


Abstract
Allele-specific expression (ASE) is a fundamental problem in studying gene regulation and diploid transcriptome profiles, with two key challenges: (i) haplotyping and (ii) estimation of ASE at the gene isoform level. Existing ASE analysis methods are limited by a dependence on haplotyping from laborious experiments or extra genome/family trio data. In addition, there is a lack of methods for gene isoform level ASE analysis. We developed a tool, IDP-ASE, for full ASE analysis. By innovative integration of Third Generation Sequencing (TGS) long reads with Second Generation Sequencing (SGS) short reads, the accuracy of haplotyping and ASE quantification at the gene and gene isoform level was greatly improved as demonstrated by the gold standard data GM12878 data and semi-simulation data. In addition to methodology development, applications of IDP-ASE to human embryonic stem cells and breast cancer cells indicate that the imbalance of ASE and non-uniformity of gene isoform ASE is widespread, including tumorigenesis relevant genes and pluripotency markers. These results show that gene isoform expression and allele-specific expression cooperate to provide high diversity and complexity of gene regulation and expression, highlighting the importance of studying ASE at the gene isoform level. Our study provides a robust bioinformatics solution to understand ASE using RNA sequencing data only.
INTRODUCTION
In diploid organisms, such as human and mouse, paternal and maternal alleles can be regulated and expressed unequally, which is termed allele-specific expression (ASE). This phenomenon includes (i) random X-chromosome inactivation (1); (ii) parent-of-origin imprinting (2,3); (iii) random monoallelic expression of autosomal genes (4); (iv) widespread ASE biases, in which one allele has a significantly higher expression level than other alleles (5) and (v) allele-specific isoform expression, in which specific isoforms from one allele are exclusively expressed or have relatively higher expression in comparison to other isoforms (6). Recent studies have established that expression of alleles is non-equal for many genes, and the expression bias between alleles varies dramatically (7). These ASE effects can vary by cell/tissue type (8), developmental stage (9) and pathological features (10). For example, the rate of ASE is remarkably higher in cancer cells as compared to normal tissues, which could be caused by a change in copy number or allelic composition (11). Since alleles from the same gene/gene isoform can provide heterozygous transcripts with distinct sequences, full analysis of ASE is necessary to achieve a thorough understanding of transcriptome profiles.
The ASE problem contains two parts: haplotyping and ASE quantification. Haplotyping refers to grouping heterozygous genetic variants (e.g. single nucleotide variants/SNVs; note that below ‘SNVs’ refers to heterozygous SNVs for conciseness) at multiple heterozygous sites into two sets. Most existing methods can only identify each SNV independently (12,13). Haplotyping is necessary to reconstruct entire alleles so that the full-length sequences of alleles can be studied as a whole. Moreover, correct haplotyping is necessary for accurate quantification of ASE. ASE quantification refers to estimating the abundance of alleles and measuring the proportion of allele expression within a gene. In addition to the gene level, ASE at the gene isoform level should be also estimated.
To analyze ASE, many experimental and bioinformatics approaches have been developed. In contrast to genome-wide genotyping arrays based on microarray hybridization (14,15) and large-scale synthetic padlock probes that capture transcripts with known exonic SNVs (16,17), next generation sequencing provides data to study genome-wide ASE with less bias while not being limited to only known SNVs (18). A number of bioinformatics tools based on high-throughput Second Generation Sequencing (SGS) data have been developed, such as AlleleSeq (19), MMSEQ (6), asSeq (20), Allim (21), MBASED (11), Allele Workbench (22), QuASAR (23), ASEQ (24), EMASE (25) and others (8,26,27). However, either available phased genotypes (e.g. MMSEQ, asSeq and EMASE) or family trio data (e.g. AlleleSeq and Allim) are required for haplotyping using most of these applications. While QuASAR uses solely RNA-seq data, it can only perform ASE analysis at the single SNV level. MBASED is the only currently available tool for ASE analysis at the gene level using only RNA-seq data. However, the false positive rate of its ‘pseudo haplotyping’ procedure is uncertain when imbalances of two alleles are not significant or when isoforms have distinct ASE profiles within a gene. These problems of SGS methods are mostly caused by the short read length (100–250 bp) because multiple SNVs cannot be covered by single short reads. Another challenging but fundamental problem is the quantification of ASE at the gene isoform level. Although MMSEQ could perform gene isoform level ASE analysis, the dependence of known haplotypes and known isoform library greatly limits its utility and quantification accuracy. Overall, a bioinformatics method that does not rely on known haplotypes or known isoform library but only requires RNA-seq data is of high demand to promote ASE research.
Third Generation Sequencing (TGS), including Pacific Biosciences (PacBio) sequencing (28,29) and Oxford Nanopore Technologies (ONT) (30) provides much longer reads (1–100 kb). TGS long reads have been used successfully to identify full-length gene isoforms and thus have the potential to overcome the haplotyping problem and ASE quantification at the gene isoform level (31–34). Single TGS long reads can cover multiple or even all SNVs within a gene, which reduces or solves the combinatorial complexity of haplotyping SNVs. However, the high error rate of TGS limits the accuracy of haplotyping, and the low throughput is not suitable for quantifying ASE. Hybrid sequencing (‘Hybrid-Seq’), which integrates TGS and SGS data, can address the limitations associated with SGS-only and TGS-only analysis and can improve the overall performance and resolution of the output data. In particular, a series of bioinformatics tools for Hybrid-Seq transcriptome data, including LSC, IDP and IDP-fusion, have been demonstrated to elucidate transcriptomes at the gene isoform level with high precision and sensitivity (31,34–36).
Here, we present a new method (termed IDP-ASE, http://www.healthcare.uiowa.edu/labs/au/IDP-ASE/) for haplotyping and quantification of ASE at both the gene and gene isoform levels requiring only RNA sequencing data. First, IDP-ASE integrates TGS and SGS data with a Bayesian model to determine haplotypes and quantify ASE at the gene level. After utilizing our previously published tool IDP to identify the expressed isoforms, we applied a Poisson model to estimate the abundance of allele-specific isoforms and further calculate ASE at the gene isoform level. The proof-of-concept application to the gold-standard data GM12878 demonstrates the superior accuracy of haplotyping by IDP-ASE with Hybrid-Seq data. In addition, we examined the haplotyping performance with respect to sequencing coverage, which established that TGS long reads are informative for haplotyping. We also evaluated the quantification performance at the gene and gene isoform levels by semi-simulation data. Applying IDP-ASE to human breast cancer cells (MCF-7 cell line) and human embryonic stem cells (hESCs, H1 cell line), we not only identified extensive ASE events, including a few tumorigenesis-relevant genes and pluripotency markers, but we also discovered distinct ASE imbalances among isoforms within single genes.
