

Abstract
Natural products are characterized by extreme structural diversity and thus they offer a unique source for the identification of novel anti-tumor agents. Herein, we report that the herbal substance acteoside being isolated by advanced phytochemical methods from Lippia citriodora leaves showed enhanced cytotoxicity against metastatic tumor cells; acted in synergy with various cytotoxic agents and it sensitized chemoresistant cancer cells. Acteoside was not toxic in physiological cellular contexts, while it increased oxidative load, affected the activity of proteostatic modules and suppressed matrix metalloproteinases in tumor cell lines. Intraperitoneal or oral (via drinking water) administration of acteoside in a melanoma mouse model upregulated antioxidant responses in the tumors; yet, only intraperitoneal delivery suppressed tumor growth and induced anti-tumor-reactive immune responses. Mass-spectrometry identification/quantitation analyses revealed that intraperitoneal delivery of acteoside resulted in significantly higher, vs. oral administration, concentration of the compound in the plasma and tumors of treated mice, suggesting that its in vivo anti-tumor effect depends on the route of administration and the achieved concentration in the tumor. Finally, molecular modeling studies and enzymatic activity assays showed that acteoside inhibits protein kinase C. Conclusively, acteoside holds promise as a chemical scaffold for the development of novel anti-tumor agents.
Abbreviations: ALP, Autophagy Lysosome Pathway; LC-MS, Liquid Chromatography Mass Spectrometry; NMR, Nuclear Magnetic Resonance; Nrf-2, NF-E2-related factor 2; PKC, Protein Kinase C; PDR, Proteome Damage Responses; PN, Proteostasis Network; ROS, Reactive Oxygen Species; STAT, Signal Transducer And Activator Of Transcription; UPP, Ubiquitin Proteasome Pathway
Keywords: Acteoside, Cancer, Natural compound, Oxidative stress, Proteostasis, Immunomodulation
Graphical abstract
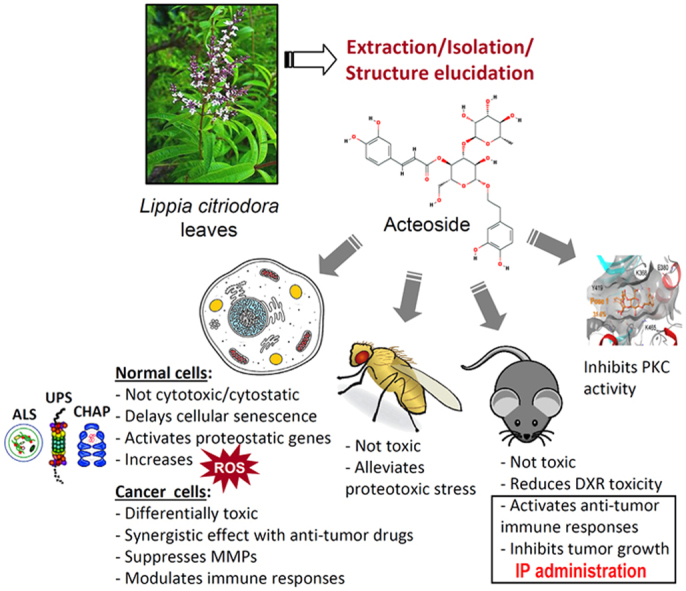
Highlights
-
•
Acteoside was not toxic in physiological cellular or tissue contexts.
-
•
This natural compound modulated antioxidant responses and proteostatic modules.
-
•
Acteoside showed in vitro and in vivo selective cytotoxicity against tumor cells.
-
•
IP administration of acteoside in a mouse tumor model activated immune responses.
-
•
Acteoside inhibited Protein Kinase C.
1. Introduction
Carcinogenesis is characterized by the deregulation of several cell signaling pathways and is associated with increased cellular oxidative, replicative, metabolic, genotoxic and proteotoxic stress [1]. In order to adapt and overcome these stress phenotypes malignant cells upregulate (apart from oncogenes) a number of non-oncogenic pathways aiming to either suppress or (at least) alleviate on-going stress. Among the various non-oncogenic pathways found to be frequently upregulated during tumorigenesis are the modules of the proteostasis network (PN) along with cellular antioxidant responses [2], [3].
Key components of the PN are the two main degradation machineries, namely the Autophagy- Lysosome pathway (ALP) and the Ubiquitin-Proteasome pathway (UPP), along with the network of the cellular molecular chaperones. ALP is mainly involved in the degradation of protein aggregates and damaged organelles [4], while UPP degrades normal short-lived proteins and non-repairable misfolded or unfolded proteins [5], [6], [7], [8]. The 26Seukaryotic proteasome is constituted from the 19S regulatory particles (RP) and the 20 S core particle (CP); the latter is comprised of four stacked heptameric rings (two of α-type surrounding two of β-type) that form a barrel-like structure. The chymotrypsin-like (CT-L), trypsin-like (T-L) and caspase-like (C-L) proteasome peptidase activities are located at the β5, β2, and β1 proteasomal subunits, respectively [6]. The demonstrated clinical efficacy of the proteasome inhibitors Bortezomib (Velcade®; also named PS-341) and Carfilzomib against various hematologic malignancies [9] has provided the “proof-of-concept” of targeting UPP as a promising strategy for the treatment of cancer.
The complex network of cellular antioxidant defense pathways that is frequently upregulated during carcinogenesis [3], [10] includes antioxidant enzymes, as well as several transcription factors that mobilize cytoprotective genomic responses; these include forkhead box O (Foxo) and the nuclear factor erythroid 2-related factor (Nrf-2). Nrf-2 is central in the protection of cells against oxidative and/or xenobiotic damage as it stimulates the expression of antioxidant and phase II genes [11], [12], [13].
We and others have recently proposed that targeting these tumor dependencies (i.e., proteostatic modules and/or antioxidant response pathways) or increasing cellular levels of ROS in the context of a transformed genotype offer a potential therapeutic window for the selective killing of tumor cells [3], [14]; in support, selective killing of cancer cells by a small molecule that upregulated cellular ROS levels has been reported [2]. It is expected that combinatorial approaches including also the activation of anti-tumor-reactive immune responses [15] will likely maximize the therapeutic window offered by inhibiting PN and antioxidant responses and/or by elevating cellular ROS levels.
Natural products (extracts or pure compounds) (NPs) from various sources (plants, marine organisms, microorganisms, etc) are screened for their ability to act as anti-tumor agents due to their extreme structural diversity and chemical complexity, as well as because they act pleiotropically modulating several cellular signaling pathways [16]. Reportedly, NPs activate anti-inflammatory, anti-tumor and/or anti-metastatic responses [16], [17] and also evade multidrug resistance [18], [19]. Among these, phenolic compounds (including phenylethanoid glycosides) have attracted significant interest because of their reported role in the prevention and/or treatment of various human diseases.
