

SUMMARY
Respiratory and arthropod-borne viral infections are a global threat due to the lack of effective antivirals and vaccines. A potential strategy is to target host proteins required for viruses but non-essential for the host. To identify such proteins, we performed a genome-wide knockout screen in human haploid cells and identified the calcium pump SPCA1. SPCA1 is required by viruses from the Paramyxo-, Flavi-, and Togaviridae families, including measles, dengue, West Nile, Zika, and chikungunya viruses. Calcium transport activity is required for SPCA1 to promote virus spread. SPCA1 regulates proteases within the trans-Golgi network that require calcium for their activity and are critical for virus glycoprotein maturation. Consistent with these findings, viral glycoproteins fail to mature in SPCA1-deficient cells preventing viral spread, which is evident even in cells with partial loss of SPCA1. Thus, SPCA1 is an attractive antiviral host target for a broad spectrum of established and emerging viral infections.
Keywords: respiratory syncytial virus, haploid cell screen, ATP2C1, SPCA1, trans-Golgi network, furin, glycoprotein processing, Hailey-Hailey disease
IN BRIEF
In this issue of Cell Host & Microbe, Hoffmann et al. determine that the calcium pump, SPCA1, is required for virus spread. Loss of SPCA1 reduces activity of calcium-dependent proteases in the trans-Golgi network and consequently, hinders viral glycoprotein maturation. Thus, SPCA1 is an attractive host target for viral infections.

INTRODUCTION
The human respiratory syncytial virus (RSV) is a major pathogen infecting the upper and lower respiratory tract, resulting in significant morbidity and mortality in children as well as the immunocompromised and elderly populations. Estimates suggest that each year, RSV leads to 3.4 million hospitalizations and at least 66,000 deaths in children under five years of age (Falsey et al., 2005; Hall et al., 2009; Nair et al., 2010; Shay et al., 1999). Despite ongoing efforts, there are no vaccines against RSV and the only treatments available are passive immunoprophylaxis (Shadman and Wald, 2011) and nebulized ribavirin (Smith et al., 1991). Clearly, new treatment strategies are needed.
One treatment strategy with therapeutic potential is to target host proteins that RSV requires for infection. To do this, however, suitable target proteins must first be identified. In recent years, genome-wide knockout (KO) screens in human haploid cells—and most recently, CRISPR KO screens in a variety of cells—have been used to identify host factors required by several viruses from different families e.g. Arenaviridae, Bunyaviridae, Filoviridae, Flaviviridae, Orthomyxoviridae and Parvoviridae (Carette et al., 2009; Carette et al., 2011; Jae et al., 2014; Jae et al., 2013; Marceau et al., 2016; Pillay et al., 2016; Riblett et al., 2016; Savidis et al., 2016; Zhang et al., 2016). In all cases, genes that are essential for N-linked glycosylation were highly enriched in the screens. This is likely because cell-surface sugars play an important role in virus attachment prior to receptor engagement. While these genome-wide KO screens were highly successful in identifying entry factors and virus receptors (Pillay and Carette, 2015), few host factors have been described and characterized that interfere with later steps in the viral life cycle such as virus maturation and egress. And unlike receptors, which are often virus specific, host genes required for late stages of the viral life cycle might affect many viruses of the same family or even viruses from different families. With their potential for broad antiviral impact, identifying these host factors may be of value.
Here, we describe a genome-wide KO screen performed in human haploid cells to identify host factors required for RSV infection. Like similar screens with other viruses, proteins involved in N-linked glycosylation were highly enriched. However, we also identified the calcium pump, SPCA1, which resides in the trans-Golgi network (TGN). Unlike many of the other hits in the screen that are likely to affect virus entry, our mechanistic studies suggest that SPCA1 is critical for late stages of the virus lifecycle. Indeed, we show that SPCA1 influences protease activity in the Golgi and is required for proper viral glycoprotein processing and virus maturation. Further, we show that in addition to RSV, a variety of RNA viruses require SPCA1 for infection. We also provide evidence that even partial reduction in SPCA1 protein levels negatively impacts virus spread. Taken together, these features make SPCA1 a promising target for therapeutic intervention against a diverse set of emerging viruses.
