

ABSTRACT
Methicillin-resistant Staphylococcus aureus (MRSA) is a threat to global health. Consequently, much effort has focused on the development of new antimicrobials that target novel aspects of S. aureus physiology. Fatty acids are required to maintain cell viability, and bacteria synthesize fatty acids using the type II fatty acid synthesis (FASII) pathway. FASII is significantly different from human fatty acid synthesis, underscoring the therapeutic potential of inhibiting this pathway. However, many Gram-positive pathogens incorporate exogenous fatty acids, bypassing FASII inhibition and leaving the clinical potential of FASII inhibitors uncertain. Importantly, the source(s) of fatty acids available to pathogens within the host environment remains unclear. Fatty acids are transported throughout the body by lipoprotein particles in the form of triglycerides and esterified cholesterol. Thus, lipoproteins, such as low-density lipoprotein (LDL), represent a potentially rich source of exogenous fatty acids for S. aureus during infection. We sought to test the ability of LDLs to serve as a fatty acid source for S. aureus and show that cells cultured in the presence of human LDLs demonstrate increased tolerance to the FASII inhibitor triclosan. Using mass spectrometry, we observed that host-derived fatty acids present in the LDLs are incorporated into the staphylococcal membrane and that tolerance to triclosan is facilitated by the fatty acid kinase A, FakA, and Geh, a triacylglycerol lipase. Finally, we demonstrate that human LDLs support the growth of S. aureus fatty acid auxotrophs. Together, these results suggest that human lipoprotein particles are a viable source of exogenous fatty acids for S. aureus during infection.
IMPORTANCE Inhibition of bacterial fatty acid synthesis is a promising approach to combating infections caused by S. aureus and other human pathogens. However, S. aureus incorporates exogenous fatty acids into its phospholipid bilayer. Therefore, the clinical utility of targeting bacterial fatty acid synthesis is debated. Moreover, the fatty acid reservoir(s) exploited by S. aureus is not well understood. Human low-density lipoprotein particles represent a particularly abundant in vivo source of fatty acids and are present in tissues that S. aureus colonizes. Herein, we establish that S. aureus is capable of utilizing the fatty acids present in low-density lipoproteins to bypass both chemical and genetic inhibition of fatty acid synthesis. These findings imply that S. aureus targets LDLs as a source of fatty acids during pathogenesis.
KEYWORDS: Staphylococcus aureus, fatty acid kinase, fatty acids, geh, lipase, lipoprotein particles
INTRODUCTION
The Gram-positive pathogen Staphylococcus aureus is the leading cause of health care-associated infections and is a significant source of morbidity and mortality in the United States (1). In keeping with this, S. aureus infects skin and soft tissue but also has the capacity to penetrate the vasculature and colonize internal organs, including the heart, bone, and liver (2). The propensity to colonize multiple vertebrate organs is due to the ability of this pathogen to survive and proliferate in the blood. Within the vasculature, S. aureus withstands the innate immune response and hijacks host-derived metabolites needed for proliferation (3). In addition to a remarkable ability to survive in vertebrate blood, the prevalence of antibiotic-resistant S. aureus isolates presents additional challenges to the efficacious treatment of infection. Consequently, the development of new therapeutic strategies is needed to combat S. aureus infections.
Bacterial synthesis of fatty acids is a focus for the design of new therapeutics to treat infections caused by S. aureus and other bacterial pathogens (4, 5). Bacteria utilize the type II fatty acid synthesis (FASII) pathway for de novo synthesis of fatty acids and maintenance of the plasma membrane (6, 7). FASII is composed of multiple enzymes, distinguishing it from the single-enzyme type I system used by eukaryotes (8). Therefore, FASII offers several ideal candidate targets for antimicrobial development (9, 10). As such, multiple FASII inhibitors are currently being developed. Many of these molecules target FabI (11–13), an NADPH-dependent, enoyl-acyl carrier protein reductase that elongates the fatty acid chain. The effectiveness of FabI inhibition is best exemplified by triclosan, which has been broadly used in consumer and medical goods for decades (14, 15). FASII inhibitors demonstrate various degrees of efficacy in murine models of S. aureus infection (11, 13, 16–19). This variability is likely due to the ability of S. aureus to resist FASII inhibition by incorporating host-derived fatty acids (20–22). Consistent with this, FASII inhibitor bypass mutants can be isolated when S. aureus is cultured in a medium supplemented with exogenous fatty acids. These FASII inhibitor-resistant isolates contain mutations within FASII initiation genes, resulting in fatty acid auxotrophy (20, 22–24). Notably, mutations in FASII initiation genes resulting in fatty acid auxotrophy have also been observed in S. aureus strains isolated from clinical specimens (24). Together, these data demonstrate that the ability of S. aureus to incorporate exogenous fatty acids obscures the efficacy of FASII inhibitors (21, 22, 25, 26), underscoring the importance of increasing our understanding of the fatty acid reservoirs exploited by S. aureus during infection.
The incorporation of exogenous fatty acids into the cell membrane by S. aureus is dependent on the fatty acid kinase FakA (also called VfrB). FakA phosphorylates exogenous fatty acids, producing acyl-PO4, which enters phospholipid synthesis (27–29). The identification of FakA substantially advanced our understanding of the mechanisms that S. aureus utilizes to incorporate exogenous fatty acids. However, the role of FakA in FASII inhibitor bypass has not been established. In addition, the source(s) of fatty acids accessible to S. aureus within the host environment is not known. Within the vasculature, free fatty acids are a minor component of the total fatty acids present (30, 31). The vast majority of fatty acids in host blood are esterified into triglycerides and cholesterol esters found within host lipoprotein particles (30–33). Low-density lipoprotein (LDL) is one type of several classes of lipoprotein particles that function as lipid transport vehicles that deliver fatty acids and cholesterol to and from host cells throughout the vasculature. The hydrophilic exterior of lipoprotein particles consists of phospholipids, cholesterol, and proteins that surround a fatty acid-rich hydrophobic core of triglycerides and cholesterol esters (33). In the host, fatty acids are released from the triglycerides and phospholipids present within lipoprotein particles by a tissue-specific family of triacylglycerol lipases (34–36). Notably, expression of secreted lipases is a clinically defining feature of S. aureus, and most strains encode multiple lipases (37, 38). Previous studies established that LDL particles bind to and sequester factors secreted by S. aureus, such as autoinducing peptides and alpha-toxin (39–43), but the capacity of LDLs to serve as an exogenous source of fatty acids for S. aureus has not been explored.
We hypothesized that S. aureus utilizes fatty acids present within lipoprotein particles. To test this hypothesis, we monitored the sensitivity of S. aureus cultured in the presence of human LDL to the FASII inhibitor triclosan. We show that LDLs act as a reservoir of exogenous fatty acids that increase S. aureus tolerance to triclosan and that this leads to incorporation of LDL-specific fatty acids in the phospholipids of S. aureus. Human LDLs also support the growth of fatty acid auxotrophs of S. aureus. Importantly, we also demonstrate that incorporation of host fatty acids from LDLs is reduced in strains lacking the major triacylglycerol lipase, Geh. Additionally, genetic inactivation of fakA significantly impairs triclosan resistance in S. aureus cells cultured in the presence of LDLs. Thus, host-derived lipoprotein particles provide exogenous fatty acids for the maintenance of the staphylococcal membrane, increasing the tolerance of S. aureus to triclosan exposure. Together, these findings show that lipoprotein particles are sources of host fatty acids for S. aureus.
RESULTS
Lipoprotein particles protect S. aureus from FASII triclosan inhibition.
S. aureus scavenges exogenous free fatty acids for incorporation into membrane phospholipids (5, 22, 44). However, little is known regarding the host-derived sources of fatty acids that are utilized by S. aureus. Host fatty acids are predominantly esterified into triglycerides or cholesterol, which are transported through the vasculature by lipoprotein particles, such as LDL (30–33). We reasoned that lipoprotein particles are a viable source of host-derived exogenous fatty acids for S. aureus. To test this, we used the FASII inhibitor triclosan to block endogenous fatty acid production (20–22, 44) and determined the capacity of LDLs to rescue staphylococcal growth. To this end, Kirby-Bauer disk diffusion assays were performed using a laboratory-derived, methicillin-resistant USA300 strain cultured on tryptic soy agar (TSA) supplemented with chicken egg yolk, a rich source of lipoprotein particles that contain esterified fatty acids (45–47). Egg yolk contains less than 0.5% free fatty acids (48). Compared to plates without supplementation, egg yolk-supplemented plates provided significant protection from triclosan (Fig. 1A). To further characterize the source of the triclosan resistance, egg yolk was separated into granule and plasma fractions. Egg yolk plasma (EYP) is enriched for low-density lipoprotein (LDL) particles, which compose ∼85% of the solution. The remaining ∼15% is soluble proteins (45, 47, 49–51). EYP provided protection from triclosan comparable to that provided by egg yolk (Fig. 1A). To determine the growth kinetics of the protection provided by EYP, S. aureus was grown in tryptic soy broth (TSB) under the following conditions: untreated, 1% EYP alone, 1 μM triclosan alone, or 1% EYP together with 1 μM triclosan. We chose to perform our growth curves with 1 μM triclosan, as this concentration is below the concentration (7 μM) that has been described to damage the S. aureus cytoplasmic membrane (52). Cells cultured in the presence of 1% EYP grew similarly to untreated cells, but triclosan treatment impaired growth for up to 12 h. The addition of EYP restored the growth of S. aureus in the presence of triclosan (Fig. 1B). These results suggest that lipoprotein particles derived from chicken egg yolk serve as a source of exogenous fatty acids for S. aureus.
FIG 1.

Lipoprotein particles protect S. aureus from FASII inhibition by triclosan. (A) S. aureus was plated as a lawn on tryptic soy agar (TSA) with the following supplements to determine triclosan sensitivity by Kirby-Bauer disk diffusion assays: no supplementation, 1% chicken egg yolk, and 1% chicken egg yolk plasma (EYP). The mean diameter of the zone of inhibition from four independent experiments is shown. Error bars represent the standard error of the mean. (B) The growth of S. aureus was monitored over time by measurement of the optical density (OD) in TSB under the following conditions: untreated, 1% EYP, 1 μM triclosan (TCS), or 1 μM triclosan with 1% EYP. The mean from six independent experiments is shown. Error bars represent the standard error of the mean. (C) The growth of S. aureus was monitored over time by measurement of the OD in TSB under the following conditions: untreated, 0.34 μg/μl purified human LDL, 1 μM triclosan, or 1 μM triclosan with 0.34 μg/μl purified human LDL. (D) S. aureus was plated as a lawn on 1% tryptone agar with or without EYP for Kirby-Bauer disk diffusion assays to determine triclosan sensitivity, as described in the legend to panel A. The mean diameter of the zone of inhibition from four independent experiments is shown. Error bars represent the standard error of the mean. (E) The growth of S. aureus was monitored over time by measurement of the OD in 1% tryptone broth under the following conditions: untreated, 1% EYP, 1 μM triclosan, or 1 μM triclosan with 1% EYP. The mean from four independent experiments is shown. Error bars represent the standard error of the mean. (F) The growth of S. aureus was monitored over time by measurement of the OD in 1% tryptone broth under the following conditions: untreated, 1 μM triclosan, or 1 μM triclosan with 0.34 μg/μl purified human LDL. The mean from four independent experiments is shown. Error bars represent the standard error of the mean.