Acteoside, also called kusagin or verbascoside [20], is a phenylethanoid glycoside isolated from many dicotyledons. Reportedly, acteoside exerts some interesting biological activities, including antioxidant, anti-inflammatory, and cell apoptosis regulating properties [21], [22]. Nevertheless, its potential anti-tumor activity has not been addressed. We report here, that acteoside showed increased cytotoxicity against mammalian cancer cells with no apparent toxic effects in physiological cellular contexts. Furthermore, acteoside suppressed melanoma tumor growth in an in vivo mouse model, by (among others) activating anti-tumor-reactive immune responses.
2. Materials and methods
2.1. Plant material and extraction
Dried Lippia citriodora (Lamiaceae) leaves (4.5 kg) were purchased from the local market in Athens, Greece. The leaves were pulverized and extracted by mechanical stirring for 12 h with methanol (2 × 20 L). The methanolic extract was evaporated to dryness and washed with a mixture of CH2Cl2/MeOH 98/2 (15 L). The insoluble residue was separated and dried, producing a green-yellow powder (450 g).
2.2. Purification of acteoside and UPLC-HRMS analysis
A portion (10 g) of the aforementioned residue was subjected to countercurrent chromatography using a fast centrifugal partition chromatograph (FCPC) apparatus (Kromaton, France); a mixture of EtOAc/EtOH/H2O at ratio 5/0.5/4.5 was used as biphasic solvent system. Collected fractions were subjected to Thin Layer Chromatography; then the chromatograms were observed under a UV lamp (254 and 365 nm) and visualized by spraying with methanol vanillin sulfate followed by heating for two minutes. A total of 2.1 g of acteoside (purity ≥ 90%) was isolated by the aforementioned process. The identification of acteoside was performed by nuclear magnetic resonance (NMR) and mass spectrometry (MS) spectra, while its purity was established by UPLC-MS and NMR analysis; for details see Suppl. Materials and Methods.
2.3. Cell lines
Human lung embryonic fibroblasts (IMR90 cells) along with the B16.F1, B16.F10, YAC-1 and WEHI-164 mouse cell lines were obtained from the American Tissue Culture Collection (ATCC). The U2 OS and Sa OS human osteosarcoma cell lines were kindly donated by Prof. V. Gorgoulis (School of Medicine, National and Kapodistrian University of Athens, Greece), while the KH OS osteosarcoma cells and the chemoresistant osteosarcoma cell lines [23] were a donation of Dr. E. Gonos (National Hellenic Research Foundation, Greece). The mouse cancer cell lines C5N and A5 belong to a multistage mouse skin carcinogenesis model [24], [25] and were donated by Prof. A. Balmein (Comprehensive Cancer Center, University of California, USA). Culturing conditions of the used cell lines are reported in Suppl. Materials and Methods.
2.4. Melanoma mouse model
Male C57BL/6 mice (25–30 g of weight, 6–8 weeks of age) were obtained from the Hellenic Pasteur Institute and housed under controlled temperature (22 °C) and photoperiod (12 h light:12 h dark) with free access to water and food. Mice were subcutaneously inoculated with 105 B16.F1 melanoma cells (in 100 μL PBS) and were randomly assigned to 3 groups (n = 5/group). When tumors became palpable (day 11) mice received acteoside via two routes; either intraperitoneally (IP) (1 mg/mouse diluted in 200 μL PBS; in total 6 doses administered every other day) or orally by drinking water (OR) (2.5 mg/mouse; in total 13 doses for 13 consecutive days). Control mice were administered PBS. Tumor growth was recorded every 2 days by measuring the major and minor axes of the formed tumors with a digital caliper. Measurements were transformed into tumor volume using the formula: tumor volume (cm3) = major axis × minor axis2 × 0.5. On day 28, animals were euthanized by cervical dislocation and spleens were aseptically removed. The experiment was repeated three times with similar findings.
Splenocytes were isolated from individually homogenized spleens and immediately tested for their cytotoxicity vs. B16.F1, YAC-1 and WEHI-164 cell targets. Cytotoxicity was evaluated based on the detection of CD107 exposure on cell surface, as a result of effector cell degranulation. Splenocytes (105 cells/well) were co-cultured with targets in 96-well U bottom microplates at an effector to target (E:T) ratio of 100:1, at 37 °C in 5% CO2. FITC-conjugated anti-CD107a and anti-CD107b monoclonal antibodies (25 μL/mL) and monensin (6 μL/mL; all from BD Biosciences) were added in each well. Cells were harvested 6 h later and analyzed using a FACSCanto II flow cytometer. In parallel, tumors were excised and processed for downstream assays as described in Suppl. Materials and Methods.
2.5. Preparation of cell or tissue protein extracts
Cell protein extracts were prepared as described previously [26], [27]. Tumor biopsies were homogenized on ice in NP-40 lysis buffer containing protease inhibitors and centrifuged for 10 min at 19, 000 ×g (4 °C). The protein content of cell or tissue lysates was adjusted by Bradford assay (Bio-Rad), and was analyzed by SDS-PAGE and immunoblotting as described previously [27], [28].
Full Materials and Methods, description of Statistical analyses and any associated References are available in Supplementary Information.
3. Results
3.1. Acteoside is increasingly toxic in mouse and human cancer cells
Initially, we sought to identify a cell model suitable for the evaluation of new anti-tumor agents. The multistage mouse skin carcinogenesis model was generated through chemical carcinogenesis [24]. Various cell lines were isolated; among which C5N cells represent an immortalized non-tumorigenic cell line, whereas the A5 cell line represents metastatic spindle carcinoma expressing mutated p53 and H-Ras proteins, as well as increased levels of members of the MAPK and AP-1 pathways and of MMP-2 and MMP-9 [25]. We found that, compared to C5N cells, malignant A5 cells are characterized by increased (~3 fold) endogenous production of ROS (Suppl. Fig. S1A), as well as by upregulation of antioxidant responses (e.g. nrf2 and its transcriptional targets txnrd1 and nqo1); of proteostatic modules (e.g. the proteasomal β5 and the autophagy-related lc3β, ctsL and hdac6) and of the foxo1 and foxo3 genes (Suppl. Fig. S1B). We also noted increased proteasomal (Suppl. Fig. S1C1) but not cathepsin B, L (Suppl. Fig. S1C2) enzymatic activities; lower levels of spontaneous apoptosis (Suppl. Fig. S1D) and higher sensitivity of A5 (as compared to C5N) cells to chemotherapeutic drugs (doxorubicin), oxidants (H2O2), lysosomal (chloroquinone) and proteasome (PS-341) inhibitors (Suppl. Fig. S1E). We also studied the basal phosphorylation pattern of proteins (see, Suppl. Table S1) involved in tumorigenesis and cellular immune responses in the A5 vs. C5N cells by using the xMAP phosphoproteomics technology. We found that A5 cells tend tohave higher phosphorylation levels of Akt, RS6 and Wnk1 (target of Akt) proteins, whereas the phosphorylation levels of Creb1, Hspb1 (hsp27), Ptn11 (Shp2) and of the immune response-related Signal Transducer and Activator of Transcription 3 (Stat3) and Stat6 proteins (Suppl. Fig. S2) tended to decline; these findings indicate that likely the functionality of pathways involved in Stats regulation has been compromised in A5 cells. Thus, A5 cells apart from alterations in oncogenic pathways are characterized by increased oxidative stress and expression levels of antioxidant/proteostatic modules.