RESULTS
A genome-wide screen identifies SPCA1 as a host factor required for RSV infection
To identify host factors required for RSV infection as potential therapeutic targets, we performed a screen utilizing a library of 1 × 108 human haploid cells (Hap1) that were mutagenized with a retroviral gene trap as reported previously (Carette et al., 2009; Carette et al., 2011; Jae et al., 2014; Jae et al., 2013; Pillay et al., 2016). Cells that survived RSV challenge were harvested and gene trap insertion sites were identified by DNA sequencing (Figure 1A and Table S1).
Figure 1. Fishtail plot of hits identified in a RSV screen.
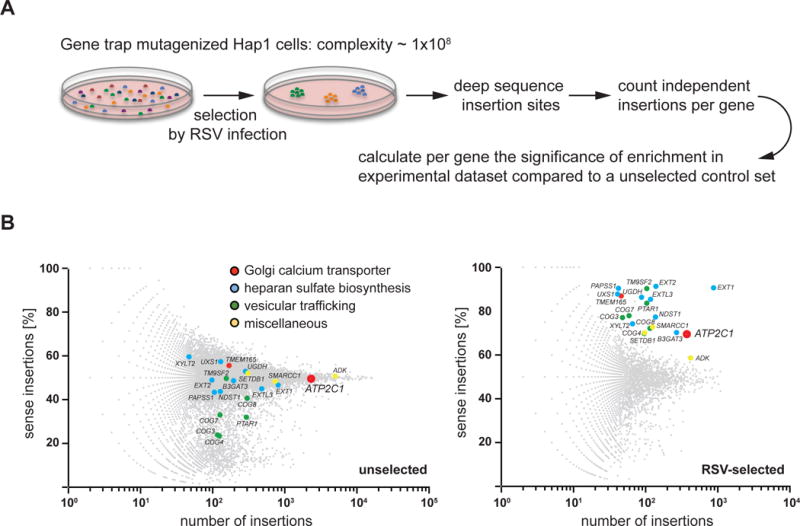
(A) Outline of a genetic haploid screen. (B) Fishtail plots of the unselected control set and the RSV selected set. Significant hits that were identified in the RSV screen are labeled by name and shown in both unselected and RSV selected sets to highlight enrichment. Distinct gene-trap insertions were mapped and counted taking into account their orientation relative to the affected genes. Per gene, the percentage of sense orientation gene-trap insertions (y axis) and the total number of insertions in a particular gene (x axis) are plotted. Hits are colored to indicate their involvement in different pathways: blue = heparan sulfate biosynthesis; green = vesicular trafficking; red = Golgi calcium transporters; and yellow = miscellaneous. See also Table S1.
Surviving cells showed significant enrichment for disruptive integrations in numerous genes compared to non-infected cells (Blomen et al., 2015). Genes involved in heparan sulfate biosynthesis were top hits, as were genes important for vesicle trafficking and protein maturation within the endoplasmic reticulum (ER) and Golgi apparatus (Figure 1B). Similar genes were recently identified in host factor screens for dengue virus (DENV), West Nile virus (WNV), Zika virus (ZIKV) and Rift Valley fever virus (Marceau et al., 2016; Riblett et al., 2016; Savidis et al., 2016; Zhang et al., 2016). We also identified SPCA1, a secretory pathway calcium (Ca2+) transport ATPase encoded by ATP2C1 (Figure 1B) that facilitates Ca2+ and Mn2+ uptake into the trans-Golgi network (TGN) (Behne et al., 2003; Lissandron et al., 2010; Micaroni et al., 2010). The TGN is rich in enzymes (e.g. glycosyl- and sulfotransferases, and proprotein convertases) that require Ca2+ and/or Mn2+ as cofactors (Kaufman et al., 1994; Oda, 1992; Vanoevelen et al., 2007), and since SPCA1 has not previously been reported to influence virus infection, we chose to further investigate its role.