We next sought to test the capacity of human-derived lipoprotein particles to rescue S. aureus from FASII inhibition. We performed a similar growth assay with commercially available, highly purified human LDL as a source of exogenous fatty acids. S. aureus cells cultured on TSB supplemented with 0.34 μg/μl of purified human LDLs grew slightly greater than untreated cells, indicating that human LDLs are not toxic to S. aureus (Fig. 1C). Notably, the addition of human LDLs restored the growth of triclosan-treated S. aureus. To control for the possible effects of the fatty acids found in the soybean-based medium used in the assay described above, we evaluated the phenotypes in a fatty acid-free broth composed of 1% tryptone (22, 53). Kirby-Bauer disk diffusion assays were performed on tryptone agar augmented with EYP or not augmented. The addition of EYP to tryptone agar had a similar effect of reducing the triclosan-induced zone of inhibition in S. aureus described above (Fig. 1D). Fatty acid-free tryptone broth was used to test the ability of EYP (Fig. 1E) or human LDLs (Fig. 1F) to be utilized as sources of exogenous fatty acids when S. aureus is grown in the presence of triclosan. The addition of EYP or human LDLs to the fatty acid-free, tryptone medium also restored the growth of S. aureus cultured in the presence of triclosan. The ability of both chicken- and human-derived lipoprotein particles to reverse triclosan growth inhibition implies that lipoprotein particles serve as a source of exogenous fatty acids for S. aureus.
Host-derived lipoprotein particles are a source of exogenous fatty acids for S. aureus.
Two possibilities may account for the ability of LDLs to rescue growth in the presence of triclosan. First, a nonspecific triclosan-LDL interaction may sequester triclosan from the cells, resulting in rescued growth (22, 26). Alternatively, S. aureus actively incorporates LDL-derived fatty acids into its phospholipid bilayer, bypassing triclosan FASII inhibition. To distinguish between these two possibilities, we directly measured the amount of LDL-derived fatty acids that are incorporated into the staphylococcal membrane. S. aureus does not synthesize unsaturated fatty acids but can incorporate them into phosphatidylglycerol (PG), the major phospholipid present in the staphylococcal membrane (22, 54, 55). In the absence of exogenous fatty acids, the fatty acid moieties of S. aureus PG are predominantly saturated, consisting of 14 to 20 carbon atoms (22, 55). Consistent with this, saturated fatty acids of 15 and 17 carbons (C15:0 and C17:0) are the most common fatty acids comprising PG (PG32:0) in S. aureus (22). Human LDL primarily contains C16:0, C18:0, C16:1, C18:1, C18:2, and C20:1 fatty acids esterified within triglycerides or to cholesterol (56, 57). Therefore, the unsaturated fatty acids C16:1, C18:1, C18:2, and C20:1 serve as distinct molecular signatures of LDL-derived fatty acids. We employed an unbiased lipidomic analysis utilizing direct-infusion high-resolution/accurate mass spectrometry (MS) and tandem mass spectrometry to monitor the fatty acid profile of S. aureus PG. S. aureus was incubated in the presence or absence of human LDLs, and the PG profile of these cells was compared to that of cells cultured in 1% tryptone broth (see Table S1 in the supplemental material). The PG profile of S. aureus grown in tryptone broth supplemented with human LDL was significantly different than the profile of cells cultured in the absence of LDL (Table 1). The altered PG profile included a large increase in the percentage of unsaturated PG (UPG) in the cell membrane (Fig. 2A). The unsaturated fatty acids that comprise the UPG are common within human LDLs, and we observed a 14.59-fold increase in these unsaturated fatty acids in the PG of cells cultured with human LDLs compared to those grown in tryptone broth alone (Fig. 2B). In keeping with these results, the four most abundant unsaturated fatty acids in human LDLs, C16:1, C18:1, C18:2, and C20:1, were all increased in S. aureus PG when the cells were cultured in the presence of human LDLs compared to when they were cultured without human LDLs (Table S1). In an independent experiment using TSB, we observed similar differences in the PG profiles of S. aureus cultures supplemented with LDL. Specifically, the relative abundance of all unsaturated fatty acids within the UPG was increased in cells cultured in the presence of human LDLs (Table S2), although the fatty acids from the soybean component of TSB (55, 58, 59) likely were also incorporated by S. aureus, making these results more difficult to interpret than the results of the analysis performed in fatty acid-free tryptone broth (55). Together, these data demonstrate that S. aureus incorporates unsaturated fatty acids present within human LDLs into the membrane PG.
TABLE 1.
Unsaturated fatty acid profile of the WT and S. aureus geh lip grown in the presence of human LDLs
| PG TC:TDBa | WT cultured in tryptone broth |
WT cultured in tryptone broth with human LDLs |
S. aureus
geh lip cultured in tryptone broth |
S. aureus
geh lip cultured in tryptone broth with human LDLs |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normalized ion abundance/mg of cells | SD | Fatty acidsc | Normalized ion abundance/mg of cells | SD | Fatty acidsc | Normalized ion abundance/mg of cells | SD | Fatty acidsc | Normalized ion abundance/mg of cells | SD | Fatty acidsc | |
| 30:1 | 0.00 | 0.00 | NDb | 0.11 | 0.08 | ND | 0.13 | 0.06 | ND | 0.00 | 0.00 | ND |
| 31:1 | 1.96 | 0.19 | 16:1_15:0, 14:1_17:0, 12:1_19:0 | 7.46 | 0.38 | 16:1_15:0, 14:1_17:0, 12:1_19:0, 11:1_20:0 | 2.64 | 0.24 | 16:1_15:0, 14:1_17:0, 12:1_19:0 | 3.02 | 0.18 | 16:1_15:0, 14:1_17:0, 12:1_19:0, 11:1_20:0 |
| 32:1 | 1.49 | 0.07 | ND | 7.79 | 0.58 | 17:1_15:0, 16:1_16:0, 18:1_14:0 | 1.99 | 0.29 | 17:1_15:0, 15:1_17:0, 13:1_19:0 | 2.18 | 0.10 | 17:1_15:0, 16:1_16:0 |
| 33:1 | 12.76 | 1.01 | 18:1_15:0 | 214.05 | 19.82 | 18:1_15:0 | 14.19 | 0.59 | 18:1_15:0 | 28.02 | 1.43 | 18:1_15:0 |
| 34:1 | 0.00 | 0.00 | ND | 16.58 | 1.53 | 18:1_16:0, 16:1_18:0 | 0.00 | 0.00 | ND | 0.00 | 0.00 | ND |
| 34:2 | 0.00 | 0.00 | ND | 5.04 | 0.37 | 16:1_18:1, 18:2_16:0 | 0.00 | 0.00 | ND | 2.00 | 0.25 | 16:1_18:1, 18:2_16:0 |
| 35:1 | 17.07 | 1.75 | 20:1_15:0, 19:1_16:0 | 61.86 | 5.26 | 20:1_15:0, 18:1_17:0, 19:1_16:0 | 19.16 | 0.22 | 20:1_15:0, 19:1_16:0 | 14.66 | 0.46 | 20:1_15:0, 19:1_16:0, 18:1_17:0 |
| 35:2 | 0.00 | 0.00 | ND | 8.04 | 0.50 | 20:2_15:0, 18:2_17:0, 16:1_19:1 | 0.03 | 0.01 | ND | 6.04 | 0.23 | 20:2_15:0, 18:2_17:0 |
| 35:4 | 0.00 | 0.00 | ND | 1.46 | 0.26 | 20:4_15:0, 17:2_18:2, 20:3_15:1 | 0.00 | 0.00 | ND | 20.13 | 0.60 | 20:4_15:0 |
| 35:5 | 0.00 | 0.00 | ND | 0.00 | 0.00 | ND | 0.00 | 0.00 | ND | 1.27 | 0.15 | 20:5_15:0, 20:4_15:1 |
| 36:2 | 0.00 | 0.00 | ND | 46.15 | 4.14 | 18:1_18:1, 18:2_18:0 | 0.00 | 0.00 | ND | 0.39 | 0.16 | 18:2_18:0, 18:1_18:1 |
| 36:3 | 0.11 | 0.05 | ND | 17.05 | 1.56 | 18:1_18:2, 20:3_16:0 | 0.00 | 0.00 | ND | 0.20 | 0.10 | 18:2_18:1 |
| 36:4 | 0.00 | 0.00 | ND | 2.34 | 0.35 | 18:2_18:2, 16:0_20:4, 18:3_18:1 | 0.00 | 0.00 | ND | 0.25 | 0.06 | 18:2_18:2, 20:3_16:1, 20:4_16:0 |
| 37:2 | 0.00 | 0.00 | ND | 1.85 | 0.11 | 19:0_18:2 | 0.00 | 0.00 | ND | 0.38 | 0.15 | 19:0_18:2 |
| 37:4 | 0.00 | 0.00 | ND | 0.00 | 0.00 | ND | 0.00 | 0.00 | ND | 0.91 | 0.05 | 20:4_17:0 |
Detected as [M-H]− ions. TC, total chain length; TDB, total number of double bonds.
ND, not determined.
Fatty acids are listed in order of isomer abundance. An underscore between fatty acid designations indicates that each fatty acid may be present in either the SN1 or SN2 position, as tandem mass spectrometry data alone cannot rule out the possibility that lipid species exist as a mixture of positional isomers (85).
FIG 2.
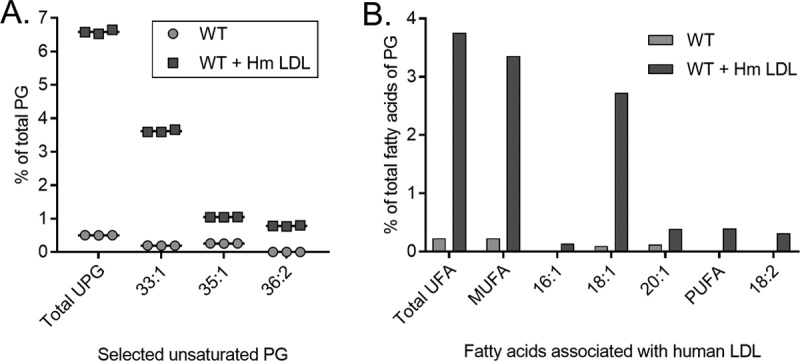
Human low-density lipoprotein (Hm LDL) particles are a source of exogenous fatty acids for synthesis of S. aureus phosphatidylglycerol. (A) Percentage of unsaturated phosphatidylglycerol (UPG) in comparison to the amount of total membrane PG of S. aureus grown in the presence (WT + Hm LDL) or absence (WT) of human LDLs. (B) Unsaturated fatty acid profile of membrane PG of S. aureus grown with (WT + Hm LDL) or without (WT) human LDLs plotted as a percentage of the amount of total PG fatty acids. UFA, MUFA, and PUFA, unsaturated, monounsaturated, and polyunsaturated fatty acids, respectively.
Strains lacking triacylglycerol lipase activity demonstrate decreased incorporation of exogenous fatty acids into the cell membrane.
We next sought to determine the mechanism by which S. aureus liberates free fatty acids from host LDLs. LDLs are composed of a structural protein and esterified fatty acids, such as triglycerides. Host lipases release fatty acids from the triglycerides of lipoprotein particles, which are then absorbed by the tissue (33). We hypothesized that S. aureus utilizes a protease or lipase to liberate fatty acids from LDLs. Thus, genetic inactivation of a protease or lipase would decrease triclosan resistance in the presence of LDLs. To test this, we examined the growth of S. aureus mutants inactivated for the protease aureolysin (aur) or V8 protease (sspA) and exposed to triclosan in the presence of lipoprotein particles (Fig. 3A). To assess the role of lipases in LDL-mediated triclosan resistance, the growth of mutants inactivated for the lipase encoded by lip or geh was quantified when the cells were treated with triclosan and lipoprotein particles (Fig. 3A). Kirby-Bauer disk diffusion assays were performed with wild-type (WT) S. aureus and protease or lipase mutants of S. aureus on tryptone agar plates supplemented with EYP. The mean diameter of the zone of inhibition produced by the aur and sspA protease mutants was approximately the same as that produced by the WT S. aureus control. Conversely, the geh mutant demonstrated an enhanced zone of inhibition, implying that this strain is more sensitive to triclosan upon supplementation with EYP. The lip mutant retained WT levels of triclosan resistance on plates supplemented with EYP. To determine if this phenotype extended to LDLs, we performed growth curves in fatty acid-free tryptone broth supplemented with human-derived LDLs in the presence of triclosan. Consistent with the Kirby-Bauer disk diffusion assay results (Fig. 3A), the capacity of human LDLs to rescue the growth of the geh mutant (Fig. 3B) exposed to triclosan was impaired, but the lip mutant (Fig. 3C) retained growth similar to that of the WT. A geh lip double mutant also demonstrated impaired growth compared to WT cultures supplemented with triclosan and human LDLs (Fig. 3D).