Given our previous findings [29], we then asked whether acteoside would display differential toxicity in A5 vs. C5N cells. For acteoside (Fig. 1A1) isolation, the residue obtained from the methanolic extract of L. citriodora leaves (after removal of the non-polar compounds) was analyzed by NMR and LC-MS techniques. The 1H NMR (Suppl. Fig. S3A) and LC-HRMS (Suppl. Fig. S3B) analyses indicated that acteoside is the major component of this mixture and its content was estimated at almost 25%. Subsequently, acteoside was isolated by normal phase dual mode elution CPC methodology using EtOAc/EtOH/H2O at ratio 5/0.5/4.5 as biphasic solvent system; 10 g of crude extract resulted to the purification of 2.1 g acteoside. The identification of acteoside was verified by NMR (Suppl. Fig. S3C1) and MS (Suppl. Fig. S3C2), and its purity was estimated at ≥ 90%.
Fig. 1.

Acteoside is increasingly cytotoxic against metastatic cancer cellsvs. normal cells. (A1) Chemical structure of acteoside. (A2) Relative (%) cell survival of C5N and A5 cells after treatment with increasing concentrations of acteoside for 24 h. (B) Flow cytometry analysis of ROS levels in C5N and A5 cells after treatment with 500 μM acteoside for 24 h; values refer to mean (n = 3) fluorescence intensity. (C) Relative mRNA expression levels of genes involved in different proteostatic modules, antioxidant responses and metabolic pathways after treatment of C5N and A5 cells with 500 μM acteoside for 24 h. (D) Relative (%) levels of the CT-L/β5, C-L/β1 and T-L/β2 proteasomal peptidase (D1) and lysosomal cathepsin B, L (D2) activities in C5N and A5 cells following treatment (+ ) or not (-) with 500 μM acteoside for 24 h. (E) Gelatin zymography in A5 metastatic cancer cells showing MMP-9 and MMP-2 enzymatic activities after treatment (+ ) or not (-) with 500 μM acteoside for 24 h. Bars, ± SD (n ≥ 2). * , P < 0.05; **, P < 0.01.
Downstream assays in order to investigate the effect(s) of acteoside to C5N and A5 cells showed that acteoside was increasingly toxic in A5 cells compared to C5N cells (Fig. 1A2) and increased cellular ROS levels in both A5 and C5N cells (Fig. 1B); nevertheless, the genomic responses in A5 cells after acteoside treatment were milder as compared to C5N cells (e.g. genes beclin, hdac6, ctsL, keap1, txnrd1, nqo1, clu, foxo1 and foxo3) (Fig. 1C) indicating that for these genes the expression levels reached in A5 cells during tumorigenesis are likely close to saturation in terms of their affordable upregulation. In support, and despite increased ROS levels (Fig. 1B), acteoside did not enhance the main proteasomal activity CT-L/β5 in A5 cells (Fig. 1D1) while it slightly increased the lysosomal cathepsins B, L activity (Fig. 1D2). In relation to phosphoproteome alterations, cell exposure to acteoside tended to differentially reduce RS6 phosphorylation and to enhance phosphorylation levels of Creb1, Fak1, Jun, p53, Stat6 and the pro-apoptotic IFN-γ signaling related Stat1 in A5 vs. C5N cells (Suppl. Fig. S4). Furthermore, we found that acteoside suppressed the MMP-2 and MMP-9 enzymatic activities in A5 cells (Fig. 1E).
We then assayed the effects of acteoside in the human osteosarcoma cell lines U2 OS (p53+/+, Rb1+/+), Sa OS (p53-/-, Rb1-/-) and KH OS (p53m/m; also express mutant H-Ras) [30], as well as in doxorubicin resistant osteosarcoma cells [23] and in the mouse melanoma cell lines B16.F1 and B16.F10. We found that independently of their genetic background acteoside was toxic in all three osteosarcoma cell lines (Suppl. Fig. S5A); exhibited synergistic effects with H2O2, doxorubicin and epoxomicin (proteasome inhibitor) (Suppl. Fig. S5B) and re-sensitized doxorubicin resistant osteosarcoma cell lines (Suppl. Fig. S5C). Acteoside was also toxic against B16.F1 and B16.F10 melanoma cells (Fig. S6A), while phosphoproteomic analyses (Fig. S6B) showed that B16.F10 cells had (compared to B16.F1 cells) increased phosphorylation levels of Akt, Creb1, Mp2k1 (Mek1), and Wnk1 proteins, but significantly reduced phosphorylation levels of Stat1 and Stat3. Exposure of melanoma cell lines to acteoside increased Creb1 phosphorylation in B16.F1 cells and Jun in B16.F10 cells, while it differentially tended to suppress phosphorylation of Fak1, Gsk3A, Mp2k1, p53, Ptn11, Rs6, Stat3, Tf65 and Wnk1 proteins in the less acteoside sensitive B16.F10 cell line.
Overall, these findings indicate that acteoside is differentially toxic to cancer cells (independently of their p53, Rb1 or H-Ras status) by pleiotropically affecting several signaling pathways; including cancer related pathways, antioxidant responses, proteostatic modules and also immune responses.
3.2. Acteoside is not cytotoxic in physiological cellular contexts
Given the reduced toxicity of acteoside in C5N cells, we exposed pre-senescent normal human diploid fibroblasts for an extended period (3 weeks) to acteoside and found that it exerted no cytotoxic effects. On the contrary, it even tended (at the doses used) to increase cellular growth (Suppl. Fig. S7A) and to slightly delay progression of cellular senescence (Suppl. Fig. S7B).
In support, acteoside was not toxic when it was orally delivered in Wistar rats, where it also alleviated the inflammatory effects of doxorubicin dermal injection. In this in vivo model, doxorubicin-mediated weight loss and animal death was alleviated when acteoside was co-administered with doxorubicin (Suppl. Fig. S8).