SPCA1-deficient cells fail to support spread of RSV and other paramyxoviruses
To confirm the importance of SPCA1 for RSV infection, we took advantage of CRISPR genome editing to generate KO Hap1 cell clones (Figures S1A and S1B). Consistent with our screen, SPCA1 was non-essential for cell survival, and loss of SPCA1 did not affect cell growth (Figure S1C) (Blomen et al., 2015). We then infected wildtype (WT) and SPCA1 KO cells with RSV at a high multiplicity of infection (MOI = 1) to monitor initial infection at 24 h post infection (hpi) or a low MOI (MOI = 0.1) to monitor virus spread at 72 hpi. There was a moderate reduction in frequency of RSV-infected cells at 24 hpi when comparing KO cells to WT cells, and in the context of strong positive selection, it may be that this moderate effect on virus entry allowed SPCA1 KO cells to survive; however, the most prominent phenotype was observed at 72 hpi, when SPCA1 KO cells showed a striking decrease in virus spread. This phenotype, observed under low MOI conditions, is likely a reflection of the need for passage through the steps of virus assembly and egress. The reduced susceptibility to RSV infection and the defect in viral spread was rescued by trans-complementation of SPCA1 (Figures 2A and 2B). These results suggest that SPCA1 may play a role during entry but is primarily involved in the later stages of the virus life cycle. We made similar observations for human parainfluenza virus 3 (hPIV-3) and measles virus (MeV): hPIV-3 failed to spread in SPCA1 KO cells (Figures 2C and 2D), and MeV glycoprotein-mediated cell-cell fusion was significantly reduced (Figure 2E). Since few host factors that effect virus spread have been studied to date, we focused on characterizing the role of SPCA1 on virus spread.
Figure 2. Impaired spread of paramyxoviruses in SPCA1 KO cells.
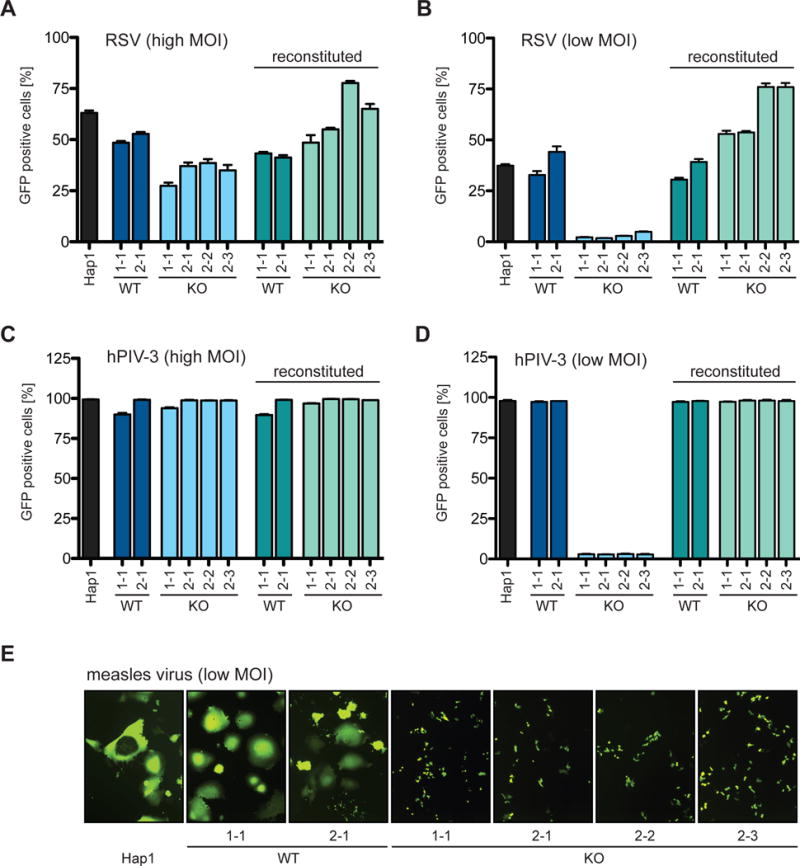
Hap1 cells, CRISPR-generated WT and KO clones of SPCA1 (indicated as 1-1 derived by sgRNA#1, and 2-1, 2-2 and 2-3 derived by sgRNA#2) and SPCA1-reconstituted clones infected with (A) RSV-GFP at a MOI of 1 and (C) hPIV-3-GFP at a MOI of 4 harvested at 24 hpi. Hap1 cells, CRISPR-generated Hap1 clones and SPCA1-reconstituted clones infected with (B) RSV-GFP at a MOI of 0.1 and (D) hPIV-3-GFP at a MOI of 0.01 harvested at 72hpi and 48hpi, respectively. (A-D) Harvested cells were analyzed by flow cytometry and plotted as a percentage of GFP positive cells. Data represent the mean and SD of 3 independent experiments. Cells are colored differently to indicate: parental WT = black; CRISPR WT clones = dark blue and green; and CRISPR KO clones = light blue and green. (E) Hap1 cells and CRISPR-generated Hap1 clones infected with MeV-GFP at a MOI of 0.1 and fixed for microscopy at 36 hpi (10× magnification). See also Figures S1, S4 and S5.
