B.; Pace J. L.; Quast K.; Shaw J. P.; Soriano E.; Trapp S. G.; Villena J. D.; Wu T. X.; Christensen B. G.; Judice J. K. (2004) Hydrophobic vancomycin derivatives with improved ADME properties: discovery of telavancin (TD-6424). J. Antibiot. 57 (5), 326–336. 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- Higgins D. L.; Chang R.; Debabov D. V.; Leung J.; Wu T.; Krause K. M.; Sandvik E.; Hubbard J. M.; Kaniga K.; Schmidt D. E. Jr.; Gao Q.; Cass R. T.; Karr D. E.; Benton B. M.; Humphrey P. P. (2005) Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 49 (3), 1127–1134. 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde C. S.; Hartouni S. R.; Janc J. W.; Mammen M.; Humphrey P. P.; Benton B. M. (2009) Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob. Agents Chemother. 53 (8), 3375–3383. 10.1128/AAC.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng-Williamson K. A.; Blick S. K. A. (2009) Drugs 69 (18), 2607–2620. 10.2165/10481380-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Hegde S. S.; Reyes N.; Wiens T.; Vanasse N.; Skinner R.; McCullough J.; Kaniga K.; Pace J.; Thomas R.; Shaw J. P.; Obedencio G.; Judice J. K. (2004) Pharmacodynamics of telavancin (TD-6424), a novel bactericidal agent, against gram-positive bacteria. Antimicrob. Agents Chemother. 48 (8), 3043–3050. 10.1128/AAC.48.8.3043-3050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepak A. J.; Zhao M.; Andes D. R. (2017) Comparative Pharmacodynamics of Telavancin and Vancomycin in the Neutropenic Murine Thigh and Lung Infection Models against Staphylococcus aureus. Antimicrob. Agents Chemother. 61 (7), e00281-17. 10.1128/AAC.00281-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A Phase 3 Telavancin Staphylococcus aureus (S. aureus) Bacteremia Trial (identifier NCT02208063), https://clinicaltrials.gov/ct2/show/NCT02208063.
- McMullen A. R.; Anderson N.; Wallace M. A.; Shupe A.; Burnham C. A. (2017) When Good Bugs Go Bad: Epidemiology and Antimicrobial Resistance Profiles of Corynebacterium striatum, an Emerging Multidrug-Resistant, Opportunistic Pathogen. Antimicrob. Agents Chemother. 61 (11), e01111-17. 10.1128/AAC.01111-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.; Hardin T. C.; Smart J. I. (2015) A review of telavancin activity in in vitro biofilms and animal models of biofilm-associated infections. Future Microbiol. 10 (8), 1325–1338. 10.2217/fmb.15.53. [DOI] [PubMed] [Google Scholar]
- Shaw J. P.; Seroogy J.; Kaniga K.; Higgins D. L.; Kitt M.; Barriere S. (2005) Pharmacokinetics, serum inhibitory and bactericidal activity, and safety of telavancin in healthy subjects. Antimicrob. Agents Chemother. 49 (1), 195–201. 10.1128/AAC.49.1.195-201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji B. T.; Leonard S. N.; Rhomberg P. R.; Jones R. N.; Rybak M. J. (2008) Evaluation of daptomycin, telavancin, teicoplanin, and vancomycin activity in the presence of albumin or serum. Diagn. Microbiol. Infect. Dis. 60 (4), 441–444. 10.1016/j.diagmicrobio.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Karlowsky J. A.; Nichol K.; Zhanel G. G. (2015) Telavancin: mechanisms of action, in vitro activity, and mechanisms of resistance. Clin. Infect. Dis. 61 (Suppl 2), S58–68. 10.1093/cid/civ534. [DOI] [PubMed] [Google Scholar]
- Hill C. M.; Krause K. M.; Lewis S. R.; Blais J.; Benton B. M.; Mammen M.; Humphrey P. P.; Kinana A.; Janc J. W. (2010) Specificity of induction of the vanA and vanB operons in vancomycin-resistant enterococci by telavancin. Antimicrob. Agents Chemother. 54 (7), 2814–2818. 10.1128/AAC.01737-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhin F. F.; Seguin D. L.; Belley A.; Moeck G. (2017) In vitro stepwise selection of reduced susceptibility to lipoglycopeptides in enterococci. Diagn. Microbiol. Infect. Dis. 89 (2), 168–171. 10.1016/j.diagmicrobio.2017.06.023. [DOI] [PubMed] [Google Scholar]
- Duncan L. R.; Sader H. S.; Smart J. I.; Flamm R. K.; Mendes R. E. (2017) Telavancin activity in vitro tested against a worldwide collection of Gram-positive clinical isolates (2014). J.Glob. Antimicrob. Resist. 10, 271–276. 10.1016/j.jgar.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Jones R. N.; Flamm R. K.; Castanheira M.; Sader H. S.; Smart J. I.; Mendes R. E. (2017) Activity of telavancin against Gram-positive pathogens isolated from bone and joint infections in North American, Latin American, European and Asia-Pacific nations. Diagn. Microbiol. Infect. Dis. 88 (2), 184–187. 10.1016/j.diagmicrobio.2017.03.003. [DOI] [PubMed] [Google Scholar]
- Mendes R. E.; Sader H. S.; Smart J. I.; Castanheira M.; Flamm R. K. (2017) Update of the activity of telavancin against a global collection of Staphylococcus aureus causing bacteremia, including endocarditis (2011–2014). Eur. J. Clin. Microbiol. Infect. Dis. 36 (6), 1013–1017. 10.1007/s10096-016-2865-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A.; Sader H. S.; Flamm R. K.; Castanheira M.; Smart J. I.; Mendes R. E. (2017) In Vitro Activity of Telavancin Against Clinically Important Gram-Positive Pathogens from 69 U.S. Medical Centers (2015): Potency Analysis by U.S. Census Divisions. Microb. Drug Resist. 23 (6), 718–726. 10.1089/mdr.2017.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz T. H.; Huprikar S.; Labombardi V.; Pinney S.; Anyanwu A.; Lee M.; Patel G. (2013) Heart transplantation in a patient with heteroresistant vancomycin-intermediate Staphylococcus aureus ventricular assist device mediastinitis and bacteremia. Transpl. Infect. Dis. 15 (5), E177–E181. 10.1111/tid.12123. [DOI] [PubMed] [Google Scholar]
- Rybak M.; Lomaestro B.; Rotschafer J. C.; Moellering R. Jr.; Craig W.; Billeter M.; Dalovisio J. R.; Levine D. P. (2009) Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health-Syst. Pharm. 66 (1), 82–98. 10.2146/ajhp080434. [DOI] [PubMed] [Google Scholar]
- Theravance Biopharma Antibiotics, Inc. Prescribing Information for VIBATIV (telavancin) for injection, for intravenous use, https://www.vibativ.com/public/pdf/PrescribingInformation.pdf (accessed Jan 10 2018).