Furthermore, lifetime oral administration of acteoside in Drosophila flies did not affect longevity (Suppl. Fig. S9A) and tended to confer mild protection (not statistically significant) against the oxidative agent tert-butyl hydroperoxide (t-BHP) (Suppl. Fig. S9B). It was also protective against proteotoxic stress induced by administration of the proteasome inhibitor PS-341 (Suppl. Fig. S9C; see, Suppl. Table S2).
Taken together, these data indicate that acteoside exerts no significant cytotoxic effects in normal cellular contexts, where it is, likely, cytoprotective against oxidative and/or proteotoxic stress.
3.3. Intraperitoneal, but not oral (via drinking water), administration of acteoside suppresses tumor growth in an in vivo melanoma mouse model and activates anti-tumor-reactive immune responses
To study the possible in vivo anti-tumor effect of acteoside, we established a melanoma model by grafting B16.F1 cells into the flank of C57BL/6 mice and acteoside was delivered either IP (1 mg/mouse) (parenteral, a standard route for drug administration) or OR via drinking water (2.5 mg/mouse) (enteral, a standard route for natural products e.g. antioxidants, administration). Gene expression analyses revealed that acteoside administration (either IP or OR) activated proteostatic modules in tumors (e.g. proteasome/autophagyrelated genes), as well as antioxidant responses, molecular chaperones and metabolic responses-related genes (Fig. 2A). Notably, the antioxidant responses-related genes induction (e.g. nrf2, keap1, txnrd1 and foxo3) tended to be more intense after OR acteoside administration. Tumors from IP- and OR-treated mice were also characterized by enhanced proteasomal (Fig. 2B1) but not lysosomal cathepsins B, L (Fig. 2B2) activity.
Fig. 2.

Acteoside induced proteostatic modules and exerted in vivo anti-tumor activity in a melanoma (B16.F1) grafted mouse model, likely via the activation of (among others) anti-tumor-reactive immune responses. (A) Relative (%) mRNA expression levels (in tumors of control and acteoside-treated mice) of genes involved in different proteostatic and antioxidant responses modules, as well as in metabolic pathways; acteoside was administered either intraperitoneally (IP) or via drinking water (OR). (B) Relative (%) proteasome (B1) and cathepsin B, L (B2) enzymatic activities in excised tumors of control (Con) and IP or OR acteoside-treated mice. (C) Average tumor volume in control mice (Con, n = 5); in mice injected every other day with 1 mg acteoside (IP) (n = 5) and in mice receiving 2.5 mg acteoside via drinking water (OR; n = 5). (D) Flow cytometry analysis of CD107 expression on splenocytes isolated from control and acteoside-treated mice. Splenocytes were used as effectors vs. the syngeneic B16.F1, YAC-1 and WEHI-164 target cells. Cells expressing CD107 are gated and the relative percentage is shown in each dot plot; representative dot plots per group with similar results are shown. Bars, ± SD; *P < 0.05; * *P < 0.01.
Protein expression analyses showed that either IP or OR administration of acteoside upregulated proteins involved in proteasome (β5), autophagy (Beclin), antioxidant (Nrf-2) and DNA damage repair (Ku70) responses; also, isolated tumors were characterized by increased levels of protein carbonylation (DNP) indicating elevated proteome oxidation (Suppl. Fig. S10A). Notably, acteoside treatment upregulated Protein Kinase C (PKC) expression levels mostly after IP administration (Suppl. Fig. S10A). Furthermore, acteoside administration via the IP route significantly suppressed tumor growth; whereas, surprisingly, OR delivery accelerated tumor growth (Fig. 2C). In support, phosphoproteomics analyses of dissected tumors showed a tendency for enhanced activating phosphorylation levels in OR vs. IP treated mice in several pro-oncogenic, e.g. Egfr, Fak, Stat3 and Stat6 proteins, as well as in Creb1, Stat1, Tf65 and Wnk1 proteins (Suppl. Fig. S10B). Furthermore, by isolating splenocytes from treated animals and assessing by flow cytometry their cytotoxicity against B16.F1, YAC-1 (NK-sensitive) and WEHI-164 (LAK-sensitive) target cells, we found that only splenocytes isolated from mice that received acteoside via the IP route exhibited increased T and NK cell-mediated cytotoxicity against B16.F1, YAC-1 and WEHI-164 target cells (Fig. 2D). Finally, by developing high resolution MS for the identification/quantification of acteoside (Fig. 3A, B) in treated mice plasma and tumors, we found dramatically higher concentrations of the compound in both the plasma (Fig. 3C) and tumors (Fig. 3D) of IP (vs. OR) treated mice.
Fig. 3.

Acteoside identification/quantification in mouse plasma and grafted tumors with the use of UHPLC-MS analyses. (A) Calibration curve of the analyte/IS vs. increasing concentrations of acteoside. (B) Representative LC-MS chromatogram of acteoside in mice plasma. (C) Representative MS spectrum of acteoside in treated mice plasma (C1) and relative quantification (C2) after OR or IP administration of the compound. (D) Quantification of acteoside in excised tumors after OR or IP administration of the compound in mice. Bars, ± SD (n ≥ 2). * , P < 0.05; * *, P < 0.01.
Overall, acteoside exerts in vivo anti-tumor properties only when administered parenterally (IP), by (among others) activating anti-tumor-reactive immune responses; on the contrary, OR administration via drinking water (even at higher doses vs. IP delivery) of this herbal substance results in very low plasma and targeted tumor concentration and enhances tumor growth.
3.4. Acteoside inhibits protein kinase C
The selective toxicity of acteoside against tumor cells, prompted us to pursue its potential intracellular targets. Based on a previous report [31], we focused on its putative inhibitory effect on PKC. We noted that acteoside inhibited the activity of purified PKC in a dose-dependent manner (Fig. 4A), while, in cell-based assays we observed that treatment of cancer cells with 500 μM acteoside for 24 h resulted in significant reduction of immunoprecipitated PKC activity in both C5N and A5 cells (Fig. 4B). Moreover, we found decreased (not reaching statistical significance) PKC activity when the activity of the enzyme was assayed in total cell lysates (Fig. 4C). Notably, PKC was expressed at higher levels in the acteoside sensitive A5 and B16.F1 cell lines (vs. C5N and B16.F10 cells, respectively) (Fig. 4D), indicating that these cell lines likely depend on high PKC activity for survival. In support, knock down of PKCα with siRNA (Suppl. Fig. S11A) decreased cell survival of C5N, A5 and B16F1 cells (Suppl. Fig. S11B); albeit less intensively (e.g. in A5 cells), as compared to acteoside likely due to remaining activity of the residually expressed enzyme.
Fig. 4.