SPCA1’s ability to transport Ca2+ is critical for viral spread
There are nine protein-coding isoforms of ATP2C1: isoforms 1A–F and 2A–C (Figure S1A). We used the predominantly expressed isoforms, 1A–F, to reconstitute SPCA1 KO cells, and all but the 1C isoform rescued viral spread (Figures 3A and 3C). It has been shown that isoform 1C does not transport Ca2+ and is rapidly degraded, which suggests that SPCA1’s ability to transport Ca2+ might be important for viral spread (Dode et al., 2005). To test this, we reconstituted the SPCA1 KO cells with a mutated form of isoform 1D that has reduced Ca2+ binding ability (Fairclough et al., 2003) and found that it failed to rescue viral spread (Figures 3B and 3D). To further confirm this, we treated SPCA1 KO cells with calcimycin, an ionophore that promotes Ca2+ flux across membranes, and found that it also rescued SPCA1 deficiency. This is illustrated by an increase in MeV glycoprotein-mediated syncytia formation upon addition of calcimycin (Figure S2A). These results suggest that SPCA1’s ability to pump Ca2+ into the TGN is critical for producing functional viral glycoproteins that permit virus spread.
Figure 3. Hap1 cells reconstituted with different SPCA1 isoforms and point mutations.
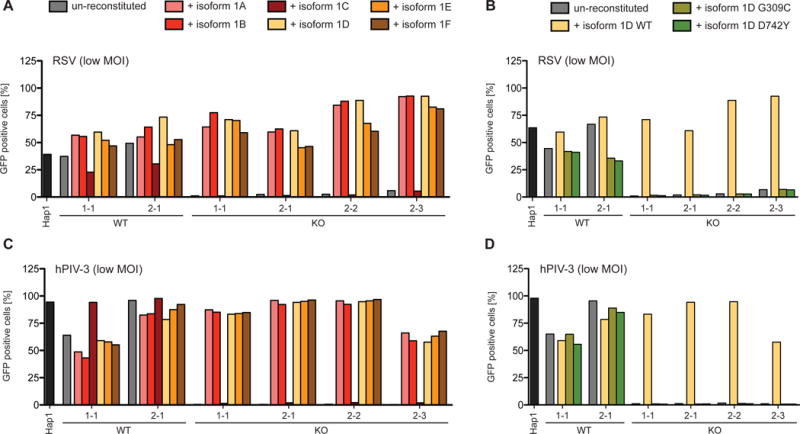
CRISPR-generated WT and KO clones of SPCA1 were reconstituted with (A and C) SPCA1 isoforms 1A-F and (B and D) isoform 1D bearing Hailey-Hailey disease-derived point mutations (G309C and D742Y) important in Ca2+-ion binding. Hap1 cells, un-reconstituted and reconstituted clones were infected with (A and B) RSV at a MOI of 0.1 and (C and D) hPIV-3-GFP at a MOI of 0.01. Cells, harvested (A and B) at 72 hpi and (C and D) at 48 hpi, were analyzed by flow cytometry and plotted as a percentage of GFP positive cells. See also Figures S1 and S2.