FIG 3.

The S. aureus lipase, encoded by geh, is required for full triclosan protection by lipoprotein particles. (A) The WT and protease (aur and sspA) and lipase (lip, geh, and lip geh) mutants were plated from overnight cultures as a lawn on 1% tryptone agar with or without 1% EYP. A triclosan-impregnated disk was placed on top of the agar. The mean diameter of the zone of inhibition from four independent experiments is shown. All error bars represent the standard error of the mean. (B to D) The growth of the WT and an isogenic geh transposon mutant (B), an isogenic lip transposon mutant (C), and an isogenic geh lip transposon mutant (D) was monitored over time by determination of the optical density (OD). Cells were grown in 1% tryptone broth under the following conditions: untreated, 1 μM triclosan (TCS), or 1 μM triclosan with 0.34 μg/μl purified human LDL. The mean results from four independent experiments are shown. ***, P < 0.005 by two-way ANOVA between the WT plus triclosan and LDL versus the geh or geh lip mutant plus triclosan and LDL; n.s., not statistically significant. All error bars represent the standard error of the mean. (E) The WT and the geh lip double mutant supernatants were incubated with human LDLs in triplicate. Free fatty acids (FFA) were detected by direct-infusion high-resolution/accurate mass spectrometry and tandem mass spectrometry, and the fold change calculated was based on the normalized number of ions per milligram. The fold change represents the amount of free fatty acids present in supernatant-treated samples compared to the amount of free fatty acids in the LDL preparation diluted in 1% tryptone broth. (F) Percentage of unsaturated PG (UPG) in comparison to the amount of total membrane PG of S. aureus grown in the presence or absence of human LDLs. (G) Unsaturated fatty acid profile of membrane PG of S. aureus grown with or without human LDLs plotted as a percentage of the amount of total PG fatty acids. SFA, UFA, MUFA, and PUFA, saturated, unsaturated, monounsaturated, and polyunsaturated fatty acids, respectively.
We hypothesized that the lipases liberate LDL-derived fatty acids. As Geh and Lip are secreted by the cell into the extracellular milieu (38), we monitored the ability of supernatants isolated from the WT and the geh lip double mutant to release free fatty acids from human LDLs using the unbiased lipidomic analysis described above. Human LDLs were mixed in tryptone broth to measure the background levels of free fatty acids. A low level of free fatty acids was present in the LDL preparation (Fig. S1). However, incubation of WT supernatants with human LDLs resulted in a 541.6-fold increase in total free fatty acids and an 829.0-fold increase in total free unsaturated fatty acids (Fig. 3E). This result supports the conclusion that an enzyme(s) secreted by S. aureus facilitates fatty acid release from human LDL. Moreover, the WT supernatant induced the liberation of the LDL-derived unsaturated fatty acids C16:1, C18:1, and C18:2, which had fold increases of 1,733.4, 1,074.9, and 1,226.7, respectively (Fig. 3E). These data are consistent with the results presented in Fig. 2A, demonstrating that culturing S. aureus in the presence of LDL increases the abundance of C16:1, C18:1, and C18:2 within UPG (Fig. 2A). The release of free fatty acids from human LDLs was lipase dependent, as supernatants isolated from the geh lip mutant demonstrated a significant reduction in the quantity of total free fatty acids after incubation with human LDLs (Fig. 3E). The amounts of the unsaturated fatty acids C16:1, C18:1, and C18:2 were markedly decreased compared to those in the WT supernatants, as denoted by 704.4-, 292.0-, and 259.8-fold reductions, respectively (Fig. 3E).
To determine if the decreased liberation of fatty acids from human LDLs leads to a reduction in LDL-derived fatty acids within the geh lip mutant PG, we performed lipodomics on the geh lip mutant cultured in the presence of human LDLs. In the absence of LDLs, WT and geh lip mutant cells had similar PG profiles (Table 1). However, upon exposure to human LDLs, the amount of UPG for the geh lip mutant was reduced 7.74-fold compared to that for the WT (Fig. 3F). In keeping with this, the levels of total unsaturated fatty acids in the membrane were reduced 5.46-fold compared to those in the membrane of the WT grown with LDLs (Fig. 3G). These data demonstrate that exogenous fatty acid source incorporation from a complex fatty source is impaired in the geh lip mutant. However, the mutant does retain some ability to incorporate exogenous fatty acids from LDLs into its PG. In keeping with this, we observed increases in the UPG species PG35:4 and PG35:5 in the geh lip mutant (Table S1). These species contain C20:4 and C20:5 unsaturated fatty acids. The incorporation of these unsaturated fatty acids in the geh lip mutant provides an explanation for why the geh lip mutant retains LDL-dependent triclosan resistance. The relatively small amount of free fatty acids present within the LDL preparation likely accounts for some of the LDL-dependent triclosan growth as well.
The methicillin-sensitive S. aureus strain Newman (NM) is lipase negative due to a prophage insertion that disrupts geh (60). Having established that the loss of geh expression in the laboratory-derived USA300 strain alters the ability of S. aureus to exploit lipoprotein particles as a source of exogenous fatty acids, we reasoned that the prophage-induced loss of geh expression in NM would similarly impair this strain's ability to utilize lipoprotein particles for triclosan protection. To assess this, WT NM and TB4, a previously described NM variant in which Geh activity was restored via prophage excision (60), were exposed to triclosan in the presence of lipoprotein particles. Kirby-Bauer disk diffusion assays were completed with WT NM and TB4 on tryptone agar plates supplemented with EYP (Fig. 4A). The mean diameter of the zone of inhibition for NM was 5.4 mm larger than that for USA300, while the mean diameter for the lipase-positive TB4 was not significantly different from that for USA300. To evaluate the contribution of geh to the difference observed between WT and TB4, we transduced a transposon-disrupted allele of geh into TB4, reducing its lipase activity. This resulted in a statistically significant increase in the triclosan-induced zone of inhibition in the TB4 geh mutant compared to that in TB4. Growth curves were conducted with NM and TB4 using purified human LDLs as the fatty acid source. Triclosan treatment completely suppressed the growth of both NM and TB4. However, NM growth was suppressed when the strain was cultured in the presence of both triclosan and human LDL (Fig. 4B). Conversely, supplementation of TB4 cells with human LDL resulted in a restoration of growth in the presence of triclosan.
FIG 4.

Lipoprotein particle protection from triclosan is reduced in lipase-negative strain Newman. (A) USA300, Newman (NM), TB4, and TB4 geh were plated as a lawn on 1% tryptone agar with or without EYP. A triclosan-impregnated disk was placed on top of the agar. The mean from four independent experiments is shown. Error bars represent the standard error of the mean. **, P = 0.0015 by Student's t test. (B) The growth of NM and TB4 was monitored over time by measurement of the OD in 1% tryptone broth under the following conditions: untreated, 1 μM triclosan (TCS), or 1 μM triclosan with 0.34 μg/μl purified human LDL. The mean results from four independent experiments are shown. ***, P < 0.005 by two-way ANOVA between NM plus triclosan and LDL versus TB4 plus triclosan and LDL. Error bars represent the standard error of the mean.
The capacity of exogenous fatty acids to increase S. aureus triclosan resistance is dependent on the fatty acid kinase FakA.
We observed that S. aureus incorporates host-derived fatty acids present in LDL into its membrane PG and that the major triacylglycerol lipase, encoded by geh, plays a role in this incorporation. Elegant studies have demonstrated that the fatty acid kinase FakA is required for the incorporation of exogenous fatty acids into the staphylococcal membrane (28, 29, 61). In keeping with this, we surmised that the fatty acid kinase FakA facilitates incorporation of the LDL-derived fatty acids into membrane PG. Inactivation of fakA has been proposed to increase triclosan sensitivity in S. aureus, despite being cultured with a source of exogenous fatty acid (20), but this has yet to be experimentally confirmed. Therefore, we sought to establish that fakA is necessary for exogenous fatty acid protection from triclosan in S. aureus. To test this, we analyzed the growth of a previously described S. aureus (USA300) fakA deletion mutant (the ΔfakA mutant) exposed to triclosan in the presence and absence of exogenous fatty acids (62). Kirby-Bauer disk diffusion assays were performed with WT S. aureus and an S. aureus ΔfakA mutant on agar plates with or without a mixture of the fatty acids palmitic acid, oleic acid, and myristic acid (20). The zone of inhibition observed for the ΔfakA mutant grown on TSA was slightly larger than that observed for WT S. aureus grown on the same agar medium (Fig. 5A). We attributed this difference to the fatty acids present in the soybean meal component of TSA being utilized by the WT but not the ΔfakA mutant (58, 63). The mean diameter of the zone of inhibition produced by the ΔfakA mutant on the fatty acid-supplemented agar was significantly larger than that produced by the WT (Fig. 5A). To further refine the role of fakA in the fatty acid-mediated tolerance of S. aureus to triclosan, we determined the half-maximal inhibitory concentration (IC50) of WT and the ΔfakA mutant cells to triclosan in the presence of an exogenous fatty acid source, Tween 80. Previous studies have shown that Tween 80 is a viable source of exogenous fatty acids that decreases sensitivity to FASII inhibitors (21, 26, 64). Compared to cells cultured in the absence of fatty acids, the IC50 of WT S. aureus grown in the presence of Tween 80 demonstrated a 54.0-fold decrease in triclosan sensitivity (Fig. 5B). Under the same conditions, the ΔfakA mutant demonstrated only a 9.39-fold reduction in the IC50 between Tween 80-supplemented TSB and TSB lacking supplementation. This difference is a substantially smaller change in triclosan sensitivity compared to that for the WT. Taken together, these data demonstrate that genetic inactivation of FakA impairs the protective effect of exogenous fatty acids shown in response to the FASII inhibitor triclosan.
FIG 5.

fakA mediates exogenous fatty acid bypass of triclosan in S. aureus. (A) WT S. aureus and an isogenic ΔfakA mutant were plated from overnight cultures separately as a lawn on TSA or TSA supplemented with a fatty acid (FA) mixture consisting of C14:0, C16:0, and C18:1. The mean diameter of the zone of inhibition from six independent experiments is shown. Error bars represent the standard error of the mean. Statistical significance was calculated by two-way ANOVA with a Tukey post hoc test for multiple comparisons; *, P = 0.0029; **, P < 0.0001. (B) The antimicrobial activities of triclosan against WT S. aureus and the ΔfakA mutant with and without 0.1% Tween 80 were compared. Cells were then suspended by pipetting, and the OD at 600 nm was measured. Results are presented as the percentage of the value for the untreated control cells. The mean from four independent experiments is shown. Error bars represent the standard error of the mean.