Acteoside inhibits protein kinase C (PKC) activity. (A) Absorbance values showing purified PKC enzymatic activity in the absence (black bars) or presence (gray bars) of increasing concentrations of acteoside. (B) Relative (%) activity of immunoprecipitated PKC from C5N and A5 cells after exposure (+) or not (-) to 500 μM acteoside for 24 h. (C) Relative (%) PKC activity in isolated C5N, A5 and B16.F1 total cell lysates after treatment (+) or not (-) with acteoside (500 μM) for 24 h. (D) Representative immunoblots of protein samples from C5N and A5 (D1), or from B16.F1 and B16.F10 cells (D2) probed with an antibody against PKC. GAPDH probing was used as reference. (E) Four dominant binding poses of acteoside in the active site of PKC accounting for 86.6% of the total Boltzmann population as predicted by docking simulations. The protein is depicted in a ribbon representation colored according to its secondary structure, while the active site is shown as a semi-transparent molecular surface colored in gray; the ligand is depicted in a ball and stick model. Bars, ± SD (n ≥ 2). * , P < 0.05; * *, P < 0.01.
To gain more insight to the putative mode of interaction between acteoside and PKC we performed molecular simulations by implementing a stepwise docking protocol. The Boltzmann distribution results indicated that the ligand likely binds in an ensemble of low-energy, physiologically-relevant poses, rather than in a single, well-defined geometry; the four dominant poses, along with their corresponding population percentages (86.6% of total) are shown in Fig. 4E. In all cases, a different part of the ligand occupies approximately the same space within the ATP-binding pocket of the kinase, thus affording a ligand solvent-accessible area that is hidden upon binding, ranging between 286 and 318 Å2. These results possibly indicate a pattern on the protein-inhibitor complex energy landscape which is characterized by more than one energetic minima separated by low barriers. This hypothesis is in agreement with the notion that the existence of multiple binding modes is usually facilitated by the occurrence of symmetries in either the ligand or the protein binding site [32]. Also, multiple alignment of PKC isoforms α, β and γ showed a high degree of similarity (e.g. the residues at positions 368, 380, 419, 465 are identical) between the three isoforms (Suppl. Fig. S12) indicating that acteoside can likely act as a potent inhibitor of all three isoforms.
Thus, according to cell-based and in silico molecular modeling/docking analyses, acteoside exerts an inhibitory effect on PKC.
4. Discussion
We report herein novel findings showing that acteoside exerted increased toxicity against mammalian cancer cells, showed synergistic effects with other anti-cancer agents and it also re-sensitized doxorubicin resistant cancer cell lines. In support, it was previously reported that acteoside was cytotoxic against a panel of human cancer cell lines [33], [34], [35], [36], while showing marginal toxicity against human peripheral blood mononuclear cells [29]. Given these findings, it is evident that acteoside acts independently of the tumor genetic background (e.g. p53, RB1, H-Ras status) and its anti-tumor effect surpasses the modulation of oncogenic signaling pathways. Mechanistically, we observed that acteoside (at the doses tested herein) increased ROS production in tumor cell lines in the absence of significant UPP (i.e. CT-L proteasomal activity) or antioxidant responses upregulation. Thus, despite the tumorigenesis related overactivation of proteostatic and antioxidant responses modules; tumor cells seem to be incapable of further inducing these cytoprotective pathways.
These properties of acteoside seem to be well tolerated in normal cells which are characterized by relatively low (as compared to tumor cells) basal ROS levels and expression levels of cytoprotective proteostatic and antioxidant responses modules. Normal cells also fully retain their ability to mount a counteracting response in the presence of increased ROS, or other types of stressors, levels. It is not thus surprising that acteoside was not toxic in different (e.g. normal human cells, flies or rodents) physiological cellular contexts, where it even exerted minor protection against a variety of exogenous stressors, likely due to its property to mobilize antioxidant responses. In support, previous studies reported that acteoside protected endothelial cells against oxidized LDL-induced cytotoxicity [37] and human skin fibroblasts or keratinocytes against X-ray or UVC irradiation-induced damage, respectively [38], [39]. Acteoside was also protective in an experimental model of intestinal inflammation or mucositis in mice [22], [40], at in vivo mouse models of acute gastric ulcers and ear edema [41] and also as a dietary supplement in swine [42]. Furthermore, acteoside was found to exert neuroprotection in the rotenone rat model of Parkinson's disease [43] and to inhibit β-amyloid aggregation [44], while it suppressed immunological liver injury induced by Bacillus Calmette-Guerin in conjunction with lipopolysaccharide, by (among others) suppressing hepatic cell apoptosis [45].
Acteoside is known (as we also report in this manuscript) to activate a strong antioxidant response; this is likely due to its indirect effects in activating endogenous detoxifying systems, rather than its direct free radical scavenging activity [46], [47], [48]. In support, we noted induction of antioxidant responses and proteostatic modules in both C5N cells and in vivo following administration of the compound in a melanoma mouse tumor model. A link between acteoside and Nrf-2 has been established before [49], [50]; nevertheless, we reveal for the first time, that acteoside also elevates proteasome activity levels. These findings suggest that acteoside may exert dose-dependent pro-oxidant effects which mobilize Nrf-2 activation. In line with this notion, it was proposed that the pro-oxidative action of polyphenols mediates their pro-apoptotic activity [51].
Our phosphoproteomics analyses showed that exposure of cells to acteoside affected the phosphorylation levels and signaling of many oncogenic pathways including higher Creb1 and Fak1 activities only in metastatic A5 cells, and Jun activation in both A5 and melanoma B16.F1 cells. Also, acteoside suppressed Stat-3 phosphorylation (in all cell lines assayed) and activated oncogenic Stat-6 in A5 cells. Some of these effects on cellular phosphoproteome may relate to our observation that acteoside inhibits PKC, a kinase with a wide range of downstream targets and a main regulator of cell survival and growth [52]. Thus, the anti-tumor activity of acteoside might be due, at least in part, to inhibition of PKC; this assumption is further supported by the finding that A5 and B16.F1 express higher PKC levels as compared to the less sensitive to acteoside C5N and B16.F10 cells.
PKC isoenzymes have been found to be involved in proliferation, anti-tumor drug resistance and apoptosis and various PKC modulators are in clinical evaluation as anti-cancer therapeutics [52], [53], [54]. The inhibitory effect of acteoside on PKC may also relate to our finding of acteoside-mediated suppression of the metastasis-promoting MMP-2 and MMP-9 activity, as it was previously reported in human fibrosarcoma HT-1080 cells that acteoside suppresses phorbol-12-myristate-13-acetate (PMA) (a known PKC activator)-mediated enhancement of MMP-9 [55]. Given the fact that our alignment studies amongst the various PKC isoforms along with the molecular docking analyses showed that acteoside will likely inhibit most of the PKC isoforms, its chemical scaffold can provide the starting point for the development of rationally optimized selective bioactive analogues.