- Durata Therapies Limited . Prescribing Information for DALVANCE (dalbavancin) for injection, for intravenous use, https://www.allergan.com/assets/pdf/dalvance_pi (accessed Jan 10 2018).
- The Medicines Company . Prescribing information for ORBACTIV (oritavancin) for injection, for intravenous use, http://www.orbactiv.com/pdfs/orbactiv-prescribing-information.pdf (accessed Jan 10 2018).
- Rybak M. J. (2006) The pharmacokinetic and pharmacodynamic properties of vancomycin. Clin. Infect. Dis. 42 (Suppl 1), S35–S39. 10.1086/491712. [DOI] [PubMed] [Google Scholar]
- Butterfield J. M.; Patel N.; Pai M. P.; Rosano T. G.; Drusano G. L.; Lodise T. P. (2011) Refining vancomycin protein binding estimates: identification of clinical factors that influence protein binding. Antimicrob. Agents Chemother. 55 (9), 4277–4282. 10.1128/AAC.01674-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A.; Kuti J. L.; Nicolau D. P. (2007) Review of dalbavancin, a novel semisynthetic lipoglycopeptide. Expert Opin. Invest. Drugs 16 (5), 717–733. 10.1517/13543784.16.5.717. [DOI] [PubMed] [Google Scholar]
- Arhin F. F.; Belley A.; McKay G.; Beaulieu S.; Sarmiento I.; Parr T. R. Jr.; Moeck G. (2010) Assessment of oritavancin serum protein binding across species. Antimicrob. Agents Chemother. 54 (8), 3481–3483. 10.1128/AAC.00271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI. (2016) Performance Standards for Antimicrobial Susceptibility Testing, 26th ed., CLSI supplement M100S, Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- Drugs.com (2018) Dalvance Approval History, https://www.drugs.com/history/dalvance.html (accessed Feb 2, 2018). [Google Scholar]
- Goldstein B. P.; Selva E.; Gastaldo L.; Berti M.; Pallanza R.; Ripamonti F.; Ferrari P.; Denaro M.; Arioli V.; Cassani G. (1987) A40926, a new glycopeptide antibiotic with anti-Neisseria activity. Antimicrob. Agents Chemother. 31 (12), 1961–1966. 10.1128/AAC.31.12.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiani G.; Abbondi M.; Borgonovi M.; Romano G.; Parenti F. (1999) In-vitro and in-vivo antibacterial activity of BI 397, a new semi-synthetic glycopeptide antibiotic. J. Antimicrob. Chemother. 44 (2), 179–192. 10.1093/jac/44.2.179. [DOI] [PubMed] [Google Scholar]
- Malabarba A.; Goldstein B. P. (2005) Origin, structure, and activity in vitro and in vivo of dalbavancin. J. Antimicrob. Chemother. 55, 15–20. 10.1093/jac/dki005. [DOI] [PubMed] [Google Scholar]
- Study on the Safety and Efficacy of Dalbavancin Versus Active Comparator in Adult Subjects With Osteomyelitis (DAL-MD-04) (identifier NCT02685033), https://clinicaltrials.gov/ct2/show/NCT02685033?term=NCT02685033&rank=1 (accessed Jan 10 2018).
- Efficacy and Safety of Dalbavancin Compared to Standard of Care Antibiotic Therapy for the Completion of Treatment of Patients With Complicated Bacteremia or Infective Endocarditis (identifier NCT03148756), https://clinicaltrials.gov/ct2/show/NCT03148756 (accessed Jan 10 2018).