MATERIALS AND METHODS
Data sources
Hybrid-Seq data from H1 cell line has been previously published (31,34) and is available in the Gene Expression Omnibus (GEO) (accession no. GSE51861). SGS data from MCF-7 cell line has been previously published (37) and is available in GEO database (accession no. GSE49831). TGS data from MCF-7 cell line has been previously published (34) and is available on the National Center of Biotechnology Information (NCBI) SRA (accession no. SRP055913). Hybrid-Seq data from GM12878 has been previously published (33) and is available in the NCBI SRA (accession no. SRP036136).
Statistical method for haplotyping and quantification of ASE at the gene level
Many bioinformatics methods (e.g. SAMtools and GATK (12,13)) based on SGS short reads provide high-accuracy SNV calling because the error rate of SGS data is very low. Therefore, we can assume that the SNVs are known (e.g. determined using SGS data). Our model is constructed only for SNVs which are nucleotide substitutions. We further assume that each variant site is biallelic and only heterozygous variants will be considered, as homozygous variants are uninformative about haplotyping (38). Suppose there are m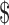 heterozygous variant sites in the gene of interest. For the jth site, let
heterozygous variant sites in the gene of interest. For the jth site, let 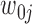 and
and 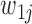 denote the two possible alleles, with
denote the two possible alleles, with 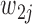 and
and 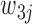 arbitrarily assigned the remaining two nucleotides. Let
arbitrarily assigned the remaining two nucleotides. Let 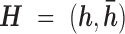 be the unordered pair of haplotypes, where
be the unordered pair of haplotypes, where 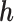 , a binary string of length
, a binary string of length 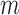 , corresponds to the phase of one of the strands, i.e.
, corresponds to the phase of one of the strands, i.e.  if the variant at site
if the variant at site 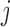 is equal to
is equal to 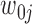 and 1 if it is equal to
and 1 if it is equal to 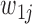 . Since the variants are heterozygous and biallelic, the phase of the other strand,
. Since the variants are heterozygous and biallelic, the phase of the other strand, 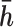 , is the bitwise complement of
, is the bitwise complement of 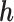 .
.
At the jth site, reads are assigned 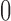 if they match
if they match 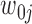 , 1 if they match
, 1 if they match 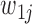 ,
, 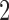 if they match
if they match 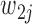 , and 3 if they match
, and 3 if they match 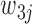 . Let
. Let 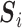 represents the
represents the 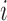 th such read where
th such read where 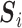 is a sequence over the set
is a sequence over the set 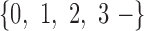 , and “
, and “ ” corresponds to the variant site not covered by the read. Assume
” corresponds to the variant site not covered by the read. Assume 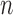 reads are uniquely mapped to the gene of interest. Then let
reads are uniquely mapped to the gene of interest. Then let 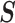 be a
be a 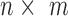 matrix whose
matrix whose 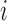 th row corresponds to
th row corresponds to 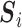 . Let
. Let 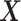 , the read matrix, denote how the
, the read matrix, denote how the 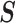 matrix aligns with the haplotype (Figure 1).
matrix aligns with the haplotype (Figure 1).
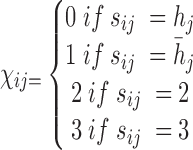 |
Figure 1.

Model for haplotyping and ASE quantification. This illustration exhibits a gene with four SNVs, two isoforms, and allele specific expression (indicated by blue and wheat color). The reference and alternative SNV calls are given by 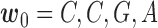 and
and 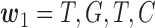 . The non-overlapping exon segments are given by
. The non-overlapping exon segments are given by 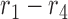 . In the gene-level model, long- and short-read alignment information, along with SNV positions, are converted into the read matrix
. In the gene-level model, long- and short-read alignment information, along with SNV positions, are converted into the read matrix 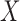 which is used by the Bayesian model for haplotyping and ASE quantification. For the isoform level model a Poisson model is used to obtain the MLE of the isoform-level allele specific abundance,
which is used by the Bayesian model for haplotyping and ASE quantification. For the isoform level model a Poisson model is used to obtain the MLE of the isoform-level allele specific abundance, 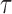 . On the right is the matrix
. On the right is the matrix 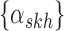 where each row corresponds to a region, each column corresponds to an isoform, and the two colors correspond to a different haplotype. The number of short reads,
where each row corresponds to a region, each column corresponds to an isoform, and the two colors correspond to a different haplotype. The number of short reads, 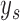 , within a region are displayed to the left of
, within a region are displayed to the left of 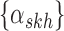 . For example, the region
. For example, the region 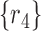 corresponds to reads that only mapped to segment
corresponds to reads that only mapped to segment 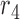 and not any other region or SNV (there are nine such reads, highlighted in pink) while region
and not any other region or SNV (there are nine such reads, highlighted in pink) while region 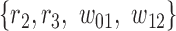 corresponds to reads that mapped to both region
corresponds to reads that mapped to both region 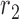 and
and 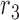 , and covered the first SNV with a sequenced nucleotide of
, and covered the first SNV with a sequenced nucleotide of 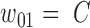 and the second SNV with a sequenced nucleotide of
and the second SNV with a sequenced nucleotide of 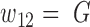 . (There is 1 such short read, highlighted in brown.)
. (There is 1 such short read, highlighted in brown.)
Each 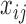 has a categorical distribution with parameter
has a categorical distribution with parameter 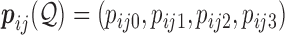 where
where 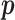 is a probability vector and
is a probability vector and 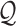 is a user input, or data derived error model. The model is general and accepts reads and the corresponding error models from different sources/platforms (e.g. different types of SGS and TGS), such as Illumina short reads, PacBio subreads and PacBio CCS reads. Below a concrete example of an error model is presented.
is a user input, or data derived error model. The model is general and accepts reads and the corresponding error models from different sources/platforms (e.g. different types of SGS and TGS), such as Illumina short reads, PacBio subreads and PacBio CCS reads. Below a concrete example of an error model is presented.