The anti-tumor activity of acteoside was also evident in an in vivo melanoma mouse model where parenteral (IP) treatment with 1 mg of acteoside resulted in significant retardation of tumor growth. Interestingly enough, enteral (OR) administration (via drinking water) of 2.5 mg of the compound accelerated tumor growth. Phosphoproteomics analyses of tumors isolated from IP- and OR-treated mice showed that OR administration tended to enhance the phosphorylation (and consequently activation) of most oncogenic proteins assayed, including Egfr, Fak1, Ptn11, Stat3 and Stat6. These observations likely relate to the significantly higher (as compared to OR) concentrations of acteoside achieved in the plasma and in the targeted tumor after IP administration. It is known that parenteral routes for drug administration offer the highest bioavailability because they avoid the first-pass effect of hepatic metabolism, which commonly occurs with orally administered chemicals and therapeutics. Indeed, it was proposed that acteoside in order to exert its full anti-tumor properties has to be delivered at high concentrations [20]. In line with the differential effects amongst IP and OR administration, and in spite of route-of-administration and eventually dose-independent proteome oxidation and mobilization of comparable anti-oxidant/proteostatic responses, we noted that only splenocytes isolated from IP-treated mice were activated in vivo and expressed CD107, which suggests the mobilization of anti-tumor immune responses. Also, PKC expression was elevated (an expected counteracting response in case of PKC inhibition) only upon IP treatment, likely indicating that only via this route the accumulated concentration of the compound was sufficient to effectively inhibit PKC. In relation to oncogenic pathways, it was previously found that PKC-epsilon interacts with Stat3 and regulates its activation that is essential for the development of skin cancer [56]. Also, it was reported that Stat3 [57], [58] and PKC-epsilon activity [58] were positively regulated by Nrf-2 suggesting that if the acteoside concentration at the tumor site does not reach the critical concentration needed to suppress PKC (as is likely the case during OR administration), the activation of solely the antioxidant responses (e.g. Nrf-2 or Foxo) would be severely pro-oncogenic. In support, the pro-oncogenic Stat3 and Stat6 were activated mostly in OR-treated mice. In conclusion, the in vivo anti-tumor effect of acteoside (and likely all other natural products) depends on the route of administration and the achieved concentration in the tumor.
5. Conclusions
Our findings indicate that acteoside exerts pleiotropic effects on mammalian cells, as by enhancing ROS levels it mobilizes antioxidant and proteostatic modules, while by inhibiting PKC it affects the signaling status of many oncogenic pathways; moreover, at high concentrations (e.g. after parenteral administration) it mobilizes anti-tumor-reactive immune responses. These data indicate that acteoside holds promise as a chemical scaffold for the development of novel selective cytotoxic agents against tumor cells. They also raise the critical issue of appropriate dosage in therapeutic interventions, as low doses of therapeutic compounds that solely mobilize antioxidant responses, will likely accelerate tumor progression and cancer cell survival. In line with this notion, it was recently reported that green tea polyphenols block the anti-cancer effects of bortezomib and other boronic acid-based proteasome inhibitors during multiple myeloma therapy [59]. In other words it is plausible that in those cases where cancer patients self-medicate “miracle antioxidant herbs”, that are commonly perceived as “innocent” or “holistic”, in hopes of augmenting the anticancer outcome of their chemotherapy the final result could be exactly the opposite.
Acknowledgements
We acknowledge Profs V. Gorgoulis and A. Balmein, as well as Dr E. Gonos for the donation of cell lines. This work was partially supported by the “EnGAGE 11ΣΥΝ-1-420” GSRT grant.
Acknowledgments
Conflict of interest
The authors declare no potential conflicts of interest.
Author contributions
IPT designed and supervised the study; CC, PS, ENT, OET and IPT conducted experiments or interpreted the data; PP, AA, NNA, TN, MH and ALS purified acteoside and performed phytochemical and NMR/mass-spectrometry analyses; VM and EM conducted molecular docking analyses; VZ and IP generated or contributed reagents, materials; TS and LGA performed the phosphoproteomics studies. All authors commented on the manuscript; IPT wrote the manuscript.
Ethics
Animal handling and experimentation were in accordance with the guidelines of the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes, and were approved by the Animal Care and Use Committee of the Hellenic Pasteur Institute and by National Authorities (Veterinary Section of the Greek Republic).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.02.015.
Appendix A. Supplementary material
Supplementary material
References
- 1.Hanahan D., Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Raj L., Ide T., Gurkar A., Foley M., Schenone M., Li X. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Trougakos I., Sesti F., Tsakiri E., Gorgoulis V. Non-enzymatic post-translational protein modifications and proteostasis network deregulation in carcinogenesis. J. Proteom. 2013;92:274–298. doi: 10.1016/j.jprot.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 4.Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Höhn A., Jung T., Grune T. Pathophysiological importance of aggregated damaged proteins. Free Radic. Biol. Med. 2014;71:70–89. doi: 10.1016/j.freeradbiomed.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 6.Tsakiri E., Trougakos I. The amazing ubiquitin-proteasome system: structural components and implication in aging. Int. Rev. Cell Mol. Biol. 2015:171–237. doi: 10.1016/bs.ircmb.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Vanhooren V., Navarrete Santos A., Voutetakis K., Petropoulos I., Libert C., Simm A., Gonos E.S., Friguet B. Protein modification and maintenance systems as biomarkers of ageing. Mech. Ageing Dev. 2015;151:71–84. doi: 10.1016/j.mad.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Reeg S., Jung T., Castro J.P., Davies K.J., Henze A., Grune T. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic. Biol. Med. 2016;99:153–166. doi: 10.1016/j.freeradbiomed.