- Steele J. M.; Seabury R. W.; Hale C. M.; Mogle B. T. (2018) Unsuccessful treatment of methicillin-resistant Staphylococcus aureus endocarditis with dalbavancin. J. Clin. Pharm. Ther. 43 (1), 101–103. 10.1111/jcpt.12580. [DOI] [PubMed] [Google Scholar]
- Leuthner K. D.; Buechler K. A.; Kogan D.; Saguros A.; Lee H. S. (2016) Clinical efficacy of dalbavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Ther. Clin. Risk Manage. 12, 931–940. 10.2147/TCRM.S86330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G.; Calic D.; Schweizer F.; Zelenitsky S.; Adam H.; Lagace-Wiens P. R. S.; Rubinstein E.; Gin A. S.; Hoban D. J.; Karlowsky J. A. (2010) New Lipoglycopeptides A Comparative Review of Dalbavancin, Oritavancin and Telavancin. Drugs 70 (7), 859–886. 10.2165/11534440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Anderson V. R.; Keating G. M. (2008) Adis drug profile - Dalbavancin. Drugs 68 (5), 639–648. 10.2165/00003495-200868050-00006. [DOI] [PubMed] [Google Scholar]
- Economou N. J.; Nahoum V.; Weeks S. D.; Grasty K. C.; Zentner I. J.; Townsend T. M.; Bhuiya M. W.; Cocklin S.; Loll P. J. (2012) A carrier protein strategy yields the structure of dalbavancin. J. Am. Chem. Soc. 134 (10), 4637–4645. 10.1021/ja208755j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M.; Ziora Z. M.; Hansford K. A.; Blaskovich M. A.; Butler M. S.; Cooper M. A. (2014) Anti-cooperative ligand binding and dimerisation in the glycopeptide antibiotic dalbavancin. Org. Biomol. Chem. 12 (16), 2568–2575. 10.1039/C3OB42428F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K. D.; Sulaiman R. M.; Rybak M. J. (2015) Dalbavancin and Oritavancin: An Innovative Approach to the Treatment of Gram-Positive Infections. Pharmacotherapy 35 (10), 935–948. 10.1002/phar.1641. [DOI] [PubMed] [Google Scholar]
- Ramdeen S.; Boucher H. W. (2015) Dalbavancin for the treatment of acute bacterial skin and skin structure infections. Expert Opin. Pharmacother. 16 (13), 2073–2081. 10.1517/14656566.2015.1075508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth B. J., Jain R., Hahn A., Cummings L., Weaver T., Waalkes A., Sengupta D., Salipante S. J., Rakita R. M., and Butler-Wu S. M. (2017) Emergence of dalbavancin non-susceptible, vancomycin-intermediate Staphylococcus aureus (VISA) after treatment of MRSA central line-associated bloodstream infection with a dalbavancin- and vancomycin-containing regimen. Clin. Microbiol. Infect., in press, 10.1016/j.cmi.2017.07.028. [DOI] [PubMed] [Google Scholar]
- Allen N. E. (2010) From Vancomycin to Oritavancin: The Discovery and Development of a Novel Lipoglycopeptide Antibiotic. Anti-Infect. Agents Med. Chem. 9 (1), 23–47. 10.2174/187152110790886745. [DOI] [Google Scholar]
- Antony S. J.; Cooper L. G. (2017) Use of Oritavancin (Novel New Lipoglycopeptide) in the Treatment of Prosthetic Joint Infections (PJI): A Possible Alternative Novel Approach to a Difficult Problem. Infect. Disord.: Drug Targets 17 (2), 77–80. 10.2174/1871526517666161108130148. [DOI] [PubMed] [Google Scholar]
- Johnson J. A.; Feeney E. R.; Kubiak D. W.; Corey G. R. (2015) Prolonged Use of Oritavancin for Vancomycin-Resistant Enterococcus faecium Prosthetic Valve Endocarditis. Open Forum Infect. Dis. 2 (4), ofv156. 10.1093/ofid/ofv156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravolatz L. D.; Stein G. E. (2015) Oritavancin: A Long-Half-Life Lipoglycopeptide. Clin. Infect. Dis. 61 (4), 627–632. 10.1093/cid/civ311. [DOI] [PubMed] [Google Scholar]
- Ambrose P. G.; Drusano G. L.; Craig W. A. (2012) In Vivo Activity of Oritavancin in Animal Infection Models and Rationale for a New Dosing Regimen in Humans. Clin. Infect. Dis. 54 (S3), S220–S228. 10.1093/cid/cis001. [DOI] [PubMed] [Google Scholar]
- Brade K. D.; Rybak J. M.; Rybak M. J. (2016) Oritavancin: A New Lipoglycopeptide Antibiotic in the Treatment of Gram-Positive Infections. Infect. Dis. Ther. 5 (1), 1–15. 10.1007/s40121-016-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G.; Schweizer F.; Karlowsky J. A. (2012) Oritavancin: Mechanism of Action. Clin. Infect. Dis. 54, S214–S219. 10.1093/cid/cir920. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Cegelski L.; Stueber D.; Singh M.; Dietrich E.; Tanaka K. S. E.; Parr T. R.; Far A. R.; Schaefer J. (2008) Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis, in Staphylococcus aureus. J. Mol. Biol. 377 (1), 281–293. 10.1016/j.jmb.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti G. J.; Kim S. J.; Yu T. Y.; Dietrich E.; Tanaka K. S. E.; Parr T. R.; Far A. R.; Schaefer J. (2009) Vancomycin and Oritavancin Have Different Modes of Action in Enterococcus faecium. J. Mol. Biol. 392 (5), 1178–1191. 10.1016/j.jmb.2009.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belley A.; Arhin F. F.; Moeck G. (2017) Evaluation of Oritavancin Dosing Strategies against Vancomycin-Resistant Enterococcus faecium Isolates with or without Reduced Susceptibility to Daptomycin in an In Vitro Pharmacokinetic/Pharmacodynamic Model. Antimicrob. Agents Chemother. 62 (1), e01873-17. 10.1128/AAC.01873-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabarba A.; Nicas T. I.; Thompson R. C. (1997) Structural modifications of glycopeptide antibiotics. Med. Res. Rev. 17 (1), 69–137. 10.1002/(SICI)1098-1128(199701)17:1<69::AID-MED3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Okano A.; Isley N. A.; Boger D. L. (2017) Total Syntheses of Vancomycin-Related Glycopeptide Antibiotics and Key Analogues. Chem. Rev. 117 (18), 11952–11993. 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim G.; Thaker M. N.; Koteva K.; Wright G. (2014) Glycopeptide antibiotic biosynthesis. J. Antibiot. 67 (1), 31–41. 10.1038/ja.2013.117. [DOI] [PubMed] [Google Scholar]
- Chen S.; Wu Q. H.; Shen Q. Q.; Wang H. (2016) Progress in Understanding the Genetic Information and Biosynthetic Pathways behind Amycolatopsis Antibiotics, with Implications for the Continued Discovery of Novel Drugs. ChemBioChem 17 (2), 119–128. 10.1002/cbic.201500542. [DOI] [PubMed] [Google Scholar]
- Cooper M. A., and Betley J. R. (10 May 2002) Antibacterial Agents Compromising Conjugates of Glycopeptide and Peptidic Membrane Associating Agents, Patent WO 02/36612 A1.