S. aureus exogenous fatty acid incorporation from lipoprotein particles is fakA dependent.
Having established that fakA is required for the fatty acid-dependent tolerance of triclosan, we next wanted to determine if fakA is also required for the LDL-dependent tolerance of triclosan. To test this, we assessed the sensitivity of the ΔfakA mutant to triclosan using three different assays. First, Kirby-Bauer disk diffusion assays revealed that the sensitivity of the ΔfakA mutant to triclosan is enhanced compared to that of the WT when the strains were grown on tryptone agar supplemented with lipoprotein particle-rich EYP (Fig. 6A). Second, we determined the IC50 of the WT and the ΔfakA mutant exposed to triclosan and supplemented with chicken egg yolk LDLs in TSB. The IC50s of triclosan for the ΔfakA mutant in the presence of egg yolk LDLs was 0.10 μM, while that for the WT was 0.29 μM (Fig. 6B). These results indicate that the ΔfakA mutant is impaired for utilizing LDLs as a source of fatty acids. Finally, growth curves were performed with the ΔfakA mutant using 0.34 μg/μl purified human LDLs as the fatty acid supplement in 1% tryptone broth. Triclosan treatment suppressed the growth of the ΔfakA mutant, despite supplementation with human LDL (Fig. 6C). Conversely, supplementation of WT cells with human LDL suppressed triclosan growth inhibition (Fig. 6C). In total, the inability of lipoprotein particles to protect the ΔfakA mutant from triclosan demonstrates that host-derived lipoprotein particles are exploited as a source of fatty acids for S. aureus.
FIG 6.

fakA is required for lipoprotein particle-dependent protection from triclosan. (A) The WT and the isogenic ΔfakA mutant were plated from overnight cultures as a lawn on TSA or TSA supplemented with 1% EYP for Kirby-Bauer disk diffusion assays to determine triclosan sensitivity. The mean diameter of the zone of inhibition from four independent experiments is shown. All error bars represent the standard error of the mean. (B) The antimicrobial activities of triclosan against the WT and the ΔfakA mutant with and without lipoprotein particles were compared by culturing cells in the presence of 0.1% purified chicken egg yolk LDL (EY-LDL) containing increasing concentrations of triclosan. The cells were then suspended, and the OD600 was measured. Results are presented as the percentage of the value for untreated control cells. The mean results from four independent experiments are shown. All error bars represent the standard error of the mean. (C) The growth of the WT and an isogenic ΔfakA mutant was monitored over time by measurement of the OD600 in 1% tryptone broth under the following conditions: untreated, 1 μM triclosan (TCS), or 1 μM triclosan with 0.34 μg/μl purified human LDL. The mean results from three independent experiments are shown. All error bars represent the standard error of the mean.
fakA is necessary for the generation of triclosan-resistant mutants of S. aureus.
The generation of spontaneous triclosan-resistant S. aureus fatty acid auxotrophs through exposure to FASII inhibitors on media supplemented with free fatty acids has previously been described by Morvan et al. (20). The mutations in the majority of these mutants map to the genes of the FASII initiation pathway (20). To determine if FakA is required for the generation of triclosan-resistant mutants, we plated WT and ΔfakA mutant cells on TSA with or without the fatty acid supplement (palmitic acid, oleic acid, and myristic acid), and a disk infused with triclosan was placed on the agar (Fig. 7). After 24 h of incubation, the number of resistant colonies that grew within the triclosan-induced zone of inhibition on fatty acid-supplemented agar generated by the WT was significantly greater than the number that grew within the zone of inhibition generated by the fakA mutant. After an additional 24 h of incubation, WT S. aureus continued to generate triclosan-resistant colonies on both supplemented agar and agar lacking supplementation, but the number of resistant colonies generated by the ΔfakA mutant was significantly reduced. These results indicate that the fatty acid-dependent generation of triclosan-resistant mutants is facilitated by FakA.
FIG 7.

Generation of triclosan-resistant mutants in the presence of exogenous fatty acids is fakA dependent. (A) WT S. aureus and the isogenic ΔfakA mutant were plated from overnight cultures as a lawn on TSA or TSA containing the fatty acid supplement, and a triclosan-impregnated disk was placed on top of the agar. Isolated colonies that grew within the zone of inhibition were counted after 1 and 2 days of growth. The mean results from four independent experiments are shown. Error bars represent the standard error of the mean. (B) Representative images of individual treatments are shown after 24 h and 48 h of incubation.
Human LDLs support the growth of fatty acid auxotrophs of S. aureus.
We sought to determine if lipoprotein particles satisfy the fatty acid requirement of S. aureus fatty acid auxotrophs. These studies allowed us to assess the ability of S. aureus to incorporate fatty acids from LDLs without relying on triclosan to inhibit endogenous fatty acid synthesis. The generation of S. aureus fatty acid auxotrophs through mutation in the genes of the FASII initiation pathway has previously been described (20, 23, 24). To ascertain if fatty acid auxotrophs are capable of utilizing human LDLs as a source of fatty acids, three resistant mutants from three independent trials were isolated from the zone of inhibition in the assays whose results are shown in Fig. 7. We sequenced the FASII initiation genes fabD, accA, accB, accC, and accD of the auxotrophs and mapped inactivating mutations in at least one of these genes in five out of nine of the strains (Table S3). In keeping with the fact that the mutants require an exogenous source of fatty acids, the strains demonstrated little to no growth on TSA but were capable of growing on plates supplemented with free fatty acids or EYP (Fig. S2). Using these auxotrophs, we performed triclosan Kirby-Bauer disk diffusion assays on TSA containing either the fatty acid supplement or 1% EYP. The mean diameter of the zone of inhibition generated by the auxotrophs was significantly smaller than that generated by the WT when the medium was supplemented with fatty acids (Fig. 8A). Additionally, the zone of inhibition generated by the fatty acid auxotrophs plated on agar supplemented with EYP was below our limit of detection of 6 mm (Fig. 8A). These data suggest that the fatty acid auxotrophs utilize lipoprotein particle-derived fatty acids to support their growth in the presence of triclosan. To determine if lipoprotein particles support the growth of the fatty acid auxotrophs, growth curves were performed using purified chicken LDLs as a source of exogenous fatty acids in fatty acid-free tryptone broth. Medium lacking chicken LDL supplementation did not support growth (Fig. 8B), whereas the auxotrophs grew upon medium with chicken LDL supplementation (Fig. 8C). Finally, we sought to extend our observation using a more relevant source of fatty acids, purified human LDLs. Human LDLs also supported the growth of the fatty acid auxotrophs in tryptone broth, while medium lacking supplementation could not (Fig. 8D). These findings show that a human lipoprotein particle, LDL, provides S. aureus with the fatty acids necessary to bypass FASII inhibition.
FIG 8.

LDL supplements the growth of triclosan-resistant, fatty acid auxotrophs. (A) Triclosan-resistant fatty acid auxotrophs were plated as a lawn on TSA with the following supplement: fatty acid mixture or 1% EYP. A triclosan-impregnated disk was placed on top of the agar. The dashed line denotes the limit of detection of 6 mm. The mean diameter of the zone of inhibition from three independent experiments is shown. All error bars represent the standard error of the mean. (B) The growth of the triclosan-resistant fatty acid auxotrophs in 1% tryptone broth was monitored over time by measurement of the OD. (C) The growth of the triclosan-resistant fatty acid auxotrophs in 1% tryptone broth with 0.1% purified egg yolk LDL (EY-LDL) was monitored over time by measurement of the OD. Squares represent mutants isolated in trial 1, triangles denote mutants isolated in trial 2, and diamonds depict mutants isolated in trial 3. The mean results from four independent experiments are shown. All error bars represent the standard error of the mean. (D) The growth of a representative triclosan-resistant fatty acid auxotroph from each trial was monitored over time by measurement of the OD600. Cells were grown in 1% tryptone broth under the following conditions: untreated or supplementation with 0.34 μg/μl purified human LDL. The mean results from three independent experiments are shown. All error bars represent the standard error of the mean.
Lipoprotein particles protect clinical isolates of S. aureus from triclosan inhibition.
We next sought to discern if the lipoprotein particle-dependent protection that we observed in laboratory-derived strains of S. aureus extended to clinical isolates. We characterized three lipase-negative isolates from a single cystic fibrosis (CF) patient across multiple time points, while isolates from the remaining six CF patients were all lipase positive (data not shown). Next, we performed Kirby-Bauer disk diffusion assays on EYP-supplemented tryptone agar utilizing lipase-positive isolates from three different CF patients and the three lipase-negative isolates from the single CF patient. The three lipase-negative strains were isolated at different times, and they also varied in their oxacillin sensitivity, implying that these were not clonal isolates. The zones of inhibition observed for the three lipase-positive isolates were nearly identical to those observed for the laboratory-derived USA300 strain (Fig. 9A). In contrast, the three lipase-negative isolates all produced a zone of inhibition larger than that produced by USA300 or any of the lipase-positive isolates. Growth curves were performed with a selected lipase-positive isolate and lipase-negative isolate, and these strains grew similarly in tryptone broth (Fig. 9B). Consistent with our previous lipase results (Fig. 3 and 4), the lipase-negative isolate demonstrated reduced growth compared to the lipase-positive isolate. Cumulatively, the inability of lipoprotein particles to protect S. aureus strains with reduced lipase activity from triclosan supports a model whereby staphylococcal lipases, such as Geh, liberate esterified fatty acids from host lipoprotein particles. The freed fatty acids are then utilized by S. aureus to synthesize membrane phospholipids in a FakA-dependent fashion (Fig. S3).
FIG 9.

Clinical isolates of S. aureus demonstrate a variable capacity to utilize lipoprotein particles as sources of exogenous fatty acids. (A) Three lipase-positive and three lipase-negative clinical isolates of S. aureus, together with USA300 as a control, were plated as a lawn on 1% tryptone agar supplemented with EYP or not supplemented. A triclosan-impregnated disk was placed on top of the agar. The mean diameter of the zone of inhibition from four independent experiments is shown. Error bars represent the standard error of the mean. (B) The growth of a selected lipase-positive and a lipase-negative clinical isolate of S. aureus was monitored over time by measurement of the OD600. Cells were grown in 1% tryptone broth under the following conditions: untreated, 1 μM triclosan (TCS), or 1 μM triclosan with 0.34 μg/μl purified human LDL. The mean results from four independent experiments are shown. ***, P < 0.005 by two-way ANOVA between CF:716 plus triclosan and LDL versus CF:720 plus triclosan and LDL. The mean from four independent experiments is shown. Error bars represent the standard error of the mean. (Inset) Overnight cultures of the selected lipase-positive and -negative clinical isolates were normalized to an OD600 of 0.5, and then 5 μl of these cultures was spotted onto a 1% tryptone agar plate containing EYP to determine lipase activity.
DISCUSSION
Antimicrobial-resistant pathogens are a public health crisis, with one of the most prevalent being S. aureus (65). This fact has precipitated research focused on developing inhibitors against the bacterial de novo fatty acid synthesis pathway, FASII (4, 5). However, S. aureus and many other Gram-positive pathogens bypass FASII inhibition via the incorporation of exogenous fatty acids into their membrane phospholipids (21, 27, 29). Several studies have established that the sensitivity of S. aureus to triclosan and other FASII inhibitors decreases when exogenous fatty acids are available (20–22, 26, 44). Thus, the therapeutic potential of FASII inhibitors for the treatment of S. aureus infection is uncertain. However, identifying the sources of host-derived fatty acids utilized by S. aureus may lead to strategies that improve the efficacy of FASII inhibitors.