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buac D., Shen M., Schmitt S., Rani Kona F., Deshmukh R., Zhang Z. From Bortezomib to other Inhibitors of the proteasome and beyond. Curr. Pharm. Des. 2013;19:4025–4038. doi: 10.2174/1381612811319220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klaunig J., Kamendulis L., Hocevar B. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 11.Sykiotis G., Bohmann D. Stress-activated Cap'n'collar transcription factors in aging and human disease. Sci. Signal. 2010;3:re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickering A.M., Staab T.A., Tower J., Sieburth D., Davies K.J. A conserved role for the 20S proteasome and Nrf2 transcription factor in oxidative stress adaptation in mammals, Caenorhabditis elegans and Drosophila melanogaster. J. Exp. Biol. 2013;15:543–553. doi: 10.1242/jeb.074757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H., Davies K.J., Forman H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015;88:314–336. doi: 10.1016/j.freeradbiomed.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo J., Solimini N., Elledge S. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernatchez C., Radvanyi L., Hwu P. Advances in the treatment of metastatic melanoma: adoptive T-Cell therapy. Semin. Oncol. 2012;39:215–226. doi: 10.1053/j.seminoncol.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Argyropoulou A., Aligiannis N., Trougakos I., Skaltsounis A. Natural compounds with anti-ageing activity. Nat. Prod. Rep. 2013;30:1412–1437. doi: 10.1039/c3np70031c. [DOI] [PubMed] [Google Scholar]
- 17.Aravindaram K., Yang N. Anti-inflammatory plant natural products for cancer therapy. Planta Med. 2010;76:1103–1117. doi: 10.1055/s-0030-1249859. [DOI] [PubMed] [Google Scholar]
- 18.Long S., Sousa E., Kijjoa A., Pinto M. Marine natural products as models to circumvent multidrug resistance. Molecules. 2016;21 doi: 10.3390/molecules21070892. (pii: E892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sklirou A., Papanagnou E., Fokialakis N., Trougakos I.P. Cancer chemoprevention via activation of proteostatic modules. Cancer Lett. 2018;413:110–121. doi: 10.1016/j.canlet.2017.10.034. [DOI] [PubMed] [Google Scholar]
- 20.Alipieva K., Korkina L., Orhan I.E., Georgiev M.I. Verbascoside-A review of its occurrence, (bio)synthesis and pharmacological significance. Biotechnol. Adv. 2014;32:1065–1076. doi: 10.1016/j.biotechadv.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Aligiannis N., Mitaku S., Tsitsa-Tsardis E., Harvala C., Tsaknis I., Lalas S. Methanolic extract of Verbascum macrurumas as a source of natural preservatives against oxidative rancidity. J. Agric. Food Chem. 2003;51:7308–7312. doi: 10.1021/jf034528+. [DOI] [PubMed] [Google Scholar]
- 22.Reinke D., Kritas S., Polychronopoulos P., Skaltsounis A.L., Aligiannis N., Tran C.D. Herbal substance, acteoside, alleviates intestinal mucositis in mice. Gastroent Res. Pract. 2015:327872. doi: 10.1155/2015/327872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lourda M., Trougakos I.P., Gonos E.S. Development of resistance to chemotherapeutic drugs in human osteosarcoma cell lines largely depends on up-regulation of clusterin/apolipoprotein. J. Int. J. Cancer. 2007;120:611–622. doi: 10.1002/ijc.22327. [DOI] [PubMed] [Google Scholar]
- 24.Buchmann A., Ruggeri B., Klein-Szanto A.J., Balmain A. Progression of squamous carcinoma cells to spindle carcinomas of mouse skin is associated with an imbalance of H-ras alleles on chromosome 7. Cancer Res. 1991;51:4097–4101. [PubMed] [Google Scholar]
- 25.Zoumpourlis V., Papassava P., Linardopoulos S., Gillespie D., Balmain A., Pintzas A. High levels of phosphorylated c-Jun, Fra-1, Fra-2 and ATF-2 proteins correlate with malignant phenotypes in the multistage mouse skin carcinogenesis model. Oncogene. 2000;19:4011–4021. doi: 10.1038/sj.onc.1203732. [DOI] [PubMed] [Google Scholar]
- 26.Sklirou A.D., Ralli M., Dominguez M., Papassideri I., Skaltsounis A.L., Trougakos I.P. Hexapeptide-11 is a novel modulator of the proteostasis network in human diploid fibroblasts. Redox Biol. 2015;5:205–215. doi: 10.1016/j.redox.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trougakos I.P., Pawelec G., Tzavelas C., Ntouroupi T., Gonos E.S. Clusterin/Apolipoprotein J up-regulation after zinc exposure, replicative senescence or differentiation of human haematopoietic cells. Biogerontol. 2006;7:375–382. doi: 10.1007/s10522-006-9052-8. [DOI] [PubMed] [Google Scholar]
- 28.Trougakos I.P., Margaritis L.H. The formation of the functional chorion structure of Drosophila virilis involves inercalation of the “middle” and “late” major chorion proteins: a general model for chorion assembly in Drosophilidae. J. Struct. Biol. 1998;123:97–110. doi: 10.1006/jsbi.1998.4999. [DOI] [PubMed] [Google Scholar]
- 29.Argyropoulou A., Samara P., Tsitsilonis O., Skaltsa H. Polar constituents of Marrubium thessalum Boiss. & Heldr. (Lamiaceae) and their cytotoxic/cytostatic activity. Phytother. Res. 2012;26:1800–1806. doi: 10.1002/ptr.4654. [DOI] [PubMed] [Google Scholar]
- 30.Trougakos I.P., Chondrogianni N., Amarantos I., Blake J., Schwager C., Wirkner U. Genome-wide transcriptome profile of the human osteosarcoma Sa OS and U-2 OS cell lines. Cancer Genet Cytogenet. 2010;196:109–118. doi: 10.1016/j.cancergencyto.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 31.Herbert J.M., Maffrand J.P., Taoubi K., Augereau J.M., Fouraste I., Gleye J. Verbascoside isolated from Lantana camara, an inhibitor of protein kinase C. J. Nat. Prod. 1991;4:1595–1600. doi: 10.1021/np50078a016. [DOI] [PubMed] [Google Scholar]
- 32.Mobley D., Dill K. Binding of small molecule ligands to proteins: what you see is not always what you get. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang F., Jia Z., Deng Z., Wei Y., Zheng R., Yu L. In vitro modulation of telomerase activity, telomere length and cell cycle in MKN45 cells by verbascoside. Planta Med. 2002;68:115–118. doi: 10.1055/s-2002-20255. [DOI] [PubMed] [Google Scholar]
- 34.Zhou L., Feng Y., Jin Y., Liu X., Sui H., Chai N. Verbascoside promotes apoptosis by regulating HIPK2-p53 signaling in human colorectal cancer. BMC Cancer. 2014;14:747. doi: 10.1186/1471-2407-14-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee K.W., Kim H.J., Lee Y.S., Park H.J., Choi J.W., Ha J. Acteoside inhibits human promyelocytic HL-60 leukemia cell proliferation via inducing cell cycle arrest at G0/G1 phase and differentiation into monocytes. Carcinogenesis. 2007;28:1928–1936. doi: 10.1093/carcin/bgm126. [DOI] [PubMed] [Google Scholar]