- Cooper M. A., and Blaskovich M. A. (13 August 2015) Antibacterial Agents, Patent WO 2015/117196 A.
- Blaskovich M. A. T.; Hansford K. A.; Gong Y.; Butler M. S.; Muldoon C.; Huang J. X.; Ramu S.; Silva A. B.; Cheng M.; Kavanagh A. M.; Ziora Z.; Premraj R.; Lindahl F.; Bradford T. A.; Lee J. C.; Karoli T.; Pelingon R.; Edwards D. J.; Amado M.; Elliott A. G.; Phetsang W.; Daud N. H.; Deecke J. E.; Sidjabat H. E.; Ramaologa S.; Zuegg J.; Betley J. R.; Beevers A. P. G.; Smith R. A. G.; Roberts J. A.; Paterson D. L.; Cooper M. A. (2018) Protein-inspired antibiotics active against vancomycin- and daptomycin-resistant bacteria. Nat. Commun. 9 (1), 22. 10.1038/s41467-017-02123-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarlagadda V.; Sarkar P.; Samaddar S.; Haldar J. (2016) A Vancomycin Derivative with a Pyrophosphate-Binding Group: A Strategy to Combat Vancomycin-Resistant Bacteria. Angew. Chem., Int. Ed. 55 (27), 7836–7840. 10.1002/anie.201601621. [DOI] [PubMed] [Google Scholar]
- Printsevskaya S. S.; Reznikova M. I.; Korolev A. M.; Lapa G. B.; Olsufyeva E. N.; Preobrazhenskaya M. N.; Plattner J. J.; Zhang Y. K. (2013) Synthesis and study of antibacterial activities of antibacterial glycopeptide antibiotics conjugated with benzoxaboroles. Future Med. Chem. 5 (6), 641–652. 10.4155/fmc.13.16. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Akkapeddi P.; Manjunath G. B.; Haldar J. (2014) Membrane Active Vancomycin Analogues: A Strategy to Combat Bacterial Resistance. J. Med. Chem. 57 (11), 4558–4568. 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- Baxter Healthcare Corporation . Vancomycin Injection, USP. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/050671s022lbl.pdf (accessed Jan 10 2018).
- European Medicines Agency . Assessment Report: Vibativ. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001240/WC500115363.pdf (accessed Jan 10 2018).
- European Medicines Agency . Assessment report for Xydalba. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002840/WC500183871.pdf (accessed Jan 20, 2018).
- European Medicines Agency . Withdrawal Assessment Report for Ramvocid (Oritavancin). http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2010/01/WC500060558.pdf (accessed Jan 20, 2018).
- Yarlagadda V.; Konai M. M.; Manjunath G. B.; Prakash R. G.; Mani B.; Paramanandham K.; Ranjan S. B.; Ravikumar R.; Chakraborty S. P.; Roy S.; Haldar J. (2015) In vivo antibacterial activity and pharmacological properties of the membrane-active glycopeptide antibiotic YV11455. Int. J. Antimicrob. Agents 45 (6), 627–634. 10.1016/j.ijantimicag.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Samaddar S.; Haldar J. (2016) Intracellular activity of a membrane-active glycopeptide antibiotic against meticillin-resistant Staphylococcus aureus infection. J. Glob. Antimicrob. Res. 5, 71–74. 10.1016/j.jgar.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Manjunath G. B.; Sarkar P.; Akkapeddi P.; Paramanandham K.; Shome B. R.; Ravikumar R.; Haldar J. (2016) Glycopeptide Antibiotic To Overcome the Intrinsic Resistance of Gram-Negative Bacteria. ACS Infect. Dis. 2 (2), 132–139. 10.1021/acsinfecdis.5b00114. [DOI] [PubMed] [Google Scholar]
- Yoganathan S.; Miller S. J. (2015) Structure Diversification of Vancomycin through Peptide-Catalyzed, Site-Selective Lipidation: A Catalysis-Based Approach To Combat Glycopeptide-Resistant Pathogens. J. Med. Chem. 58 (5), 2367–2377. 10.1021/jm501872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarlagadda V.; Konai M. M.; Manjunath G. B.; Ghosh C.; Haldar J. (2015) Tackling vancomycin-resistant bacteria with ’lipophilic-vancomycin-carbohydrate conjugates’. J. Antibiot. 68 (5), 302–312. 10.1038/ja.2014.144. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Konai M. M.; Paramanandham K.; Nimita V. C.; Shome B. R.; Haldar J. (2015) In vivo efficacy and pharmacological properties of a novel glycopeptide (W4465) against vancomycin-intermediate Staphylococcus aureus. Int. J. Antimicrob. Agents 46 (4), 446–450. 10.1016/j.ijantimicag.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Samaddar S.; Paramanandham K.; Shome B. R.; Haldar J. (2015) Membrane Disruption and Enhanced Inhibition of Cell-Wall Biosynthesis: A Synergistic Approach to Tackle Vancomycin-Resistant Bacteria. Angew. Chem., Int. Ed. 54 (46), 13644–13649. 10.1002/anie.201507567. [DOI] [PubMed] [Google Scholar]
- Gu W.; Chen B.; Ge M. (2014) Design and synthesis of new vancomycin derivatives. Bioorg. Med. Chem. Lett. 24 (10), 2305–2308. 10.1016/j.bmcl.2014.03.093. [DOI] [PubMed] [Google Scholar]