Denote 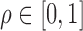 as the gene level relative ASE. Given the read data and the error, the goal of IDP-ASE is to obtain the most likely pair of haplotypes and the corresponding
as the gene level relative ASE. Given the read data and the error, the goal of IDP-ASE is to obtain the most likely pair of haplotypes and the corresponding 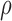 :
:
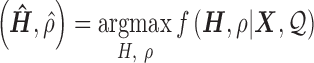 |
Since 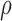 is continuous and unknown, it will not be possible to enumerate all possibilities. Instead, samples from the joint distribution of
is continuous and unknown, it will not be possible to enumerate all possibilities. Instead, samples from the joint distribution of 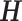 and
and 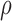 will be drawn. By Bayes' rule we have
will be drawn. By Bayes' rule we have
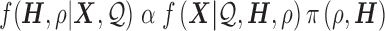 |
(1) |
A flat prior for the joint distribution of 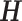 and
and 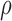 yields
yields
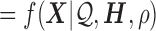 |
(2) |
Suppose the reads are conditionally independent given the haplotype 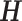 and error model
and error model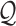
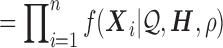 |
(3) |
To account for allele-specific expression, each read 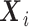 is modelled as a mixture distribution with mixture weights
is modelled as a mixture distribution with mixture weights 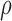 and
and 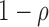 corresponding to the originating strand of the transcript to which the read mapped
corresponding to the originating strand of the transcript to which the read mapped
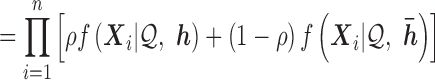 |
(4) |
Recall that the 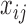 have a categorical distribution. Assuming variant calls in a read are independent of each other conditional on the haplotype and error model, and that variants not covered by a read contribute nothing to likelihood
have a categorical distribution. Assuming variant calls in a read are independent of each other conditional on the haplotype and error model, and that variants not covered by a read contribute nothing to likelihood
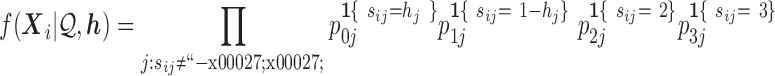 |
(5) |
Here, in the proof-of-concept application below, the error model can be derived from quality scores reported in FASTQ file. Sequence quality scores 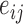 are integer values related to the error probabilities as
are integer values related to the error probabilities as 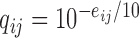 . Then
. Then 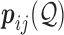 can simply be defined to be
can simply be defined to be
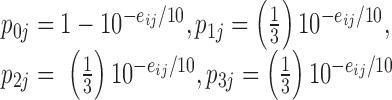 |
Where the probability the sequenced nucleotide is correct is given by 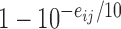 and the probability it is wrong is split evenly between the other three possible nucleotides.
and the probability it is wrong is split evenly between the other three possible nucleotides.
Slice sampling (39) will be used to sample 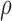 . A Metropolis–Hastings type of sampler is used to sample the haplotype (40). The MCMC (Markov chain Monte Carlo) sampler is initially run for 1500 iterations, with 1000 iterations used as burn-in. The convergence is determined by performing the Gelman–Rubin diagnostic (see Supporting Information). Once the MCMC samples have been obtained, the maximum-a-posteriori (MAP) estimate,
. A Metropolis–Hastings type of sampler is used to sample the haplotype (40). The MCMC (Markov chain Monte Carlo) sampler is initially run for 1500 iterations, with 1000 iterations used as burn-in. The convergence is determined by performing the Gelman–Rubin diagnostic (see Supporting Information). Once the MCMC samples have been obtained, the maximum-a-posteriori (MAP) estimate, 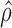 and
and 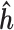 , for
, for 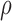 and
and 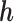 are calculated.
are calculated.
In the aforementioned model, SGS short reads and TGS long reads are used in the same way. The utility of the long reads is their ability to cover multiple SNVs. In the likelihood for each read (each row of the read matrix in Figure 1), only the loci that are covered by a read can contribute to the likelihood. Thus, long reads can contribute more information to the likelihood than short reads. As the MCMC explores the same space of 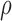 and
and 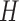 , it will tend to favor haplotypes which correspond with the long reads. Another notable point for the usage of long reads is that raw sequencing long reads should be used instead of corrected long reads. This is important because any correction for raw long reads will eliminate the SNV information embedded in long reads.
, it will tend to favor haplotypes which correspond with the long reads. Another notable point for the usage of long reads is that raw sequencing long reads should be used instead of corrected long reads. This is important because any correction for raw long reads will eliminate the SNV information embedded in long reads.
Statistical model for quantifying ASE at gene isoform level
Although ASE at the gene level can be estimated as above, these data represent a pooled mixture of gene isoforms that can have heterogeneous ASE. Estimating ASE for each gene isoform within a gene is necessary to truly quantify the final transcriptional products. Given the relatively low throughput and sequencing bias of TGS data, only SGS short reads are used in the statistical model below to estimate ASE at the gene isoform level.
Consider a gene with 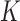 isoforms. Without loss of generality, we assume each isoform contains a SNV. For the
isoforms. Without loss of generality, we assume each isoform contains a SNV. For the 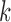 th isoform, let
th isoform, let 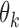 be the abundance in the observed sample.
be the abundance in the observed sample. 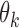 can further be decomposed into
can further be decomposed into 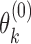 and
and 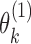 , which are the allele-specific abundance of isoform
, which are the allele-specific abundance of isoform 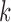 corresponding to haplotype
corresponding to haplotype 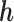 and
and 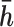 respectively. To obtain estimates of
respectively. To obtain estimates of 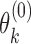 and
and 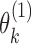 , we proceed with a two-stage procedure. In Stage 1, we identify the set of expressed isoforms from the Hybrid-Seq data by our previously published tool, IDP (Isoform Detection and Prediction) (31). Although a reference annotation library can be used instead, a sample-specific annotation library can provide more accurate abundance estimation of isoform (Additional file 1: Supporting information). Next, Stage 2 of IDP-ASE uses
, we proceed with a two-stage procedure. In Stage 1, we identify the set of expressed isoforms from the Hybrid-Seq data by our previously published tool, IDP (Isoform Detection and Prediction) (31). Although a reference annotation library can be used instead, a sample-specific annotation library can provide more accurate abundance estimation of isoform (Additional file 1: Supporting information). Next, Stage 2 of IDP-ASE uses 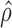 and
and 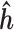 obtained above and extends Jiang and Wong's Poisson model of short read coverage to estimate
obtained above and extends Jiang and Wong's Poisson model of short read coverage to estimate 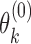 and
and 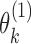 by Maximum Likelihood Estimation (MLE) (41).
by Maximum Likelihood Estimation (MLE) (41).