In the current work, we monitored the capacity of host lipoprotein particles to serve as reservoirs of exogenous fatty acids for S. aureus. Through the combined approaches of chemical and genetic inhibition of FASII, together with mass spectrometry-based analysis of the fatty acid composition of the bacterial membrane, we determined that human LDLs are a viable source of exogenous fatty acids for laboratory-derived and clinical isolates of S. aureus. LDLs are abundant in multiple types of fatty acids that are esterified as phospholipids, cholesterol esters, and triglycerides (33). LDLs and the other lipoprotein particles are prevalent in the serum fraction of the blood and account for the majority of the host's fatty acids (30–32, 66). Several studies have noted that the MIC of various FASII inhibitors for S. aureus increases when serum is added to the FASII inhibitor-treated cultures (21, 22, 67–69). Additionally, it has been demonstrated that the culture of S. aureus with serum leads to incorporation of host-derived fatty acids into the cell membrane phospholipids (55). Given this, we asked if the observed increase in triclosan tolerance is due to incorporation of LDL-derived fatty acids into the membrane phospholipids of S. aureus. Using mass spectrometry, we established that the fatty acid profile of the membrane phospholipids is altered in S. aureus cultured in the presence of human LDLs. Specifically, the staphylococcal membrane included PG with C16:1, C18:1, C18:2, and other unsaturated fatty acids. S. aureus is not capable of synthesizing unsaturated fatty acids, but they are abundant in human lipoprotein particles (22). Consistent with these facts, we conclude that S. aureus liberates fatty acids from the LDL particle for incorporation into its phospholipids. We observed that unsaturated fatty acids that are commonly integrated into the cell membrane after exposure to human LDLs were among the free fatty acids with the largest fold increase and were some of the most abundant free fatty acids liberated after the human LDLs were incubated with S. aureus supernatant. These results are consistent with previous reports showing that the serum fraction of blood serves as a source of exogenous fatty acids for S. aureus phospholipid synthesis (55). Additional support for S. aureus modification of fatty acids from LDLs is that the levels of free fatty acids, including C16:1, C18:1, and C18:2, within the commercial preparation of human LDLs was minimal. Importantly, significant increases in the levels of free fatty acids were observed when incubating culture supernatant with these LDLs, including individual increases of over 1,000-fold for C16:1, C18:1, and C18:2. Importantly, the changes that we observed were dependent on the expression of staphylococcal lipases (Fig. 3E), and these data are consistent with those from multiple growth analyses demonstrating that lipase-deficient strains are impaired for LDL protection from triclosan (Fig. 3, 4, and 9). Therefore, we posit that accumulation of LDL-derived fatty acids in S. aureus PG species is primarily due to modification and incorporation of the fatty acids from human LDL. Genetic inactivation of the fatty acid kinase FakA also decreased the ability of S. aureus to resist FASII inhibition in the presence of LDLs, lending further support to our model. Together, these results demonstrate that S. aureus utilizes a complex, host-derived source of fatty acids for incorporation into the membrane phospholipid.
An ongoing debate in regard to S. aureus incorporation of exogenous fatty acids is the proposed requirement for endogenous fatty acid synthesis. Phospholipids comprise two fatty acid moieties, each occupying a specific position, sn1 or sn2, in the glycerol backbone. Elegant biochemical studies have led to the suggestion that fatty acids synthesized by the FASII pathway must occupy the sn2 position (22, 23, 27). However, the generation of fatty acid auxotrophs via the inhibition of endogenous fatty acid synthesis reported herein and by others brings into question the requirement of endogenous fatty acids populating the sn2 position (20, 22, 23). These mutants fail to grow in the absence of a source of fatty acids; therefore, it is likely that exogenous fatty acids populate both the sn1 and sn2 positions. Unfortunately, a limitation of our analysis is that we cannot discern which position the LDL-derived fatty acid populates. However, we observed PG species that contained only host-derived unsaturated fatty acids when analyzing the fatty acid content of S. aureus cultured with human LDLs, implying that an exogenous fatty acid can populate the sn2 position (see Table S4 in the supplemental material). Our finding that exogenous fatty acids can be placed in either position of PG is consistent with several recent reports, although the mechanism for this remains elusive (20, 24). Clearly, additional experimentation is required to resolve these conflicting models. However, the results reported herein provide additional evidence that (i) fatty acid auxotrophs can be generated by selecting for FASII inhibitor-resistant mutants, (ii) generation of these mutant auxotrophs is dependent on FakA activity, and (iii) human LDLs serve as a source of fatty acids for these auxotrophs.
Fatty acid auxotrophs of S. aureus have been previously reported and primarily result from mutations in accABCD and fabD (20, 22–24). Therefore, we sought to demonstrate that LDLs can supply the fatty acids required to support the growth of a fatty acid auxotroph. We generated fatty acid auxotrophs of S. aureus following a previously described method (20). Similarly, the mutants that we isolated grew only upon supplementation with free fatty acids. Importantly, supplementation of the growth medium with LDLs also supported the growth of the fatty acid auxotrophs, demonstrating that LDLs can be utilized as an exogenous source of fatty acids. Sequencing of the fabD, accA, accB, accC, and accD open reading frames of the auxotrophs identified at least one mutation in the FASII initiation pathway for five of the nine mutants (Table S3). Presumably, the remaining four fatty acid auxotrophs have mutations outside the coding regions of the FASII initiation genes that limit their expression, and genome sequencing would more conclusively locate these mutations. Nonetheless, it is clear that the growth of these mutants is dependent on an exogenous source of fatty acids and that human LDLs satisfy this requirement. These findings are consistent with the supposition that S. aureus liberates LDL-derived fatty acids during colonization of the host. On the basis of our results, it is tempting to speculate that host LDLs may support the growth of fatty acid auxotrophs of S. aureus during infection, providing a mechanism for the bypass of FASII inhibition in vivo. Consistent with this, spontaneous triclosan-resistant fatty acid auxotrophs have previously been isolated from clinical samples (24). Previous studies focused on the interactions between S. aureus and lipoprotein particles showed that LDLs defended against S. aureus infection by sequestering autoinducing peptide and phenol-soluble modulins (40–43, 70). Our studies expand on these findings by demonstrating that S. aureus is also capable of utilizing LDLs as a source of fatty acids.
Having established that S. aureus utilizes human LDLs as an exogenous fatty acid source, we turned our efforts to defining the molecular mechanism of this observation. Given that fatty acids of lipoprotein particles are esterified, we surveyed S. aureus lipase mutants for increased sensitivity to triclosan in the presence of LDLs. We observed that genetic inactivation of the lipase Geh reduced the ability of multiple strains of S. aureus to utilize LDLs as a source of exogenous fatty acids. Based on the decreased liberation of fatty acids from LDLs incubated with the geh lip mutant supernatant, we propose that the growth reduction of the geh lip mutant is due to an inability to liberate a sufficient quantity of fatty acids from the LDLs to overcome the effects of triclosan. Inactivation of geh does not ablate the ability of S. aureus to utilize LDL-derived fatty acids, indicating other potential mechanisms of fatty acid liberation from LDLs. Consistent with this, supernatants devoid of Geh and Lip still liberated fatty acids from human LDL, and a geh lip double mutant incorporated exogenous fatty acids into the cell membrane. Several hypothetical lipases encoded by the S. aureus genome remain uncharacterized and may also play a role in the liberation of fatty acids from lipoprotein particles (Table S5). Notably, purified bacterial lipases have been used in several atherosclerosis studies to liberate fatty acids from lipoprotein particles, and these bacterium-derived lipases have effects on lipoprotein particles similar to those of host lipases (71, 72). We explored the clinical relevance of our finding by evaluating the ability of lipase-positive and -negative clinical isolates of S. aureus to resist triclosan in the presence of lipoprotein particles. The loss of lipase activity in clinical isolates correlated with a reduced ability to use LDLs as an exogenous fatty acid source. The culmination of these studies supports the conclusion that S. aureus lipases liberate fatty acids from human LDLs.
FakA facilitates S. aureus incorporation of exogenous fatty acids into the membrane phospholipids (29). We reasoned that FakA is also necessary for LDLs to be used as an exogenous fatty acid source and found that the ΔfakA mutant is more sensitive to triclosan, when grown in the presence of exogenous fatty acids, than the WT. Moreover, genetic inactivation of fakA significantly impaired the ability of S. aureus to generate triclosan-resistant colonies on agar supplemented with free fatty acids. These results confirm that the molecular mechanism by which S. aureus utilizes exogenous fatty acids to resist triclosan treatment is FakA dependent. Additional support for this conclusion is based on the finding that fakA is necessary for LDL-dependent protection from triclosan. Together with the results obtained using geh and geh lip mutants, this finding implies that an active modification of human LDL-derived fatty acids is required for S. aureus triclosan resistance, reducing the possibility that protection occurs via a nonspecific triclosan-LDL interaction. A recent study using a murine systemic infection model with a fakA mutant found that this mutant is attenuated for colonization of the liver but not the kidneys (73). This finding is intriguing, considering our results demonstrating that FakA is necessary for the ability of S. aureus to utilize LDLs, as the biogenesis of host lipoprotein particles occurs predominantly in the liver. In total, our results demonstrate that S. aureus lipases liberate fatty acids from human LDLs, which are then integrated into membrane PG in a fakA-dependent fashion.
In summary, we demonstrate that S. aureus scavenges fatty acids from human LDLs. While the ability of S. aureus to acquire fatty acids from its environment was well understood prior to this work, the fatty acid reservoirs present in the host exploited by S. aureus remained uncertain. LDLs and other lipoprotein particles are particularly relevant sources, as they are highly abundant in the blood and other tissues that S. aureus infects (74). The inhibition of FASII for treatment of S. aureus is the focus of much research, but debate continues regarding its clinical potential. This is because S. aureus and many other Gram-positive pathogens compensate for the loss of fatty acid synthesis by importing exogenous fatty acids. While our work is limited to in vitro studies, our results indicate that LDLs could support two possible routes to increase FASII inhibitor tolerance by S. aureus: (i) LDLs can supply fatty acids to WT cells via Geh and FakA in the presence of triclosan, which allows for the continued production of membrane phospholipids, and (ii) LDLs support the growth of fatty acid auxotrophs which, in a fakA-dependent manner, are insensitive to the effects of FASII inhibitors. Thus, this work also demonstrates that FakA is at the center of both possible mechanisms of FASII resistance in S. aureus and could possibly be a target for small-molecule inhibitors for use in conjunction with FASII inhibitors.
MATERIALS AND METHODS
Reagents.
Most chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO), including triclosan (catalog number 72779), Tween 80 (catalog number P1454), palmitic acid (catalog number P5585), sodium oleate (catalog number O7501), sodium myristate (catalog number M8005), sodium chloride (catalog number S9625), and purified human LDL (catalog number L7914; lot number SLBT1781 at 6.73 mg of protein per ml, lot number SLBV0530 at 6.26 mg of protein per ml, lot number SLBW4532 at 6.37 mg of protein per ml, and lot number SLBV8079 at 6.3 mg of protein per ml). Ammonium sulfate was purchased from Thermo Fisher Scientific (catalog number BP212R; Waltham, MA). Egg yolk plasma separation was based on previously described protocols (75, 76). Chicken egg yolks were separated from the egg whites and rinsed twice using sterile phosphate-buffered saline (PBS). The egg yolk was transferred to filter paper, and the vitelline membrane was punctured using a sterile pipette tip. The contents of the vitelline membrane were drained into a sterile conical tube. The egg yolk was then mixed vigorously with an equal volume of sterile PBS. Centrifugation of diluted egg yolk was executed at 4°C for 1 h at 15,344 × g in a F14-6x250 rotor from Thermo Fisher Scientific (Waltham, MA). The plasma supernatant was removed, placed in a sterile test tube, and stored at 4°C. Egg yolk LDL isolation was performed as described by Moussa et al. (77). Briefly, the egg yolk was diluted in 2 volumes of 0.17 M NaCl and stirred for 1 h at 4°C. The diluted egg yolk was then centrifuged at 10°C for 45 min at 10,000 × g. The supernatant was then removed, placed in a sterile test tube, and mixed with 40% ammonium sulfate for 1 h at 4°C. The pH of the supernatant was adjusted to 8.7. The supernatant was then centrifuged at 4°C for 45 min at 10,000 × g. The resulting supernatant was dialyzed overnight in ultrapure water to remove the ammonium sulfate. Dialyzed supernatant was then centrifuged at 4°C for 45 min at 10,000 × g, and an LDL-rich floating residue was removed into a sterile tube and stored at 4°C.