- Crowley B. M.; Boger D. L. (2006) Total synthesis and evaluation of [Psi[CH2NH]Tpg4]vancomycin aglycon: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc. 128 (9), 2885–2892. 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Pierce J. G.; James R. C.; Okano A.; Boger D. L. (2011) A redesigned vancomycin engineered for dual D-Ala-D-ala And D-Ala-D-Lac binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J. Am. Chem. Soc. 133 (35), 13946–13949. 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Okano A.; Pierce J. G.; James R. C.; Stamm S.; Crane C. M.; Boger D. L. (2012) Total Synthesis of [Psi[C(=S)NH]Tpg(4)]Vancomycin Aglycon, [Psi[C(=NH)NH]Tpg(4)]Vancomycin Aglycon, and Related Key Compounds: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J. Am. Chem. Soc. 134 (2), 1284–1297. 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A.; James R. C.; Pierce J. G.; Xie J.; Boger D. L. (2012) Silver(I)-Promoted Conversion of Thioamides to Amidines: Divergent Synthesis of a Key Series of Vancomycin Aglycon Residue 4 Amidines That Clarify Binding Behavior to Model Ligands. J. Am. Chem. Soc. 134 (21), 8790–8793. 10.1021/ja302808p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A.; Nakayama A.; Schammel A. W.; Boger D. L. (2014) Total Synthesis of [Psi[C(=NH)NH]Tpg(4)]Vancomycin and its ((4)-Chlorobiphenyl)methyl Derivative: Impact of Peripheral Modifications on Vancomycin Analogues Redesigned for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J. Am. Chem. Soc. 136 (39), 13522–13525. 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A.; Nakayama A.; Wu K. J.; Lindsey E. A.; Schammel A. W.; Feng Y. Q.; Collins K. C.; Boger D. L. (2015) Total Syntheses and Initial Evaluation of [Psi[C(=S)NH]Tpg(4)]vancomycin, [Psi[C(=NH)NH]Tpg(4)]vancomycin, [Psi[CH2NH]Tpg(4)]vancomycin, and Their (4-Chlorobiphenyl)methyl Derivatives: Synergistic Binding Pocket and Peripheral Modifications for the Glycopeptide Antibiotics. J. Am. Chem. Soc. 137 (10), 3693–3704. 10.1021/jacs.5b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A.; Isley N. A.; Boger D. L. (2017) Peripheral modifications of [Psi[CH2NH]Tpg(4)]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc. Natl. Acad. Sci. U. S. A. 114 (26), E5052–E5061. 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinchman J. R.; Boger D. L. (2013) Probing the Role of the Vancomycin E-Ring Aryl Chloride: Selective Divergent Synthesis and Evaluation of Alternatively Substituted E-Ring Analogues. J. Med. Chem. 56 (10), 4116–4124. 10.1021/jm4004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinchman J. R.; Boger D. L. (2013) Investigation into the functional impact of the vancomycin C-ring aryl chloride. Bioorg. Med. Chem. Lett. 23 (17), 4817–4819. 10.1016/j.bmcl.2013.06.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakama Y.; Yoshida O.; Yoda M.; Araki K.; Sawada Y.; Nakamura J.; Xu S.; Miura K.; Maki H.; Arimoto H. (2010) Discovery of a Novel Series of Semisynthetic Vancomycin Derivatives Effective against Vancomycin-Resistant Bacteria. J. Med. Chem. 53 (6), 2528–2533. 10.1021/jm9017543. [DOI] [PubMed] [Google Scholar]
- Wadzinski T. J.; Gea K. D.; Miller S. J. (2016) A stepwise dechlorination/cross-coupling strategy to diversify the vancomycin ’in-chloride’. Bioorg. Med. Chem. Lett. 26 (3), 1025–1028. 10.1016/j.bmcl.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak T. P.; Miller S. J. (2012) Site-Selective Bromination of Vancomycin. J. Am. Chem. Soc. 134 (14), 6120–6123. 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak T. P.; Miller S. J. (2013) Chemical Tailoring of Teicoplanin with Site-Selective Reactions. J. Am. Chem. Soc. 135 (22), 8415–8422. 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D.; Chen F.; Xiong L.; Tang F.; Faridoon; Qiu Y.; Zhang N.; Gong L.; Li J.; Lan L.; Huang W. (2018) Extra Sugar on Vancomycin: New Analogues for Combating Multidrug-Resistant Staphylococcus aureus and Vancomycin-Resistant Enterococci. J. Med. Chem. 61, 286. 10.1021/acs.jmedchem.7b01345. [DOI] [PubMed] [Google Scholar]
- Han S.; Miller S. J. (2013) Asymmetric Catalysis at a Distance: Catalytic, Site-Selective Phosphorylation of Teicoplanin. J. Am. Chem. Soc. 135 (33), 12414–12421. 10.1021/ja406067v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane C. M.; Pierce J. G.; Leung S. S. F.; Tirado-Rives J.; Jorgensen W. L.; Boger D. L. (2010) Synthesis and Evaluation of Selected Key Methyl Ether Derivatives of Vancomycin Aglycon. J. Med. Chem. 53 (19), 7229–7235. 10.1021/jm100946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. J.; Yang Q.; Xu L.; Chang J.; Sun X. (2012) Synthesis and antibacterial activity against Clostridium difficile of novel demethylvancomycin derivatives. Bioorg. Med. Chem. Lett. 22 (15), 4942–4945. 10.1016/j.bmcl.2012.06.039. [DOI] [PubMed] [Google Scholar]
- Chang J.; Zhang S. J.; Jiang Y. W.; Xu L.; Yu J. M.; Zhou W. J.; Sun X. (2013) Design, Synthesis, and Antibacterial Activity of Demethylvancomycin Analogues against Drug-Resistant Bacteria. ChemMedChem 8 (6), 976–984. 10.1002/cmdc.201300011. [DOI] [PubMed] [Google Scholar]
- Crane C. M.; Boger D. L. (2009) Synthesis and Evaluation of Vancomycin Aglycon Analogues That Bear Modifications in the N-Terminal D-Leucyl Amino Acid. J. Med. Chem. 52 (5), 1471–1476. 10.1021/jm801549b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucs Z.; Bereczki I.; Csavas M.; Roth E.; Borbas A.; Batta G.; Ostorhazi E.; Szatmari R.; Herczegh P. (2017) Lipophilic teicoplanin pseudoaglycon derivatives are active against vancomycin- and teicoplanin-resistant enterococci. J. Antibiot. 70 (5), 664–670. 10.1038/ja.2017.2. [DOI] [PubMed] [Google Scholar]
- Szucs Z.; Csavas M.; Roth E.; Borbas A.; Batta G.; Perret F.; Ostorhazi E.; Szatmari R.; Vanderlinden E.; Naesens L.; Herczegh P. (2017) Synthesis and biological evaluation of lipophilic teicoplanin pseudoaglycon derivatives containing a substituted triazole function. J. Antibiot. 70 (2), 152–157. 10.1038/ja.2016.80. [DOI] [PubMed] [Google Scholar]