For the gene of interest, define the exon regions of the gene as the non-overlapping set of exons that comprise the isoforms of the gene. Let 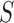 be the number of exon regions and junction regions spanning multiple exons. Furthermore, each region can be distinguished by the SNVs that it contains and whether these SNVs are consistent with haplotype
be the number of exon regions and junction regions spanning multiple exons. Furthermore, each region can be distinguished by the SNVs that it contains and whether these SNVs are consistent with haplotype 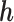 or
or 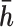 . So, there can be a total of
. So, there can be a total of 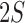 exon/junction regions. Define effective length as the number of positions from which a read could map to the region. Let
exon/junction regions. Define effective length as the number of positions from which a read could map to the region. Let 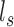 denote the effective length of the
denote the effective length of the 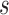 th region (Figure 1). Any junction region with non-positive effective length is not considered to be part of the model.
th region (Figure 1). Any junction region with non-positive effective length is not considered to be part of the model.
Let 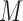 be the total number of short read sequences that map to the gene of interest. Each read will fall into either an exon region or a junction region. This model assumes short read sequencing is a simple random process, in which every read is sampled independently and uniformly from every possible position in the sample. Denote the number of short reads that fall into the
be the total number of short read sequences that map to the gene of interest. Each read will fall into either an exon region or a junction region. This model assumes short read sequencing is a simple random process, in which every read is sampled independently and uniformly from every possible position in the sample. Denote the number of short reads that fall into the 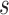 th region as
th region as 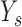 and assume
and assume 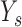 follows a Poisson distribution. Then
follows a Poisson distribution. Then
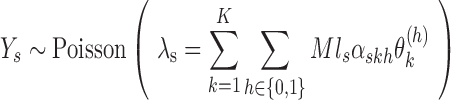 |
where 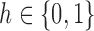 corresponds to the haplotype,
corresponds to the haplotype, 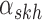 is 1 if region
is 1 if region 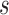 is contained in isoform
is contained in isoform 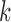 (with haplotype
(with haplotype 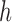 ) and 0 otherwise, and
) and 0 otherwise, and 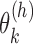 is the abundance of the
is the abundance of the 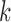 th isoform under haplotype
th isoform under haplotype 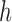 .
.
When distributing reads into regions, we calculate 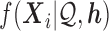 and
and 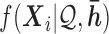 if the
if the 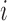 th read covers a SNV. Then this read is assigned to haplotype
th read covers a SNV. Then this read is assigned to haplotype 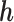 with probability
with probability
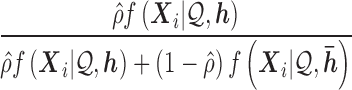 |
As the concavity of the Poisson likelihood was shown by Jiang and Wong, IDP-ASE uses the Newton–Raphson algorithm to obtain the MLE for 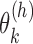 . Let
. Let 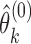 and
and 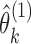 correspond to the estimates, respectively. The isoform specific relative ASE,
correspond to the estimates, respectively. The isoform specific relative ASE, 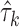 is then calculated as
is then calculated as
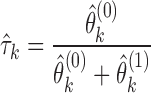 |
RESULTS
Haplotyping performance
To evaluate the haplotyping performance, IDP-ASE was applied to the gold standard GM12878 (33), the haplotypes of which have been well determined by 1000 Genome Project and Illumina Platinum Genomes Project (42). The Hybrid-Seq transcriptome data of GM12878 includes 715 902 PacBio long reads (median length is 1081 bp and up to 6217 bp) and 106 675 299 paired-end Illumina short reads (101 bp). Based on short reads, 19,907 heterozygous exonic SNVs from 5841 genes were called by GATK, 82.40% (16,383) of which were consistent with gold standard in 1000 Genome Project or Illumina Platinum Genomes Project (Additional file 1: Supplementary Figure S1). Among 5841 genes, we found that 58.60% of genes had multiple SNVs requiring phasing, and a significant proportion (15.56%, 909) of genes contain five or more SNVs, in which haplotyping is very difficult (Figure 2A).
Figure 2.

Performance of haplotyping by GM12878 data. (A) Gene distribution with different number of heterozygous exonic SNVs in exon regions, including known genes annotated by RefSeq and novel genes predicted by IDP. (B) Average CHPR and (C) CSPR of genes grouped by numbers of SNVs in a gene. Gray solid line represents randomly phasing process. Red and blue colors indicate Hybrid-Seq data and SGS-only data, respectively. Solid and dashed lines indicate IDP-ASE and MBASED, respectively.
Two metrics were designed to measure the haplotyping accuracy: (i) Correct Haplotype Phasing Rate (CHPR): proportion of the whole haplotypes correctly determined and (ii) Correct SNV Phasing Rate (CSPR): proportion of SNVs correctly phased within a gene. IDP-ASE with Hybrid-Seq data obtained very high CHPR and CSPR with an average of 62.96% and 88.37%, respectively, for multi-SNV genes. We found that 49.06% of genes with five or more SNVs could be phased perfectly, while the successful rate by random haplotyping was 6.25% or lower (Figure 2B). In addition, 85.78% of SNVs in the genes with five or more SNVs could be properly phased, which means only about one SNV on average was incorrectly phased when a gene was not perfectly phased (Figure 2C).
Overall, IDP-ASE with Hybrid-Seq data provided the best haplotyping results. Both CHPR and CSPR dropped dramatically to a similar level as random haplotyping when only SGS data was used in IDP-ASE. These data established the useful haplotyping information provided by TGS long reads but not SGS short reads. In contrast, for MBASED, there was a negligible difference between Hybrid-Seq and SGS-only data. The similarity of MBASED with Hybrid-Seq input and IDP-ASE with SGS-only input also established that MBASED did not make use of the valuable information of the TGS long reads. Therefore, IDP-ASE provides an appropriate data analysis method required to fully utilize long reads in haplotyping.
The influences of sequencing coverage on haplotyping
To elucidate the influence of TGS long reads on haplotyping, we examined the changes of CHPR and CSPR with respect to the adjusted long read coverage (see definition in Supporting Information). Briefly, adjusted long read coverage represents the depth of long reads (i.e. the number of long read mapped to the gene) as well as the length (i.e. the maximum number of SNVs covered by single long reads). The depth measures the data size and the length is a metric of how well long reads can link multiple SNVs. When the log of adjusted long read coverage was 0, IDP-ASE performed similarly with MBASED with CHPR around 0.4 and CSPR around 0.8. As the log of adjusted long read coverage increased, CHPR and CSPR of IDP-ASE output improved linearly and approached 0.8 and 0.95, respectively (Figures 3A and 4B). However, neither CHPR nor CSPR improved with MBASED because this tool was not developed to use the SNV linkage information from long reads but only utilizes the marginal allele counts of the read matrix (11). Therefore, an increase in sequencing depth with longer read length can improve haplotyping, which the statistical approach of IDP-ASE can take advantage.