- 36.Sitarek P., Skała E., Toma M., Wielanek M., Szemraj J., Nieborowska-Skorska M. A preliminary study of apoptosis induction in glioma cells via alteration of the Bax/Bcl-2-p53 axis by transformed and non-transformed root extracts of Leonurus sibiricus L. Tumour Biol. 2016;37:8753–8764. doi: 10.1007/s13277-015-4714-2. [DOI] [PubMed] [Google Scholar]
- 37.Martin-Nizard F., Sahpaz S., Furman C., Fruchart J.C., Duriez P., Bailleul F. Natural phenylethanoids protect endothelial cells against oxidized LDL-induced cytotoxicity. Planta Med. 2003;69:207–211. doi: 10.1055/s-2003-38474. [DOI] [PubMed] [Google Scholar]
- 38.Yang J., Yan Y., Liu H., Wang J., Hu J. Protective effects of acteoside against X-ray-induced damage in human skin fibroblasts. Mol. Med. Rep. 2015;12:2301–2306. doi: 10.3892/mmr.2015.3630. [DOI] [PubMed] [Google Scholar]
- 39.Kostyuk V.A., Potapovich A., Suhan T., De Luca C., Pressi G., Dal Toso R. Plant polyphenols against UV-C-induced cellular death. Planta Med. 2008;74:509–514. doi: 10.1055/s-2008-1074499. [DOI] [PubMed] [Google Scholar]
- 40.Hausmann M., Obermeier F., Paper D.H., Balan K., Dunger N., Menzel K. In vivo treatment with the herbal phenylethanoid acteoside ameriliorates intestinal inflammation in dextran sulphate sodium-induced colitis. Clin. Exp. Immunol. 2007;148:373–381. doi: 10.1111/j.1365-2249.2007.03350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanchez P.M., Villarreal M.L., Herrera-Ruiz M., Zamilpa A., Jiménez-Ferrer E., Trejo-Tapia G. In vivo anti-inflammatory and anti-ulcerogenic activities of extracts from wild growing and in vitro plants of Castilleja tenuiflora Benth. (Orobanchaceae) J. Ethnopharmacol. 2013;150:1032–1037. doi: 10.1016/j.jep.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Di Giancamillo A., Rossi R., Vitari F., Carollo V., Deponti D., Corino C. Changes in nitrosative stress biomarkers in swine intestine following dietary intervention with verbascoside. Histol. Histopathol. 2013;28:715–723. doi: 10.14670/HH-28.715. [DOI] [PubMed] [Google Scholar]
- 43.Yuan J., Ren J., Wang Y., He X., Zhao Y. Acteoside binds to caspase-3 and exerts neuroprotection in the rotenone rat model of parkinson's disease. PLoS One. 2016;11:e0162696. doi: 10.1371/journal.pone.0162696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurisu M., Miyamae Y., Murakami K., Han J., Isoda H., Irie K. Inhibition of amyloid β aggregation by acteoside, a phenylethanoid glycoside. Biosci. Biotechnol. Biochem. 2013;77:1329–1332. doi: 10.1271/bbb.130101. [DOI] [PubMed] [Google Scholar]
- 45.Zhao J.1, Liu T., Ma L., Yan M., Zhao Y., Gu Z. Protective effect of acteoside on immunological liver injury induced by Bacillus Calmette-Guerin plus lipopolysaccharide. Planta Med. 2009;75:1463–1469. doi: 10.1055/s-0029-1185796. [DOI] [PubMed] [Google Scholar]
- 46.Funes L., Fernandez-Arroyo S., Laporta O., Pons A., Roche E., Segura-Carretero A. Correlation between plasma antioxidant capacity and verbascoside levels in rats after oral administration of lemon verbena extract. Food Chem. 2009;117:589–598. [Google Scholar]
- 47.Di Giancamillo A., Rossi R., Pastorelli G., Deponti D., Carollo V., Casamassima D. The effects of dietary verbascoside on blood and liver oxidative stress status induced by a high n-6 polyunsaturated fatty acids diet in piglets. J. Anim. Sci. 2015;93:2849–2859. doi: 10.2527/jas.2014-8607. [DOI] [PubMed] [Google Scholar]
- 48.Martino N.A., Ariu F., Bebbere D., Uranio M.F., Chirico A., Marzano G. Supplementation with nanomolar concentrations of verbascoside during in vitro maturation improves embryo development by protecting the oocyte against oxidative stress: a large animal model study. Reprod. Toxico. 2016;65:204–211. doi: 10.1016/j.reprotox.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 49.Pastore S., Lulli D., Fidanza P., Potapovich A.I., Kostyuk V.A., De Luca C. Plant polyphenols regulate chemokine expression and tissue repair in human keratinocytes through interaction with cytoplasmic and nuclear components of epidermal growth factor receptor system. Antioxid. Redox Signal. 2012;16:314–328. doi: 10.1089/ars.2011.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kostyuk V.A., Potapovich A.I., Suhan T.O., De Luca C., Korkina L.G. Antioxidant and signal modulation properties of plant polyphenols in controlling vascular inflammation. Eur. J. Pharmacol. 2011;658:248–256. doi: 10.1016/j.ejphar.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 51.Lecci R.M., Logrieco A., Leone A. Pro-oxidative action of polyphenols as action mechanism for their pro-apoptotic activity. Anticancer Agents Med Chem. 2014;14:1363–1375. doi: 10.2174/1871520614666140922121014. [DOI] [PubMed] [Google Scholar]
- 52.Tarafdar A., Michie A.M. Protein kinase C in cellular transformation: a valid target for therapy? Biochem. Soc. Trans. 2014;42:1556–1562. doi: 10.1042/BST20140255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Serova M., Ghoul A., Benhadji K.A., Cvitkovic E., Faivre S., Calvo F., Lokiec F., Raymond E. Preclinical and clinical development of novel agents that target the protein kinase C family. Semin. Oncol. 2006;33:466–478. doi: 10.1053/j.seminoncol.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 54.Martin-Liberal J., Cameron A.J., Claus J., Judson I.R., Parker P.J., Linch M. Targeting protein kinase C in sarcoma. Biochim Biophys. Acta. 2014;1846:547–559. doi: 10.1016/j.bbcan.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Hwang Y.P., Kim H.G., Choi J.H. Acteoside inhibits PMA-induced matrix metalloproteinase-9 expression via CaMK/ERK- and JNK/NF-κB-dependent signaling. Mol. Nutr. Food Res. 2011;55(Suppl. 1):S103–S116. doi: 10.1002/mnfr.201000336. [DOI] [PubMed] [Google Scholar]
- 56.Aziz M.H., Manoharan H.T., Sand J.M., Verma A.K. Protein kinase Cepsilon interacts with Stat3 and regulates its activation that is essential for the development of skin cancer. Mol. Carcinog. 2007;46:646–653. doi: 10.1002/mc.20356. [DOI] [PubMed] [Google Scholar]
- 57.Türei D., Papp D., Fazekas D., Földvári-Nagy L., Módos D., Lenti K., Csermely P. NRF2-ome: an integrated web resource to discover protein interaction and regulatory networks of NRF2. Oxid. Med. Cell Longev. 2013:737591. doi: 10.1155/2013/737591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calvert J.W., Jha S., Gundewar S., Elrod J.W., Ramachandran A., Pattillo C.B. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Golden E.B., Lam P.Y., Kardosh A., Gaffney K.J., Cadenas E., Louie S.G. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113:5927–5937. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material