Bacterial strains and growth conditions.
The strains used in this study are described in Table 2. AH1263 is derived from a methicillin-resistant human clinical isolate of S. aureus, community-acquired methicillin-resistant S. aureus (CA-MRSA) USA300 LAC (78). AH1263 ΔfakA is an in-frame deletion mutant previously described by Bose et al. (62). Newman is a laboratory-derived methicillin-sensitive human isolate of S. aureus (79). The prophage-repaired variant of Newman, TB4, has been described by Bae et al. (60). The aur (NE163), sspA (NE1506), geh (NE1775), and lip (NE338) transposon mutants were obtained from the Nebraska Transposon Mutant Library (80). The backcrossed strains used in the assays described here were produced via phage-mediated transduction (80). For the AH1263 geh lip double mutant, the erythromycin resistance cassette was replaced with a tetracycline resistance cassette using a previously described allelic replacement strategy (81). The tetracycline-resistant geh allele was transduced into AH1263 lip harboring an erythromycin resistance allele. PCR was used to confirm that the transposon insertion disrupted the correct genes. Cells were grown in tryptic soy broth (catalog number 211822; BD) or tryptone broth at 37°C. Tryptone broth consisted of 1% tryptone (catalog number 211705; BD), 0.8% NaCl, and ultrapure water; for agar, 1.5% agarose (catalog number 214010; BD) was added to the broth. Mannitol salt agar (catalog number 211407; BD) was prepared following the manufacturer's directions. Baird-Parker agar (catalog number R452342; Remel) was prepared by following the manufacturer's directions.
TABLE 2.
S. aureus strains used in this study
| Straina | Relevant characteristic(s) | Reference or source |
|---|---|---|
| MRSA USA300 strains | ||
| Wild type | AH1263 | 78 |
| fakA mutant | ΔfakA | 62 |
| aur mutant | aur::erm | This study |
| sspA mutant | sspA::erm | This study |
| geh mutant | geh::erm | This study |
| lip mutant | lip::erm | This study |
| geh lip mutant | geh::tet lip::erm | This study |
| AH1263 triclosan-resistant strains | ||
| Trial 1, isolate A (1A) | accB fatty acid auxotroph | This study |
| Trial 1, isolate B (1B) | Fatty acid auxotroph | This study |
| Trial 1, isolate C (1C) | accC fatty acid auxotroph | This study |
| Trial 2, isolate A (2A) | accA fatty acid auxotroph | This study |
| Trial 2, isolate B (2B) | Fatty acid auxotroph | This study |
| Trial 2, isolate C (2C) | accD fatty acid auxotroph | This study |
| Trial 3, isolate A (3A) | Fatty acid auxotroph | This study |
| Trial 3, isolate B (3B) | Fatty acid auxotroph | This study |
| Trial 3, isolate C (3C) | fadD accC fatty acid auxotroph | This study |
| MSSA NM strains | ||
| Wild type | Lipase negative | 79 |
| TB4 | Prophage repaired, lipase positive | 60 |
| TB4 geh mutant | geh::erm | This study |
| Cystic fibrosis isolates | ||
| CF:716 | Lipase positive | This study |
| CF:720 | Lipase negative | This study |
| CF:769 | Lipase negative | This study |
| CF:781 | Lipase negative | This study |
| CF:789 | Lipase positive | This study |
| CF:796 | Lipase positive | This study |
MRSA, methicillin-resistant S. aureus; MSSA, methicillin-sensitive S. aureus.
Growth curves for all strains, except for the fatty acid auxotrophs (see below), were performed by diluting overnight cultures 1:100 into fresh broth and growing the diluted cells to an optical density (OD) at 600 nm (OD600) of 0.5. Cells were then added to 96-well round-bottom plates at a final OD600 of 0.01. Growth was monitored as the optical density at 600 nm using an H1 BioTek plate reader set at 37°C with continuous, linear shaking.
Genomic DNA isolation and gene sequencing.
Fatty acid auxotrophs were grown overnight in TSB supplemented with EYP. Cells were pelleted, resuspended in buffer, and incubated with lysostaphin from ABMI (Lawrence, NY) to remove the cell wall. Genomic DNA was isolated using a Promega Wizard genomic DNA purification kit (Madison, WI) following the manufacturer's directions. Amplification was performed using New England BioLabs Phusion HF DNA polymerase (Ipswich, MA) following the manufacturer's directions. Primers for fabD and accA, accB, accC, and accD were previously described by Morvan et al. (20). The following primers were added to our study to increase sequencing coverage: AccA-R (5′-CAGAATTTTTATTAGAGCATGGAC-3′) and AccD-F (5′-CTTCTTGTAAATCCACATCATTT-3′). Sanger sequencing was performed by the Michigan State University Research Technology Support Facility (East Lansing, MI).
Isolation of S. aureus from cystic fibrosis patient sputum.
The collection of patient sputum samples was reviewed and approved by the Institutional Review Board of the Michigan State University Human Research Protection Program and performed according to NIH guidelines and U.S. federal law by Martha Mulks in the Department of Microbiology and Molecular Genetics at Michigan State University. Sputum samples are collected as part of the routine care of cystic fibrosis patients. The sputum samples were given a deidentification study number, mixed with an equal volume of 4% tryptone–40% glycerol freezing medium, and then archived at −80°C. Frozen sputum samples were streaked onto an S. aureus selective medium, mannitol salt agar. Isolated colonies of suspected S. aureus isolates were picked and streaked for a second time to mannitol salt agar and to the S. aureus selective medium Baird-Parker. Isolated colonies were lastly confirmed through positive Gram staining. These samples were characterized for lipase activity by spotting 5 μl of an overnight culture grown in TSB onto a TSA plate with or without 1% EYP and mannitol salt agar. The plates were incubated overnight at 37°C. On the following day, the colonies were inspected for the formation of a zone of clearing around the colony and compared to USA300 and Newman as controls.
Kirby-Bauer disk diffusion assay.
Overnight cultures were normalized to an OD600 of 0.5, and a sterile cotton applicator was used to plate a lawn of cells on agar plates consisting of 20 ml of TSA or 1% tryptone agar with or without fatty acid supplementation. A sterile 6-mm filter disk was impregnated with 10 μl of 1.7 mM triclosan and allowed to dry. The sterile disk was then placed on top of the agar plate containing the cells. The plates were incubated for ∼14 h at 37°C. The diameter of the zone of inhibition (in millimeters) was determined by measurement of the distance between the edges of the solid bacterial lawn using a digital caliper. The chemical fatty acid supplement consisted of 170 μM (each) palmitic acid, sodium oleate, and sodium myristate, as previously described by Morvan et al. (20). Additionally, 1% EYP was also used as a source of fatty acids.
IC50 assay.
Overnight cultures of WT S. aureus or the S. aureus fakA mutant were diluted 1:100 into 96-well flat-bottom plates containing fresh TSB supplemented with 0.1% Tween 80 or 0.1% chicken LDLs or not supplemented and increasing concentrations of triclosan. Cultures were grown statically for ∼16 h at 37°C. Cells were suspended by pipetting, and the optical density at 600 nm was measured. IC50s were calculated with the aid of the computer program Prism (version 6.07; GraphPad Software, La Jolla, CA).
Generation and maintenance of triclosan-resistant mutants.
Fatty acid-based triclosan-resistant mutants were generated by plating overnight cultures of WT strain AH1263 as a lawn on TSA with a fatty acid supplement (20). A 6-mm filter disk steeped with 10 μl of 1.7 mM triclosan was placed on top of the agar. The plates were incubated for 2 days at 37°C, and resistant colonies were counted each day. On day 2, colonies that grew in the zone of clearing were patched onto TSA or TSA containing the fatty acid supplement and incubated overnight at 37°C. To confirm that the triclosan-resistant mutants were fatty acid auxotrophs, the cells were patched onto TSA supplemented with fatty acids or not supplemented. Cells that grew on fatty acid-supplemented TSA were selected for further characterization. Fatty acid auxotrophs were maintained by streaking onto agar plates containing the fatty acid supplement or in broth cultures supplemented with 0.1% EYP. For growth assays, fatty acid auxotrophs were washed once with equal volumes of fresh TSB. Cells were then pelleted and resuspended in an equal volume of fresh TSB before being subcultured at 1:100 into a 96-well round-bottom plate containing TSB or TSB supplemented with LDLs.
Lipid extraction for mass spectrometry analysis of membrane phospholipid fatty acids of cells and culture supernatants.
The culturing of cells for mass spectrometry analysis of exogenous fatty acid incorporation into the plasma membrane of S. aureus was based on a previously described protocol (22). Overnight cultures of AH1263 and AH1263 geh lip in tryptone broth were subcultured at 1:100 into 10 ml of fresh tryptone broth. The cultures were grown to mid-log phase, pelleted, and resuspended into 300 μl of fresh tryptone broth with or without 5% human LDLs. Each treatment condition was performed in triplicate. The cultures were grown for an additional 4 h before the cells were pelleted and washed twice in 2 volumes of sterile PBS. Mass spectrometry analysis of cultures performed in TSB was completed as described above, except that all culturing was performed with TSB.
To measure the lipase activity of the culture supernatants on human LDLs, 5-ml cultures of AH1263 and AH1263 geh lip were grown overnight in tryptone broth. These cultures were then pelleted twice. Each time, the top half of the supernatant was then carefully removed. Supernatants were then incubated briefly with 15 μg/ml chloramphenicol. Next, 300 μl of chemically sterilized supernatants was transferred into fresh sterile tubes and 5% human LDLs were added to each tube. The tubes were then incubated at 37°C for 2 h. Each treatment condition was performed in triplicate.
Monophasic lipid extraction in methanol-chloroform-water (2:1:0.74) was performed as previously described (82) using 15 to 20 mg (wet weight) of freshly grown S. aureus. Prior to lipid extraction, each sample was spiked with 10 nmol of synthetic phosphatidylserine (PS; 14:0/14:0) obtained from Avanti Polar Lipids (Alabaster, AL) for relative lipid quantitation. Dried lipid extracts were washed three times with 10 mM ammonium bicarbonate, dried under vacuum, and resuspended in methanol containing 0.01% butylated hydroxytoluene. For each analysis, 3 μl of lipid extract was transferred to an Eppendorf Twin-Tec 96-well plate (Sigma-Aldrich, St. Louis, MO) and evaporated under nitrogen. The dried lipid film was then resuspended in 30 μl isopropanol-methanol-chloroform (4:2:1, vol/vol/vol) containing 20 mM ammonium formate and sealed with Teflon ultrathin sealing tape (Analytical Sales and Services, Pompton Plains, NJ).
High-resolution/accurate mass spectrometry and tandem mass spectrometry.
Untargeted lipidomic analysis utilized direct-infusion high-resolution/accurate mass spectrometry and tandem mass spectrometry. Samples were directly infused into the mass spectrometer by nanoelectrospray ionization (nESI) using a Triversa Nanomate nESI source (Advion, Ithaca, NY) with a spray voltage of 1.4 kV and a gas pressure of 0.3 lb/in2. The mass spectrometry platform was a Thermo Scientific LTQ-Orbitrap Velos system (San Jose, CA). High-resolution MS and MS/MS spectra were acquired in the negative ion mode using an Fourier transform analyzer operating at 100,000 mass resolving power. Ion mapping higher-energy collision induced dissociation (HCD-MS/MS) product ion spectra were acquired to confirm lipid head groups and the acyl chain compositions.
Peak and lipid identification and quantification.