Figure 3.

Influence of sequencing coverage on haplotyping. (A–D) Average CHPR and CSPR versus the log of adjusted long reads coverage (A and B) and the log of adjusted short reads coverage (C and D). Genes are grouped by the 10 percentiles of the log adjusted coverage and a smooth loess curve is fit to the data. Red and blue colors indicate Hybrid-Seq data and SGS-only data, respectively. Solid and dashed lines indicate IDP-ASE and MBASED, respectively.
Figure 4.

Performance of ASE quantification at gene level by simulation data. (A) It shows the performance of 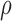 estimation in simulation data, measured by Euclidean distance of the true value to the estimated value in genes with same number of SNVs. (B) and (C) show the density distribution of
estimation in simulation data, measured by Euclidean distance of the true value to the estimated value in genes with same number of SNVs. (B) and (C) show the density distribution of 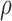 estimates in semi-simulation data by IDP-ASE (B) and MBASED (C). The dotted line indicates the simulation density. Red, blue and green colors indicate Hybrid-Seq data, SGS-only data and TGS-only data, respectively. Solid and dashed lines indicate IDP-ASE and MBASED, respectively.
estimates in semi-simulation data by IDP-ASE (B) and MBASED (C). The dotted line indicates the simulation density. Red, blue and green colors indicate Hybrid-Seq data, SGS-only data and TGS-only data, respectively. Solid and dashed lines indicate IDP-ASE and MBASED, respectively.
When investigating the influences of adjusted short read coverage on haplotyping, we found adjusted short read coverage can also improve CHPR and CSPR of IDP-ASE with Hybrid-Seq input data (Figure 3C and D). However, using SGS-only data, minimal improvement was obtained for either tool as adjusted short read coverage increase. Therefore, the improvement in haplotyping with an increase in adjusted short read coverage likely results from the increase of long read depth, considering that depths of long reads and short reads are correlated via gene abundance (Additional file 1: Supplementary Figure S2).
Since differences in gene abundance result in differences in sequencing coverage, haplotyping by transcriptome sequencing data can cause a large variability in accuracy. Based on the gold standard GM12878 as training data, IDP-ASE can predict CHPR and CSPR using the adjusted long reads coverage, which will be very informative for estimating the haplotyping accuracy of a gene of interest and to select the candidate genes for follow-up research (Additional file 1: Supporting information and Supplementary Figure S3).
Quantification of ASE at the gene level
To evaluate the estimate of ASE at the gene level, we generated a semi-simulation data based on GM12878 data as described before (see Supporting Information) (11). We retained information about total sequencing coverage of each heterozygous SNV detected and discarded the observed reference and alternative allele counts. Next, ASE patterns were artificially generated at different genes at various allele preferences and expression levels. The simulated data set has realistic distributions of both the number of heterozygous SNVs per gene and the read coverage per SNV.
The estimated errors of both IDP-ASE and MBASED were largest when only TGS long reads were input (Figure 4A), likely due to the relatively low-throughput and sequencing bias of TGS. In contrast, both tools provided smaller errors from SGS-only data, which was more suitable for quantitative analysis. Though the high throughput and less sequencing bias of SGS data is useful for ASE quantification, proper haplotyping is key for deconvolution of SGS coverage of alleles. As a result, IDP-ASE with Hybrid-Seq data provided the best estimates of ASE at the gene level. Moreover, IDP-ASE outperformed MBASED in analysis of all data (Figure 4A).
The distribution of the ASE estimate using IDP-ASE corresponds closely to the density of the simulated values, which were truncated Gaussian (truncated from 0 to 1 and centered at 0.5) (Figure 4B). In contrast, MBASED missed a significant proportion of ASE at the 0.5 vicinity (Figure 4B and C). This suggests a better ASE estimation performance by IDP-ASE around 0.5, where the ASE bias is so small that MBASED failed to estimate.
Quantification of ASE at the gene isoform level
We tested the quantification performance of ASE at the gene isoform level using semi-simulation data that retained the realistic sequencing coverage distribution of GM12878 but simulated allele-specific isoform abundance by Gamma distribution (see Supporting Information). The estimate of allele-specific isoform abundance highly correlated with the true values (R2 = 85.94) (Figure 5A). Moreover, the estimates of allele isoform abundance was unbiased since the difference between the true value and the estimate was centered at 0 with a standard deviation of 0.37 (Figure 5B). In addition, the estimate of isoform level ASE 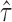 was also unbiased (Figure 5D). Therefore, IDP-ASE can estimate ASE at the gene isoform level with high accuracy.
was also unbiased (Figure 5D). Therefore, IDP-ASE can estimate ASE at the gene isoform level with high accuracy.
Figure 5.

Performance of estimations of FPKM and 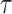 in simulation data. (A) True FPKM is plotted against estimated FPKM. (B) The gene density distribution of the difference between true FPKM and estimated FPKM. The horizontal axis represents the difference between true FPKM and estimated FPKM, and the vertical axis represents gene density. (C) True
in simulation data. (A) True FPKM is plotted against estimated FPKM. (B) The gene density distribution of the difference between true FPKM and estimated FPKM. The horizontal axis represents the difference between true FPKM and estimated FPKM, and the vertical axis represents gene density. (C) True 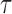 is plotted against estimated
is plotted against estimated 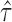 . (D) The gene density distribution of the difference between true
. (D) The gene density distribution of the difference between true 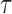 to estimated
to estimated 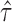 . The horizontal axis represents the difference between true
. The horizontal axis represents the difference between true 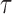 and estimated
and estimated 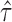 , and the vertical axis represents gene density.
, and the vertical axis represents gene density.
It is important to quantify isoform level ASE with correct haplotyping and sample-specific isoform library, because of the complex cooperation of gene isoform expression and ASE. For example, in MCF-7, we discovered two novel isoforms in gene PPP2R3C (Protein Phosphatase 2, Regulatory Subunit B, Gamma), which also expressed an annotated isoform NM_017917. The haplotypes TG/CA predicted by IDP-ASE was supported by 75 PacBio long reads (Figure 6). Due to the expression of two novel isoforms that did not contain SNV2, the coverage ratios at SNV2 (G = 24/A = 17) were opposite to SNV1 (T = 42/C = 84). Based the reads count ratio of major allele and minor allele, the ‘pseudo phasing’ procedure used by MBASED called an incorrect haplotype (CG/TA) and subsequently incorrectly estimated ASE. Correct isoform identification and haplotyping by long reads allows IDP-ASE to interpret the sequencing coverage properly and find distinct ASE at three isoforms of PPP2R3C (the corresponding 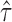 are 0.59, 0.23 and 0.16). In addition, it also suggests that the coverage ratios at single SNVs cannot represent the true ASE at the gene or gene isoform level.
are 0.59, 0.23 and 0.16). In addition, it also suggests that the coverage ratios at single SNVs cannot represent the true ASE at the gene or gene isoform level.