- Sundram U. N.; Griffin J. H.; Nicas T. I. (1996) Novel vancomycin dimers with activity against vancomycin-resistant enterococci. J. Am. Chem. Soc. 118 (51), 13107–13108. 10.1021/ja9621298. [DOI] [Google Scholar]
- Nicolaou K. C.; Hughes R.; Cho S. Y.; Winssinger N.; Labischinski H.; Endermann R. (2001) Synthesis and biological evaluation of vancomycin dimers with potent activity against vancomycin-resistant bacteria: target-accelerated combinatorial synthesis. Chem. - Eur. J. 7 (17), 3824–3843. 10.1002/1521-3765(20010903)7:17<3824::AID-CHEM3824>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Hughes R.; Cho S. Y.; Winssinger N.; Smethurst C.; Labischinski H.; Endermann R. (2000) Target-accelerated combinatorial synthesis and discovery of highly potent antibiotics effective against vancomycin-resistant bacteria. Angew. Chem., Int. Ed. 39 (21), 3823–3828. 10.1002/1521-3773(20001103)39:21<3823::AID-ANIE3823>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rao J. H.; Lahiri J.; Isaacs L.; Weis R. M.; Whitesides G. M. (1998) A trivalent system from vancomycin - D-Ala-D-Ala with higher affinity than avidin - biotin. Science 280 (5364), 708–711. 10.1126/science.280.5364.708. [DOI] [PubMed] [Google Scholar]
- Rao J.; Whitesides G. M. (1997) Tight Binding of a Dimeric Derivative of Vancomycin with Dimeric l-Lys-d-Ala-d-Ala. J. Am. Chem. Soc. 119 (43), 10286–10290. 10.1021/ja971225l. [DOI] [Google Scholar]
- Jain R. K.; Trias J.; Ellman J. A. (2003) d-Ala-d-Lac Binding Is Not Required for the High Activity of Vancomycin Dimers against Vancomycin Resistant Enterococci. J. Am. Chem. Soc. 125 (29), 8740–8741. 10.1021/ja0359761. [DOI] [PubMed] [Google Scholar]
- Staroske T.; Williams D. H. (1998) Synthesis of covalent head-to-tail dimers of vancomycin. Tetrahedron Lett. 39 (27), 4917–4920. 10.1016/S0040-4039(98)00895-8. [DOI] [Google Scholar]
- Adamczyk M.; Moore J. A.; Rege S. D.; Yu Z. G. (1999) Investigations into self-association of vancomycin covalent dimers using surface plasmon resonance technology. Bioorg. Med. Chem. Lett. 9 (16), 2437–2440. 10.1016/S0960-894X(99)00402-3. [DOI] [PubMed] [Google Scholar]
- Griffin J. H.; Linsell M. S.; Nodwell M. B.; Chen Q.; Pace J. L.; Quast K. L.; Krause K. M.; Farrington L.; Wu T. X.; Higgins D. L.; Jenkins T. E.; Christensen B. G.; Judice J. K. (2003) Multivalent drug design. Synthesis and in vitro analysis of an array of vancomycin dimers. J. Am. Chem. Soc. 125 (21), 6517–6531. 10.1021/ja021273s. [DOI] [PubMed] [Google Scholar]
- Lu J.; Yoshida O.; Hayashi S.; Arimoto H. (2007) Synthesis of rigidly-linked vancomycin dimers and their in vivo efficacy against resistant bacteria. Chem. Commun. 3, 251–253. 10.1039/B613319C. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Sarkar P.; Manjunath G. B.; Haldar J. (2015) Lipophilic vancomycin aglycon dimer with high activity against vancomycin-resistant bacteria. Bioorg. Med. Chem. Lett. 25 (23), 5477–5480. 10.1016/j.bmcl.2015.10.083. [DOI] [PubMed] [Google Scholar]
- Silverman S. M.; Moses J. E.; Sharpless K. B. (2017) Reengineering Antibiotics to Combat Bacterial Resistance: Click Chemistry [1,2,3]-Triazole Vancomycin Dimers with Potent Activity against MRSA and VRE. Chem. - Eur. J. 23 (1), 79–83. 10.1002/chem.201604765. [DOI] [PubMed] [Google Scholar]
- Stryjewski M. E.; Potgieter P. D.; Li Y. P.; Barriere S. L.; Churukian A.; Kingsley J.; Corey G. R.; Group T. D. I. (2012) TD-1792 versus vancomycin for treatment of complicated skin and skin structure infections. Antimicrob. Agents Chemother. 56 (11), 5476–5483. 10.1128/AAC.00712-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell K. L.; Citron D. M.; Warren Y. A.; Goldstein E. J. (2012) In vitro activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic, against 377 strains of anaerobic bacteria and 34 strains of Corynebacterium species. Antimicrob. Agents Chemother. 56 (4), 2194–2197. 10.1128/AAC.06274-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais J.; Lewis S. R.; Krause K. M.; Benton B. M. (2012) Antistaphylococcal activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic. Antimicrob. Agents Chemother. 56 (3), 1584–1587. 10.1128/AAC.05532-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde S. S.; Okusanya O. O.; Skinner R.; Shaw J. P.; Obedencio G.; Ambrose P. G.; Blais J.; Bhavnani S. M. (2012) Pharmacodynamics of TD-1792, a novel glycopeptide-cephalosporin heterodimer antibiotic used against Gram-positive bacteria, in a neutropenic murine thigh model. Antimicrob. Agents Chemother. 56 (3), 1578–1583. 10.1128/AAC.05382-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuthner K. D.; Vidaillac C.; Cheung C. M.; Rybak M. J. (2010) In vitro activity of the new multivalent glycopeptide-cephalosporin antibiotic TD-1792 against vancomycin-nonsusceptible Staphylococcus isolates. Antimicrob. Agents Chemother. 54 (9), 3799–3803. 10.1128/AAC.00452-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R-Pharm. (Press Release 18 March 2015) R-Pharm Group of Companies Continues Development of New Generation Antibiotic, http://www.r-pharm.com/en/press-center/news/197 (accessed 20 November 2017).