Lipids were identified using the Lipid Mass Spectrum Analysis (LIMSA; version 1.0) software linear fit algorithm, in conjunction with an in-house database of hypothetical S. aureus and human-derived lipid compounds, for automated peak finding and correction of 13C isotope effects. Values were reported only for molecular species that exhibited a full-scan MS signal-to-noise ratio of at least 3 in all biological replicates of each control or treatment group. The S. aureus lipid classes and molecular species used for database searches were derived from the published literature (54, 83). Relative quantification of abundance between samples was performed by normalization of target lipid ion peak areas to the PS (14:0/14:0) internal standard as previously described (84). For this untargeted analysis, no attempt was made to correct for differences in lipid species ionization due to head group polarity or the length or degree of unsaturation of the esterified fatty acids. Therefore, lipid abundance values are reported only as ion abundances after normalization against the internal standard rather than as absolute values.
Statistical analyses.
Statistical significance was determined using Student's t test or a two-way analysis of variance (ANOVA) with a Tukey post hoc test for multiple comparisons and was performed with Prism software (version 6.07; GraphPad Software, La Jolla, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the N. D. Hammer laboratory for their critical evaluation of the manuscript and overall support of this work. Alex Horswill of the University of Colorado and Jeffrey L. Bose of the University of Kansas Medical Center kindly provided AH1263 and the ΔfakA mutant used in this study, respectively. Taeok Bae of Indiana University generously contributed the prophage-repaired variant of Newman, TB4. Undergraduate assistants Jesse Rizor and Alicia Webb of Michigan State University aided in the isolation and characterization of S. aureus from patient sputum samples. Peter C. Lucas of the University of Pittsburgh kindly provided technical advice. The Victor DiRita, Chris Waters, and Martha Mulks laboratories at Michigan State University provided reagents and technical assistance. We also thank the Michigan State University Poultry Teaching and Research Center for the donation of chicken eggs. Strains NE163, NE338, NE1506, and NE1775 were obtained through the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) for distribution by BEI Resources, NIAID, NIH: Nebraska Transposon Mutant Library (NTML) Screening Array, NR-48501.
This work was supported by American Heart Association grant 16SDG30170026 and start-up funds provide by Michigan State University.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00728-17.
REFERENCES
- 1.Laible BR. 2014. Antimicrobial resistance: CDC releases report prioritizing current threats. S D Med 67:30–31. [PubMed] [Google Scholar]
- 2.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 3.Hammer ND, Skaar EP. 2011. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu Rev Microbiol 65:129–147. doi: 10.1146/annurev-micro-090110-102851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsons JB, Rock CO. 2011. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery? Curr Opin Microbiol 14:544–549. doi: 10.1016/j.mib.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao J, Rock CO. 2015. How bacterial pathogens eat host lipids: implications for the development of fatty acid synthesis therapeutics. J Biol Chem 290:5940–5946. doi: 10.1074/jbc.R114.636241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang YM, Rock CO. 2008. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol 6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 7.Fan F, Yan K, Wallis NG, Reed S, Moore TD, Rittenhouse SF, DeWolf WE Jr, Huang J, McDevitt D, Miller WH, Seefeld MA, Newlander KA, Jakas DR, Head MS, Payne DJ. 2002. Defining and combating the mechanisms of triclosan resistance in clinical isolates of Staphylococcus aureus. Antimicrob Agents Chemother 46:3343–3347. doi: 10.1128/AAC.46.11.3343-3347.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asturias FJ, Chadick JZ, Cheung IK, Stark H, Witkowski A, Joshi AK, Smith S. 2005. Structure and molecular organization of mammalian fatty acid synthase. Nat Struct Mol Biol 12:225–232. doi: 10.1038/nsmb899. [DOI] [PubMed] [Google Scholar]
- 9.Zhang YM, White SW, Rock CO. 2006. Inhibiting bacterial fatty acid synthesis. J Biol Chem 281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- 10.Sohlenkamp C, Geiger O. 2016. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol Rev 40:133–159. doi: 10.1093/femsre/fuv008. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, Dorsey M, Hafkin B, Ramnauth J, Romanov V, Schmid MB, Thalakada R, Yethon J, Pauls HW. 2012. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob Agents Chemother 56:5865–5874. doi: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yum JH, Kim CK, Yong D, Lee K, Chong Y, Kim CM, Kim JM, Ro S, Cho JM. 2007. In vitro activities of CG400549, a novel FabI inhibitor, against recently isolated clinical staphylococcal strains in Korea. Antimicrob Agents Chemother 51:2591–2593. doi: 10.1128/AAC.01562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Escaich S, Prouvensier L, Saccomani M, Durant L, Oxoby M, Gerusz V, Moreau F, Vongsouthi V, Maher K, Morrissey I, Soulama-Mouze C. 2011. The MUT056399 inhibitor of FabI is a new antistaphylococcal compound. Antimicrob Agents Chemother 55:4692–4697. doi: 10.1128/AAC.01248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heath RJ, Li J, Roland GE, Rock CO. 2000. Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J Biol Chem 275:4654–4659. doi: 10.1074/jbc.275.7.4654. [DOI] [PubMed] [Google Scholar]
- 15.Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. 1998. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J Biol Chem 273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 16.Miller WH, Seefeld MA, Newlander KA, Uzinskas IN, Burgess WJ, Heerding DA, Yuan CC, Head MS, Payne DJ, Rittenhouse SF, Moore TD, Pearson SC, Berry V, DeWolf WE Jr, Keller PM, Polizzi BJ, Qiu X, Janson CA, Huffman WF. 2002. Discovery of aminopyridine-based inhibitors of bacterial enoyl-ACP reductase (FabI). J Med Chem 45:3246–3256. doi: 10.1021/jm020050+. [DOI] [PubMed] [Google Scholar]
- 17.Payne DJ, Miller WH, Berry V, Brosky J, Burgess WJ, Chen E, DeWolf WE Jr, Fosberry AP, Greenwood R, Head MS, Heerding DA, Janson CA, Jaworski DD, Keller PM, Manley PJ, Moore TD, Newlander KA, Pearson S, Polizzi BJ, Qiu X, Rittenhouse SF, Slater-Radosti C, Salyers KL, Seefeld MA, Smyth MG, Takata DT, Uzinskas IN, Vaidya K, Wallis NG, Winram SB, Yuan CC, Huffman WF. 2002. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob Agents Chemother 46:3118–3124. doi: 10.1128/AAC.46.10.3118-3124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplan N, Hafkin B. 2014. Preclinical pharmacokinetics and efficacy of Debio 1450 (previously AFN-1720), a prodrug of the staphylococcal-specific antibiotic Debio 1452 (previously AFN-1252), poster P1717. Abstr ECCMID 2014, Barcelona, Spain. [Google Scholar]
- 19.Takhi M, Sreenivas K, Reddy CK, Munikumar M, Praveena K, Sudheer P, Rao BN, Ramakanth G, Sivaranjani J, Mulik S, Reddy YR, Narasimha Rao K, Pallavi R, Lakshminarasimhan A, Panigrahi SK, Antony T, Abdullah I, Lee YK, Ramachandra M, Yusof R, Rahman NA, Subramanya H. 2014. Discovery of azetidine based ene-amides as potent bacterial enoyl ACP reductase (FabI) inhibitors. Eur J Med Chem 84:382–394. doi: 10.1016/j.ejmech.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 20.Morvan C, Halpern D, Kenanian G, Hays C, Anba-Mondoloni J, Brinster S, Kennedy S, Trieu-Cuot P, Poyart C, Lamberet G, Gloux K, Gruss A. 2016. Environmental fatty acids enable emergence of infectious Staphylococcus aureus resistant to FASII-targeted antimicrobials. Nat Commun 7:12944. doi: 10.1038/ncomms12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. 2009. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458:83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 22.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. 2011. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc Natl Acad Sci U S A 108:15378–15383. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons JB, Frank MW, Rosch JW, Rock CO. 2013. Staphylococcus aureus fatty acid auxotrophs do not proliferate in mice. Antimicrob Agents Chemother 57:5729–5732. doi: 10.1128/AAC.01038-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gloux K, Guillemet M, Soler C, Morvan C, Halpern D, Pourcel C, Vu Thien H, Lamberet G, Gruss A. 2017. Clinical relevance of type II fatty acid synthesis bypass in Staphylococcus aureus. Antimicrob Agents Chemother 61:e02515-. doi: 10.1128/AAC.02515-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altenbern RA. 1977. Cerulenin-inhibited cells of Staphylococcus aureus resume growth when supplemented with either a saturated or an unsaturated fatty acid. Antimicrob Agents Chemother 11:574–576. doi: 10.1128/AAC.11.3.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balemans W, Lounis N, Gilissen R, Guillemont J, Simmen K, Andries K, Koul A. 2010. Essentiality of FASII pathway for Staphylococcus aureus. Nature 463:E3. doi: 10.1038/nature08667. [DOI] [PubMed] [Google Scholar]
- 27.Parsons JB, Frank MW, Jackson P, Subramanian C, Rock CO. 2014. Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol Microbiol 92:234–245. doi: 10.1111/mmi.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broussard TC, Miller DJ, Jackson P, Nourse A, White SW, Rock CO. 2016. Biochemical roles for conserved residues in the bacterial fatty acid-binding protein family. J Biol Chem 291:6292–6303. doi: 10.1074/jbc.M115.706820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parsons JB, Broussard TC, Bose JL, Rosch JW, Jackson P, Subramanian C, Rock CO. 2014. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc Natl Acad Sci U S A 111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopf T, Schmitz G. 2013. Analysis of non-esterified fatty acids in human samples by solid-phase-extraction and gas chromatography/mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 938:22–26. doi: 10.1016/j.jchromb.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 31.Gordon RS, Cherkes A. 1956. Unesterified fatty acid in human blood plasma. J Clin Invest 35:206–212. doi: 10.1172/JCI103265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fredrickson DS, Gordon RS. 1958. Transport of fatty acids. Physiol Rev 38:585–630. doi: 10.1152/physrev.1958.38.4.585. [DOI] [PubMed] [Google Scholar]
- 33.Feingold KR, Grunfeld C. 2000. Introduction to lipids and lipoproteins. In De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A (ed), Endotext https://www.ncbi.nlm.nih.gov/books/NBK305896/. Last updated 10 June 2015.