Figure 6.

Influence of correct haplotyping and isoform identification on ASE analysis at isoform level by one example from MCF-7 data. PPP2R3C gene with phased TG/CA haplotype has SNV1 (T = 42/C = 84, number represents Illumina short reads, same in Figure 8) and SNV2 (G = 24/A = 17). Of two SNVs, SNV1 is shared by three isoforms and SNV2 is used only by known isoform NM_017917 annotated by RefSeq but not two novel isoforms (Novel isoform 1 and Novel isoform 2) identified by IDP. Three isoforms show distinct ASE patterns which would be missed by ‘pseudo phasing’ procedure of MBASED.
Presence of pseudogenes may impact the performance of our method. If a gene has a pseudogene pair then many of the reads will be aligned to both regions. Since our analysis only uses uniquely mapped reads, these multiply mapped reads will not be considered in our analysis. This can become an issue in genes with low coverage, potentially resulting in an underestimate of the abundance of these genes (Additional file 1: Supporting information and Supplementary Figure S4).
ASE analysis of human embryonic stem cells and breast cancer cells
To demonstrate the utility of IDP-ASE, we analyzed the ASE events in human embryonic stem cells (H1 cell line) and breast cancer cells (MCF-7 cell line) as both were reported to have diverse transcript expression (31,34). In H1, 6508 SNVs from 3078 genes, including 1480 genes with multiple SNVs, were called from 93 880 208 101 bp Illumina short reads. In MCF-7, 5588 SNVs from 2523 genes, including 1270 genes with multiple SNVs, were called from 84 439 179 89 bp Illumina short reads. 2 289 890 and 6 170 149 PacBio long reads from H1 and MCF-7 were input to IDP-ASE, respectively.
The corresponding standard deviations of the gene-level ASE estimate 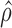 were 0.09 and 0.14 in H1 and MCF-7, respectively (Figure 7A and B), indicating more extensive ASE events detected in MCF-7 than H1. A total of 2461 genes (34.36%) in MCF-7 had significant ASE (
were 0.09 and 0.14 in H1 and MCF-7, respectively (Figure 7A and B), indicating more extensive ASE events detected in MCF-7 than H1. A total of 2461 genes (34.36%) in MCF-7 had significant ASE (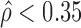 or
or 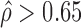 ) at the gene level as compared to only 649 genes (8.67%) in H1.
) at the gene level as compared to only 649 genes (8.67%) in H1.
Figure 7.

Density distribution of genes with different ASE levels at both gene and isoform levels. The horizontal axis represents estimated 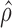 (gene level) and
(gene level) and 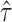 (isoform level). The vertical axis represents gene density. Red color shows significant ASE genes/isoforms (
(isoform level). The vertical axis represents gene density. Red color shows significant ASE genes/isoforms (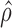 < 0.35 or
< 0.35 or 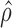 > 0.65 for gene level, and
> 0.65 for gene level, and 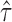 < 0.35 or
< 0.35 or 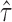 > 0.65 for isoform level). (A) and (C) show the analysis results in MCF-7. (B) and (D) show the analysis results in H1.
> 0.65 for isoform level). (A) and (C) show the analysis results in MCF-7. (B) and (D) show the analysis results in H1.
In addition, the variance in ASE at the gene isoform level was larger than at the gene level (Figure 7). That is, significant ASE at the gene isoform level may be concealed by the pooled gene-level ASE. 1083 gene isoforms (15.98%) in H1 and 2,500 (39.33%) gene isoforms in MCF-7 had significant allele-specific expression (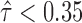 or
or 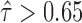 ). Among these genes in H1, four genes (CGGBP1, LARS, ZNF138 and ZNF43) are associated with embryonic stem cell identity based on a previous study (43). In addition, TDGF1 (Teratocarcinoma-Derived Growth Factor 1), which plays an essential role in embryonic development and tumor growth (44), also shows significant allele-specific expression at the gene isoform level but not at the gene level (Figure 8A). Notably, in MCF-7, nine ASE genes (BARD1, CASP8, CCND3, KRAS, MAPEK4, NF2, TET2, TP53 and ZFP36L1) are considered as driver genes in breast cancer (45). In particular, p53 is widely recognized as a tumor suppressor in many tumor types, and BARD1 interacts with N-terminal region of BRCA1 (46,47). We next categorized and exemplified the complexity of ASE at the gene isoform level (Figure 8).
). Among these genes in H1, four genes (CGGBP1, LARS, ZNF138 and ZNF43) are associated with embryonic stem cell identity based on a previous study (43). In addition, TDGF1 (Teratocarcinoma-Derived Growth Factor 1), which plays an essential role in embryonic development and tumor growth (44), also shows significant allele-specific expression at the gene isoform level but not at the gene level (Figure 8A). Notably, in MCF-7, nine ASE genes (BARD1, CASP8, CCND3, KRAS, MAPEK4, NF2, TET2, TP53 and ZFP36L1) are considered as driver genes in breast cancer (45). In particular, p53 is widely recognized as a tumor suppressor in many tumor types, and BARD1 interacts with N-terminal region of BRCA1 (46,47). We next categorized and exemplified the complexity of ASE at the gene isoform level (Figure 8).
Isoforms with mutually exclusive sets of SNVs: CROT (Carnitine O-octanoyltransferase) had two SNVs that were exclusively expressed by isoforms NM_021151 and NM_001243745, respectively.
Isoforms that share SNVs but also have mutually exclusive SNVs: two isoforms of KLC1 (Kinesin light chain 1) shared SNV1, while SNV2 and SNV3 were exclusively expressed in NM_182923 and NM_005552, respectively.
Isoforms that share all SNVs but some SNVs are expressed exclusively with isoform-specific junctions: three isoforms of LETMD1 (LETM1 domain containing protein 1) expressed all three SNVs, yet SNV1 located at the flanking region of isoform-specific junctions. Although three isoforms contained the same SNVs, their imbalance of ASE was distinct: NM_015416 had almost equal expression of the two alleles (
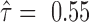 ), while NM_045018 was biased slightly to one allele (
), while NM_045018 was biased slightly to one allele ( 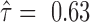 ) and NM_045020 biased to the other (
) and NM_045020 biased to the other ( 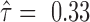 ).