- Sader H. S., Rhomberg P. R., Farrell D. J., Flamm R. K., and Jones R. N. (2014) Antimicrobial activity of TD-1607 tested against contemporary (2010–2012) methicillin-resistant Staphylococcus aureus (MRSA) strains in 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) 2014, Poster F-970, ASM, Washington, DC, USA.
- Theravance Biopharma Inc. (31 Dec 2014) FORM 10-K United States securities and exchange commission annual report, http://investor.theravance.com/static-files/ab0a84d1-2aa7-4fb1-a682-c456f59477bb (accessed 20 November 2017).
- Theravance Biopharma Inc. (1 Mar 2017) FORM 10-K United States securities and exchange commission annual report, http://investor.theravance.com/static-files/cb4506bc-5f24-4abd-9622-1e50676e440d (accessed 20 November 2017).
- Arnusch C. J.; Bonvin A. M. J. J.; Verel A. M.; Jansen W. T. M.; Liskamp R. M. J.; de Kruijff B.; Pieters R. J.; Breukink E. (2008) The Vancomycin-Nisin(1–12) Hybrid Restores Activity against Vancomycin Resistant Enterococci. Biochemistry 47 (48), 12661–12663. 10.1021/bi801597b. [DOI] [PubMed] [Google Scholar]
- Cochrane S. A.; Li X. F.; He S. S.; Yu M.; Wu M.; Vederas J. C. (2015) Synthesis of Tridecaptin-Antibiotic Conjugates with in Vivo Activity against Gram-Negative Bacteria. J. Med. Chem. 58 (24), 9779–9785. 10.1021/acs.jmedchem.5b01578. [DOI] [PubMed] [Google Scholar]
- Ghosh M.; Miller M. J. (1996) Synthesis and in vitro antibacterial activity of spermidine-based mixed catechol- and hydroxamate-containing siderophore-vancomycin conjugates. Bioorg. Med. Chem. 4 (1), 43–48. 10.1016/0968-0896(95)00161-1. [DOI] [PubMed] [Google Scholar]
- Varisco M.; Khanna N.; Brunetto P. S.; Fromm K. M. (2014) New Antimicrobial and Biocompatible Implant Coating with Synergic Silver-Vancomycin Conjugate Action. ChemMedChem 9 (6), 1221–1230. 10.1002/cmdc.201400072. [DOI] [PubMed] [Google Scholar]
- Karau M. J.; Schmidt-Malan S. M.; Greenwood-Quaintance K. E.; Mandrekar J.; Cai J.; Pierce W. M.; Merten K.; Patel R. (2013) Treatment of Methicillin-resistant Staphylococcus aureus experimental Osteomyelitis with bone-targeted Vancomycin. SpringerPlus 2, 329. 10.1186/2193-1801-2-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albayati Z. A. F.; Sunkara M.; Schmidt-Malan S. M.; Karau M. J.; Morris A. J.; Steckelberg J. M.; Patel R.; Breen P. J.; Smeltzer M. S.; Taylor K. G.; Merten K. E.; Pierce W. M.; Crooks P. A. (2016) Novel Bone-Targeting Agent for Enhanced Delivery of Vancomycin to Bone. Antimicrob. Agents Chemother. 60 (3), 1865–1868. 10.1128/AAC.01609-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson M. C.; Shoemaker R.; Hoth K. B.; Bowman C. N.; Anseth K. S. (2009) Polymerizable Vancomycin Derivatives for Bactericidal Biomaterial Surface Modification: Structure-Function Evaluation. Biomacromolecules 10 (8), 2221–2234. 10.1021/bm900410a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson M. C.; Hoth K. C.; DeForest C. A.; Bowman C. N.; Anseth K. S. (2010) Inhibition of Staphylococcus epidermidis Biofilms Using Polymerizable Vancomycin Derivatives. Clin. Orthop. Relat. Res. 468 (8), 2081–2091. 10.1007/s11999-010-1266-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosten M.; Schafer T.; Gazendam J. A.; Ohlsen K.; Tsompanidou E.; de Goffau M. C.; Harmsen H. J.; Crane L. M.; Lim E.; Francis K. P.; Cheung L.; Olive M.; Ntziachristos V.; van Dijl J. M.; van Dam G. M. (2013) Real-time in vivo imaging of invasive- and biomaterial-associated bacterial infections using fluorescently labelled vancomycin. Nat. Commun. 4, 2584. 10.1038/ncomms3584. [DOI] [PubMed] [Google Scholar]
- Tiyanont K.; Doan T.; Lazarus M. B.; Fang X.; Rudner D. Z.; Walker S. (2006) Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc. Natl. Acad. Sci. U. S. A. 103 (29), 11033–11038. 10.1073/pnas.0600829103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonyarattanakalin S.; Hu J. F.; Dykstra-Rummel S. A.; August A.; Peterson B. R. (2007) Endocytic delivery of vancomycin mediated by a synthetic cell surface receptor: Rescue of bacterially infected mammalian cells and tissue targeting in vivo. J. Am. Chem. Soc. 129 (2), 268–269. 10.1021/ja067674f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koteva K.; Hong H. J.; Wang X. D.; Nazi I.; Hughes D.; Naldrett M. J.; Buttner M. J.; Wright G. D. (2010) A vancomycin photoprobe identifies the histidine kinase VanSsc as a vancomycin receptor. Nat. Chem. Biol. 6 (5), 327–329. 10.1038/nchembio.350. [DOI] [PubMed] [Google Scholar]