- 34.Goldberg IJ. 1996. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J Lipid Res 37:693–707. [PubMed] [Google Scholar]
- 35.Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. 1999. A novel endothelial-derived lipase that modulates HDL metabolism. Nat Genet 21:424–428. doi: 10.1038/7766. [DOI] [PubMed] [Google Scholar]
- 36.He PP, Jiang T, OuYang XP, Liang YQ, Zou JQ, Wang Y, Shen QQ, Liao L, Zheng XL. 2018. Lipoprotein lipase: biosynthesis, regulatory factors, and its role in atherosclerosis and other diseases. Clin Chim Acta 480:126–137. doi: 10.1016/j.cca.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 37.Gotz F, Verheij HM, Rosenstein R. 1998. Staphylococcal lipases: molecular characterisation, secretion, and processing. Chem Phys Lipids 93:15–25. doi: 10.1016/S0009-3084(98)00025-5. [DOI] [PubMed] [Google Scholar]
- 38.Cadieux B, Vijayakumaran V, Bernards MA, McGavin MJ, Heinrichs DE. 2014. Role of lipase from community-associated methicillin-resistant Staphylococcus aureus strain USA300 in hydrolyzing triglycerides into growth-inhibitory free fatty acids. J Bacteriol 196:4044–4056. doi: 10.1128/JB.02044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhakdi S, Tranum-Jensen J, Utermann G, Fussle R. 1983. Binding and partial inactivation of Staphylococcus aureus alpha-toxin by human plasma low density lipoprotein. J Biol Chem 258:5899–5904. [PubMed] [Google Scholar]
- 40.Manifold-Wheeler BC, Elmore BO, Triplett KD, Castleman MJ, Otto M, Hall PR. 2016. Serum lipoproteins are critical for pulmonary innate defense against Staphylococcus aureus quorum sensing. J Immunol 196:328–335. doi: 10.4049/jimmunol.1501835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elmore BO, Triplett KD, Hall PR. 2015. Apolipoprotein B48, the structural component of chylomicrons, is sufficient to antagonize Staphylococcus aureus quorum-sensing. PLoS One 10:e0125027. doi: 10.1371/journal.pone.0125027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall PR, Elmore BO, Spang CH, Alexander SM, Manifold-Wheeler BC, Castleman MJ, Daly SM, Peterson MM, Sully EK, Femling JK, Otto M, Horswill AR, Timmins GS, Gresham HD. 2013. Nox2 modification of LDL is essential for optimal apolipoprotein B-mediated control of agr type III Staphylococcus aureus quorum-sensing. PLoS Pathog 9:e1003166. doi: 10.1371/journal.ppat.1003166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peterson MM, Mack JL, Hall PR, Alsup AA, Alexander SM, Sully EK, Sawires YS, Cheung AL, Otto M, Gresham HD. 2008. Apolipoprotein B is an innate barrier against invasive Staphylococcus aureus infection. Cell Host Microbe 4:555–566. doi: 10.1016/j.chom.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. 2010. Essentiality of FASII pathway for Staphylococcus aureus reply. Nature 463:E4–E5. doi: 10.1038/nature08668. [DOI] [PubMed] [Google Scholar]
- 45.McCully KA, Common RH, Mok CC. 1962. Paper electrophoretic characterization of proteins and lipoproteins of hens egg yolk. Can J Biochem Physiol 40:937. [Google Scholar]
- 46.Vieira PM, Vieira AV, Sanders EJ, Steyrer E, Nimpf J, Schneider WJ. 1995. Chicken yolk contains bona fide high density lipoprotein particles. J Lipid Res 36:601–610. [PubMed] [Google Scholar]
- 47.Anton M, Martinet V, Dalgalarrondo M, Beaurnal V, David-Briand E, Rabesona H. 2003. Chemical and structural characterisation of low-density lipoproteins purified from hen egg yolk. Food Chem 83:175–183. doi: 10.1016/S0308-8146(03)00060-8. [DOI] [Google Scholar]
- 48.Parkinson TL. 1966. The chemical composition of eggs. J Sci Food Agric 17:101–111. doi: 10.1002/jsfa.2740170301. [DOI] [PubMed] [Google Scholar]
- 49.Hillyard LA, White HM, Pangburn SA. 1972. Characterization of apolipoproteins in chicken serum and egg yolk. Biochemistry 11:511–518. doi: 10.1021/bi00754a005. [DOI] [PubMed] [Google Scholar]
- 50.Anton M, Gandemer G. 1997. Composition, solubility and emulsifying properties of granules and plasma of egg yolk. J Food Sci 62:484–487. doi: 10.1111/j.1365-2621.1997.tb04411.x. [DOI] [Google Scholar]
- 51.Burley RW, Cook WH. 1961. Isolation and composition of avian egg yolk granules and their constituent alpha- and beta-lipovitellins. Can J Biochem Physiol 39:1295. doi: 10.1139/o61-136. [DOI] [PubMed] [Google Scholar]
- 52.Suller MT, Russell AD. 2000. Triclosan and antibiotic resistance in Staphylococcus aureus. J Antimicrob Chemother 46:11–18. [DOI] [PubMed] [Google Scholar]
- 53.Parsons JB, Yao J, Frank MW, Jackson P, Rock CO. 2012. Membrane disruption by antimicrobial fatty acids releases low-molecular-weight proteins from Staphylococcus aureus. J Bacteriol 194:5294–5304. doi: 10.1128/JB.00743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hewelt-Belka W, Nakonieczna J, Belka M, Baczek T, Namiesnik J, Kot-Wasik A. 2016. Untargeted lipidomics reveals differences in the lipid pattern among clinical isolates of Staphylococcus aureus resistant and sensitive to antibiotics. J Proteome Res 15:914–922. doi: 10.1021/acs.jproteome.5b00915. [DOI] [PubMed] [Google Scholar]
- 55.Sen S, Sirobhushanam S, Johnson SR, Song Y, Tefft R, Gatto C, Wilkinson BJ. 2016. Growth-environment dependent modulation of Staphylococcus aureus branched-chain to straight-chain fatty acid ratio and incorporation of unsaturated fatty acids. PLoS One 11:e0165300. doi: 10.1371/journal.pone.0165300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonanome A, Pagnan A, Biffanti S, Opportuno A, Sorgato F, Dorella M, Maiorino M, Ursini F. 1992. Effect of dietary monounsaturated and polyunsaturated fatty acids on the susceptibility of plasma low density lipoproteins to oxidative modification. Arterioscler Thromb 12:529–533. doi: 10.1161/01.ATV.12.4.529. [DOI] [PubMed] [Google Scholar]
- 57.Shepherd J, Packard CJ, Grundy SM, Yeshurun D, Gotto AM Jr, Taunton OD. 1980. Effects of saturated and polyunsaturated fat diets on the chemical composition and metabolism of low density lipoproteins in man. J Lipid Res 21:91–99. [PubMed] [Google Scholar]
- 58.Azimova SS, Glushenkova AI. 2012. Glycine hispida Moench. (Maxima) (Soya glycine L.), p 580–582. In Azimova SS, Glushenkova AI (ed), Lipids, lipophilic components and essential oils from plant sources. Springer London, London, United Kingdom. [Google Scholar]
- 59.Tattrie NH, Veliky IA. 1973. Fatty-acid composition of lipids in various plant-cell cultures. Can J Bot 51:513–516. [Google Scholar]
- 60.Bae T, Baba T, Hiramatsu K, Schneewind O. 2006. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol Microbiol 62:1035–1047. doi: 10.1111/j.1365-2958.2006.05441.x. [DOI] [PubMed] [Google Scholar]
- 61.Parsons JB, Frank MW, Eleveld MJ, Schalkwijk J, Broussard TC, de Jonge MI, Rock CO. 2015. A thioesterase bypasses the requirement for exogenous fatty acids in the plsX deletion of Streptococcus pneumoniae. Mol Microbiol 96:28–41. doi: 10.1111/mmi.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bose JL, Daly SM, Hall PR, Bayles KW. 2014. Identification of the Staphylococcus aureus vfrAB operon, a novel virulence factor regulatory locus. Infect Immun 82:1813–1822. doi: 10.1128/IAI.01655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu K. 1997. Chemistry and nutritional value of soybean components, soybeans. Springer, Boston, MA. [Google Scholar]
- 64.Dragan K, Caron K, Ian TC, Ben C, David TM. 2014. Bacterial fatty acid synthesis: effect of Tween 80 on antibiotic potency against Streptococcus agalactiae and methicillin-resistant Staphylococcus aureus. Anti-Infective Agents 12:80–84. doi: 10.2174/22113525113119990116. [DOI] [Google Scholar]
- 65.Lowy FD. 2003. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 111:1265–1273. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuchinskiene Z, Carlson LA. 1982. Composition, concentration, and size of low density lipoproteins and of subfractions of very low density lipoproteins from serum of normal men and women. J Lipid Res 23:762–769. [PubMed] [Google Scholar]
- 67.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. 2006. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 68.Wang J, Kodali S, Lee SH, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L, Tinney E, Colletti SL, Herath K, Cummings R, Salazar O, Gonzalez I, Basilio A, Vicente F, Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully DF, Singh SB. 2007. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc Natl Acad Sci U S A 104:7612–7616. doi: 10.1073/pnas.0700746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lizenko MV, Regerand TI, Bakhirev AM, Lizenko EI. 2008. Comparative biochemical analysis of blood serum lipoproteins from human and various animal species. Zh Evol Biokhim Fiziol 44:492–500. (In Russian.) [PubMed] [Google Scholar]
- 70.Surewaard BG, Nijland R, Spaan AN, Kruijtzer JA, de Haas CJ, van Strijp JA. 2012. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog 8:e1002606. doi: 10.1371/journal.ppat.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beisiegel U, Weber W, Bengtsson-Olivecrona G. 1991. Lipoprotein lipase enhances the binding of chylomicrons to low density lipoprotein receptor-related protein. Proc Natl Acad Sci U S A 88:8342–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skottova N, Savonen R, Lookene A, Hultin M, Olivecrona G. 1995. Lipoprotein lipase enhances removal of chylomicrons and chylomicron remnants by the perfused rat liver. J Lipid Res 36:1334–1344. [PubMed] [Google Scholar]
- 73.Lopez MS, Tan IS, Yan D, Kang J, McCreary M, Modrusan Z, Austin CD, Xu M, Brown EJ. 2017. Host-derived fatty acids activate type VII secretion in Staphylococcus aureus. Proc Natl Acad Sci U S A 114:11223–11228. doi: 10.1073/pnas.1700627114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rasmussen RV, Fowler VG, Skov R, Bruun NE. 2011. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol 6:43–56. doi: 10.2217/fmb.10.155 (Erratum, 6:250, doi:.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guilmineau F, Krause I, Kulozik U. 2005. Efficient analysis of egg yolk proteins and their thermal sensitivity using sodium dodecyl sulfate polyacrylamide gel electrophoresis under reducing and nonreducing conditions. J Agric Food Chem 53:9329–9336. doi: 10.1021/jf050475f. [DOI] [PubMed] [Google Scholar]
- 76.Mcbee LE, Cotterill OJ. 1979. Ion-exchange chromatography and electrophoresis of egg-yolk proteins. J Food Sci 44:656–660. doi: 10.1111/j.1365-2621.1979.tb08469.x. [DOI] [Google Scholar]
- 77.Moussa M, Marinet V, Trimeche A, Tainturier D, Anton M. 2002. Low density lipoproteins extracted from hen egg yolk by an easy method: cryoprotective effect on frozen-thawed bull semen. Theriogenology 57:1695–1706. doi: 10.1016/S0093-691X(02)00682-9. [DOI] [PubMed] [Google Scholar]
- 78.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duthie ES. 1952. Variation in the antigenic composition of staphylococcal coagulase. J Gen Microbiol 7:320–326. doi: 10.1099/00221287-7-3-4-320. [DOI] [PubMed] [Google Scholar]
- 80.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bose JL, Fey PD, Bayles KW. 2013. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl Environ Microbiol 79:2218–2224. doi: 10.1128/AEM.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lydic TA, Busik JV, Reid GE. 2014. A monophasic extraction strategy for the simultaneous lipidome analysis of polar and nonpolar retina lipids. J Lipid Res 55:1797–1809. doi: 10.1194/jlr.D050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hewelt-Belka W, Nakonieczna J, Belka M, Baczek T, Namiesnik J, Kot-Wasik A. 2014. Comprehensive methodology for Staphylococcus aureus lipidomics by liquid chromatography and quadrupole time-of-flight mass spectrometry. J Chromatogr A 1362:62–74. doi: 10.1016/j.chroma.2014.08.020. [DOI] [PubMed] [Google Scholar]
- 84.Lydic TA, Townsend S, Adda CG, Collins C, Mathivanan S, Reid GE. 2015. Rapid and comprehensive ‘shotgun’ lipidome profiling of colorectal cancer cell derived exosomes. Methods 87:83–95. doi: 10.1016/j.ymeth.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liebisch G, Vizcaíno JA, Köfeler H, Trötzmüller M, Griffiths WJ, Schmitz G, Spener F, Wakelam MJO. 2013. Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res 54:1523–1530. doi: 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.