).
Figure 8.

Complexity of ASE at isoform level by four genes. (A) Two isoforms of TDGF1 in H1 can be distinguished by their unique junction reads which cover SNV1, show opposite ASE patterns. (B) Two isoforms of CROT in MCF-7 contain one specific SNV, respectively. Two unique SNVs are used to estimate ASE, respectively. (C) Two isoforms of KLC1 in MCF-7 share SNV1, while SNV2 and SNV3 are exclusively expressed in NM_182923 and NM_005552, respectively. (D) Three isoforms of LETMD1 express all three SNVs, yet junction reads which cover SNV1 can be used to indicate different ASE patterns.
The extensiveness and complexity of ASE events in MCF-7 may be caused by the complicated gene regulation and expression in tumor transcriptome as well as the abnormal genome composition, such as structural variance and copy number variance. These results all indicate the importance of ASE analysis, especially at gene isoform level, which is particularly necessary for tumor transcriptome research.
Computational performance
We evaluate the computational performance of IDP-ASE based on the gold standard GM12878 dataset (33). For the gene level analysis, the total running time using a single process for the MCMC is 30.53 h for IDP-ASE using Hybrid-Seq, 8.19 h using TGS-only and 54.06 h using SGS-only data. The longer running time for SGS-only data is likely due to the difficulty of MCMC convergence as long reads are not available. Since each gene is independent, all of the genes can be run in parallel so that it only takes a few hours to run IDP-ASE. For distribution of running times for each gene, please see supporting information (Additional file 1: Supplementary Figure S5). MCMC output (trace plots, density estimates, and autocorrelation) is available for a few example genes (Additional file 1: Supplementary Figure S6).
The running time for the isoform level MLE program is much faster. The total running time is 0.22 h if the genes are run sequentially on a single processor. The median running time for a gene is 1.59 s.
DISCUSSION
The great advantage of IDP-ASE is the ability to study gene isoform ASE using only transcriptome data. In contrast, the existing tools either only study ASE at a single SNV site or at the gene level. The existing tools also require known haplotypes. The known haplotypes are always generated from laborious experiments or sequencing data from extra sources, such as genome data and family trio data, which greatly limit the capability of studying ASE. Our approach of integrating the complementary information in TGS (i.e. long read length) and SGS (i.e. high throughput and accuracy) by IDP-ASE can characterize ASE events with single sequencing materials (i.e. RNA). Without requiring the known haplotypes, IDP-ASE greatly extends our capability of studying ASE. The reliable sample-specific isoform identification by Hybrid-Seq data further allows us to study ASE at the gene isoform level. We compared IDP-ASE to an existing method and demonstrated superior performance, in particular the use of linkage information provided by long reads. However, haplotyping accuracy may depend on sequencing coverage and thus varies with respect to gene abundance. Using GM12878 as training data, the predictions of CHPR and CSPR exclusively provided by IDP-ASE can be very helpful for biologists to select better target candidates for follow-up characterization. Moreover, the complexity of ASE events in breast cancer cells and hESCs revealed by IDP-ASE highlights that ASE studies must take into consideration the gene isoform level.
Furthermore, with simple modifications, IDP-ASE can be generalized for the other proposes: (i) various genetic variants (short indels and structural variants) rather than only substitution; (ii) multiploid ASE analysis and (iii) copy number variants for genome data. Instead of sequencing quality score, customized error pattern could be input to better estimate the error probabilities. IDP-ASE is also compatible with the other TGS platforms (e.g. ONT, 10X genomics and Moleculo). In addition, the step of haplotyping can be skipped if a well-phased haplotype is available. A caveat of IDP-ASE is the analysis of only RNA sequencing data, which precludes identification of somatic mutations or RNA editing sites. A paired analysis of genome/exome sequencing data would resolve this issue.
To the best of our knowledge, IDP-ASE is the first method to quantify genome-wide ASE at the gene isoform level and solve haplotyping simultaneously, using only transcriptome data. As new TGS platforms (e.g. PacBio Sequel and ONT PromethION) with much lower costs have become more prevalent, the corresponding applications and publications have been increasing rapidly. Taking advantage of the exclusive information from TGS appropriately, IDP-ASE provides a timely method to achieve the gene isoform level analysis of diploid transcriptomes.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Kristina Thiel for critical reading of the manuscript.
Author contribution: K.F.A. conceived research; K.F.A. and B.D. developed methods. B.D. implemented software and performed tests; K.F.A., B.D., Y.W. and J.W. analyzed data; K.F.A., B.D. and Y.W. wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
KFA, YW and JW are supported by the National Human Genome Research Institute (R01HG008759). JW is supported by the Multidisciplinary Lung Research Career Development Program (T32HL007638). BD is supported by the Presidential Graduate Research Fellowship, University of Iowa. XW is supported by National Natural Science Foundation of China (No. 91540204). KFA, YW, JW, and BD are supported by the institutional fund of Department of Internal Medicine, University of Iowa.
Conflict of interest statement. None declared.