- Park O. K.; Choi H. Y.; Kim G. W.; Kim W. G. (2016) Generation of New Complestatin Analogues by Heterologous Expression of the Complestatin Biosynthetic Gene Cluster from Streptomyces chartreusis AN1542. ChemBioChem 17 (18), 1725–1731. 10.1002/cbic.201600241. [DOI] [PubMed] [Google Scholar]
- Yim G.; Wang W. L.; Thaker M. N.; Tan S.; Wright G. D. (2016) How To Make a Glycopeptide: A Synthetic Biology Approach To Expand Antibiotic Chemical Diversity. ACS Infect. Dis. 2 (9), 642–650. 10.1021/acsinfecdis.6b00105. [DOI] [PubMed] [Google Scholar]
- Yim G.; Kalan L.; Koteva K.; Thaker M. N.; Waglechner N.; Tang I.; Wright G. D. (2014) Harnessing the Synthetic Capabilities of Glycopeptide Antibiotic Tailoring Enzymes: Characterization of the UK-68,597 Biosynthetic Cluster. ChemBioChem 15 (17), 2613–2623. 10.1002/cbic.201402179. [DOI] [PubMed] [Google Scholar]
- Peschke M.; Brieke C.; Goode R. J. A.; Schittenhelm R. B.; Cryle M. J. (2017) Chlorinated Glycopeptide Antibiotic Peptide Precursors Improve Cytochrome P450-Catalyzed Cyclization Cascade Efficiency. Biochemistry 56 (9), 1239–1247. 10.1021/acs.biochem.6b01102. [DOI] [PubMed] [Google Scholar]
- Brieke C.; Kratzig V.; Haslinger K.; Winkler A.; Cryle M. J. (2015) Rapid access to glycopeptide antibiotic precursor peptides coupled with cytochrome P450-mediated catalysis: towards a biomimetic synthesis of glycopeptide antibiotics. Org. Biomol. Chem. 13 (7), 2012–2021. 10.1039/C4OB02452D. [DOI] [PubMed] [Google Scholar]
- Peschke M.; Brieke C.; Cryle M. J. (2016) F-O-G Ring Formation in Glycopeptide Antibiotic Biosynthesis is Catalysed by OxyE. Sci. Rep. 6, 35584. 10.1038/srep35584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittila T.; Kittel C.; Tailhades J.; Butz D.; Schoppet M.; Buttner A.; Goode R. J. A.; Schittenhelm R. B.; van Pee K.-H.; Sussmuth R. D.; Wohlleben W.; Cryle M. J.; Stegmann E. (2017) Halogenation of glycopeptide antibiotics occurs at the amino acid level during non-ribosomal peptide synthesis. Chemical Science 8 (9), 5992–6004. 10.1039/C7SC00460E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theravance Biopharma, Inc. (2017) Theravance Biopharma, Inc. Reports Second Quarter 2017 Financial Results and Provides Business Update, http://investor.theravance.com/news-releases/news-release-details/theravance-biopharma-inc-reports-second-quarter-2017-financial (accessed 20 November 2017).
- The Medicines Company (MDCO) Inc. (31 Dec 2016) FORM 10-K United States securities and exchange commission annual report, https://seekingalpha.com/filing/3435713 (accessed 20 November 2017).
- Allergan . (2016) Allergan Reports Third Quarter 2016 Continuing Operations Performance with GAAP Net Revenues of $3.6 Billion; Announces Accelerated Share Repurchase, Initiation of Cash Dividend, https://www.allergan.com/NEWS/News/Thomson-Reuters/Allergan-Reports-Third-Quarter-2016-Continuing-Ope (accessed 20 November 2017).
- Allergan . (2017) Allergan Reports Strong 2016 Finish with 7% Increase in GAAP Net Revenues to $3.9 Billion in Fourth Quarter 2016, https://www.prnewswire.com/news-releases/allergan-reports-strong-2016-finish-with-7-increase-in-gaap-net-revenues-to-39-billion-in-fourth-quarter-2016-300403996.html (accessed 20 November 2017).
- Allergan . (2017) Allergan Reports Solid Start to 2017 with 5% Increase in First Quarter GAAP Net Revenues to $3.6 Billion, https://www.allergan.com/news/news/thomson-reuters/allergan-reports-solid-start-to-2017-with-5-increa (accessed 20 November 2017).
- Allergan . (2017) Allergan Reports Continued Strong Execution in Second Quarter 2017 with 9% Increase in GAAP Net Revenues to $4.0 Billion, https://www.prnewswire.com/news-releases/allergan-reports-continued-strong-execution-in-second-quarter-2017-with-9-increase-in-gaap-net-revenues-to-40-billion-300498858.html (accessed 20 November 2017).
- Andersson D. I.; Hughes D. (2012) Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist. Updates 15 (3), 162–172. 10.1016/j.drup.2012.03.005. [DOI] [PubMed] [Google Scholar]