REFERENCES
- 1. Carrel L., Willard H.F.. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005; 434:400–404. [DOI] [PubMed] [Google Scholar]
- 2. Baran Y., Subramaniam M., Biton A., Tukiainen T., Tsang E.K., Rivas M.A., Pirinen M., Gutierrez-Arcelus M., Smith K.S., Kukurba K.R. et al. . The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015; 25:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giannoukakis N., Deal C., Paquette J., Goodyer C.G., Polychronakos C.. Parental genomic imprinting of the human Igf2 gene. Nat. Genet. 1993; 4:98–101. [DOI] [PubMed] [Google Scholar]
- 4. Chess A. Mechanisms and consequences of widespread random monoallelic expression. Nat. Rev. Genet. 2012; 13:421–428. [DOI] [PubMed] [Google Scholar]
- 5. Knight J.C. Allele-specific gene expression uncovered. Trends Genet. 2004; 20:113–116. [DOI] [PubMed] [Google Scholar]
- 6. Turro E., Su S.Y., Goncalves A., Coin L.J.M., Richardson S., Lewin A.. Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Genome Biol. 2011; 12:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gregg C. Known unknowns for allele-specific expression and genomic imprinting effects. F1000Prime Rep. 2014; 6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pirinen M., Lappalainen T., Zaitlen N.A., Dermitzakis E.T., Donnelly P., McCarthy M.I., Rivas M.A., Consortium G.. Assessing allele-specific expression across multiple tissues from RNA-seq read data. Bioinformatics. 2015; 31:2497–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eckersley-Maslin M.A., Thybert D., Bergmann J.H., Marioni J.C., Flicek P., Spector D.L.. Random monoallelic gene expression increases upon embryonic stem cell differentiation. Dev. Cell. 2014; 28:351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lowe W.L., Reddy T.E.. Genomic approaches for understanding the genetics of complex disease. Genome Res. 2015; 25:1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mayba O., Gilbert H.N., Liu J., Haverty P.M., Jhunjhunwala S., Jiang Z., Watanabe C., Zhang Z.. MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biol. 2014; 15:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. . The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Proc G.P.D.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ge B., Pokholok D.K., Kwan T., Grundberg E., Morcos L., Verlaan D.J., Le J., Koka V., Lam K.C., Gagne V. et al. . Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat. Genet. 2009; 41:1216–1222. [DOI] [PubMed] [Google Scholar]
- 15. Gimelbrant A., Hutchinson J.N., Thompson B.R., Chess A.. Widespread monoallelic expression on human autosomes. Science. 2007; 318:1136–1140. [DOI] [PubMed] [Google Scholar]
- 16. Lee J.H., Park I.H., Gao Y., Li J.B., Li Z., Daley G.Q., Zhang K., Church G.M.. A robust approach to identifying tissue-specific gene expression regulatory variants using personalized human induced pluripotent stem cells. PLoS Genet. 2009; 5:e1000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang K., Li J.B., Gao Y., Egli D., Xie B., Deng J., Li Z., Lee J.H., Aach J., Leproust E.M. et al. . Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat. Methods. 2009; 6:U613–U690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pastinen T. Genome-wide allele-specific analysis: insights into regulatory variation. Nat. Rev. Genet. 2010; 11:533–538. [DOI] [PubMed] [Google Scholar]
- 19. Rozowsky J., Abyzov A., Wang J., Alves P., Raha D., Harmanci A., Leng J., Bjornson R., Kong Y., Kitabayashi N. et al. . AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol. 2011; 7:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun W. A statistical framework for eQTL mapping using RNA-seq data. Biometrics. 2012; 68:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pandey R.V., Franssen S.U., Futschik A., Schlotterer C.. Allelic imbalance metre (Allim), a new tool for measuring allele-specific gene expression with RNA-seq data. Mol. Ecol. Resour. 2013; 13:740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soderlund C.A., Nelson W.M., Goff S.A.. Allele Workbench: transcriptome pipeline and interactive graphics for allele-specific expression. PLoS One. 2014; 9:e115740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harvey C.T., Moyerbrailean G.A., Davis G.O., Wen X., Luca F., Pique-Regi R.. QuASAR: quantitative allele-specific analysis of reads. Bioinformatics. 2015; 31:1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Romanel A., Lago S., Prandi D., Sboner A., Demichelis F.. ASEQ: fast allele-specific studies from next-generation sequencing data. BMC Med. Genomics. 2015; 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baker C.L., Kajita S., Walker M., Saxl R.L., Raghupathy N., Choi K., Petkov P.M., Paigen K.. PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype-specific initiation of meiotic recombination. PLoS Genet. 2015; 11:e1004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinn A., Juneja P., Jiggins F.M.. Estimates of allele-specific expression in Drosophila with a single genome sequence and RNA-seq data. Bioinformatics. 2014; 30:2603–2610. [DOI] [PubMed] [Google Scholar]
- 27. Skelly D.A., Johansson M., Madeoy J., Wakefield J., Akey J.M.. A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res. 2011; 21:1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rhoads A., Au K.F.. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics. 2015; 13:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. English A.C., Richards S., Han Y., Wang M., Vee V., Qu J.X., Qin X., Muzny D.M., Reid J.G., Worley K.C. et al. . Mind the gap: upgrading genomes with pacific biosciences RS long-read sequencing technology. PLoS One. 2012; 7:e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laver T., Harrisona J., O'Neill P.A., Moorea K., Farbos A., Paszkiewicz K., Studholmea D.J.. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015; 2015:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Au K.F., Sebastiano V., Afshar P.T., Durruthy J.D., Lee L., Williams B.A., van Bakel H., Schadt E.E., Reijo-Pera R.A., Underwood J.G. et al. . Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E4821–E4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharon D., Tilgner H., Grubert F., Snyder M.. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013; 31:1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tilgner H., Grubert F., Sharon D., Snyder M.P.. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:9869–9874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weirather J.L., Afshar P.T., Clark T.A., Tseng E., Powers L.S., Underwood J.G., Zabner J., Korlach J., Wong W.H., Au K.F.. Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res. 2015; 43:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Au K.F., Underwood J.G., Lee L., Wong W.H.. Improving PacBio long read accuracy by short read alignment. PLoS One. 2012; 7:e46679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koren S., Schatz M.C., Walenz B.P., Martin J., Howard J.T., Ganapathy G., Wang Z., Rasko D.A., McCombie W.R., Jarvis E.D. et al. . Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012; 30:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schueler M., Munschauer M., Gregersen L.H., Finzel A., Loewer A., Chen W., Landthaler M., Dieterich C.. Differential protein occupancy profiling of the mRNA transcriptome. Genome Biol. 2014; 15:R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Epstein M.P., Kwee L.C.. Haplotype association analysis. Handb. Anal. Hum. Genet. Data. 2010; 241–276. [Google Scholar]
- 39. Neal R.M. Slice sampling. Ann. Stat. 2003; 31:705–741. [Google Scholar]
- 40. Bansal V., Halpern A.L., Axelrod N., Bafna V.. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 2008; 18:1336–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang H., Wong W.H.. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009; 25:1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Altshuler D.M., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., Eichler E.E., Flicek P., Gabriel S.B. et al. . A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chia N.Y., Chan Y.S., Feng B., Lu X.Y., Orlov Y.L., Moreau D., Kumar P., Yang L., Jiang J.M., Lau M.S. et al. . A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature. 2010; 468:U316–U207. [DOI] [PubMed] [Google Scholar]
- 44. Kruithof-de Julio M., Alvarez M.J., Galli A., Chu J.H., Price S.M., Califano A., Shen M.M.. Regulation of extra-embryonic endoderm stem cell differentiation by Nodal and Cripto signaling. Development. 2011; 138:3885–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C. et al. . Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stracquadanio G., Wang X.T., Wallace M.D., Grawenda A.M., Zhang P., Hewitt J., Zeron-Medina J., Castro-Giner F., Tomlinson I., Goding C.R. et al. . The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat. Rev. Cancer. 2016; 16:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fackenthal J.D., Olopade O.I.. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer. 2007; 7:937–948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.