SPCA1 deficiency decreases the abundance of furin
The TGN contains proteases such as furin, which are important for maturation of viral glycoproteins and require appropriate pH and Ca2+ concentration for activity (Vanoevelen et al., 2007). Reduced Ca2+ in the TGN of SPCA1 KO cells may therefore affect protease activity. Indeed, we found that furin protein levels are decreased in SPCA1 KO cells and are restored upon SPCA1 trans-complementation, suggesting that furin is destabilized under reduced Ca2+ conditions (Figures 4F and 4G). Furthermore, treating WT cells with a proprotein convertase inhibitor that inhibits furin, Dec-RVKR-CMK, phenocopied the hPIV-3 and MeV spread defect in SPCA1 KO cells (Figures S2B and S2C). Interestingly, while the spread of both RSV and hPIV-3 is impaired in SPCA1 KO cells, in Lovo cells, which are devoid of furin activity, only the spread of RSV is impaired (Figures S3A–S3D) (Takahashi et al., 1995). This suggests that in addition to furin, hPIV-3 can utilize other proprotein convertases, which are also impaired in SPCA1 KO cells.
Figure 4. Paramyxoviruses produced in SPCA1 KO cells are less infectious.
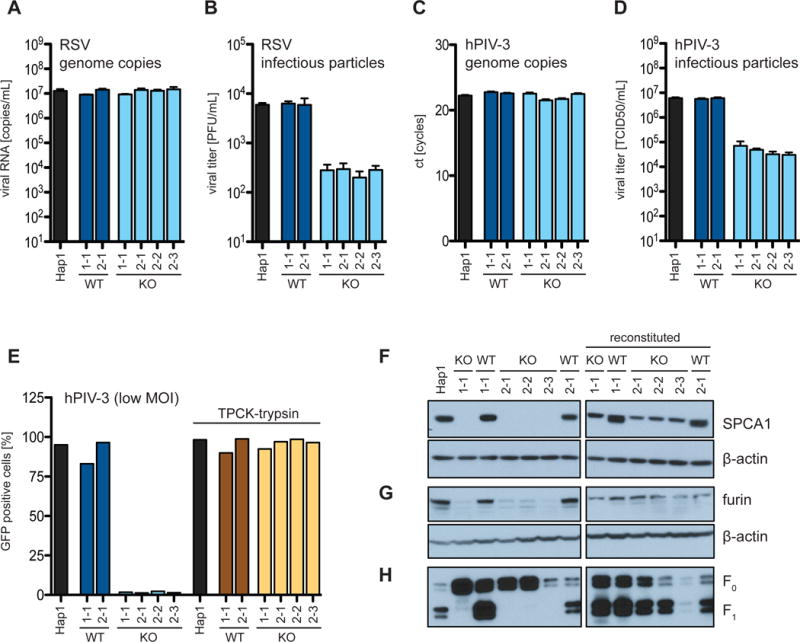
(A and B) Hap1 cells infected with RSV-GFP at a MOI of 1 and (C and D) with hPIV-3-GFP at a MOI of 4. The supernatants were harvested at 24 hpi and viral genome copies determined by (A and C) qPCR and infectious particles by (B) plaque assay and (D) TCID50. (E) Hap1 cells were infected with hPIV-3-GFP at a MOI of 0.01. Cells were cultured in low serum conditions (0.1% FBS) and supplemented as indicated with TPCK-trypsin (1 μg/ml). (A-D) Data represent the mean and SD of 3 independent experiments. Cells are colored differently to indicate: parental WT = black; CRISPR WT clones = dark blue and brown; and CRISPR KO clones = light blue and yellow. (F and G) Western blot analysis of Hap1 cells, CRISPR-generated Hap1 clones and SPCA1-reconstituted clones for (F) SPCA1 and (G) furin. (H) Infection of Hap1 cells, CRISPR-generated Hap1 clones and SPCA1-reconstituted clones with hPIV-3-GFP at a MOI of 2. Viral particles released into supernatants were harvested at 24 hpi and probed by western blot for the viral fusion protein F in its uncleaved F0 and cleaved F1 conformation.
Like furin, all known proprotein convertases undergo a series of orchestrated autoproteolytic cleavage steps (Anderson et al., 2002). For different convertases, these maturation events can occur in distinct compartments modulated by pH and Ca2+ concentration (Vanoevelen et al., 2007). Imbalances in these ion concentrations can affect proprotein stability and activation. SPCA1 resides in the TGN and is the sole provider of Ca2+ for this compartment. Therefore loss of SPCA1 may impact multiple proprotein convertases in the TGN. Indeed, furin is localized to the TGN and furin-dependent viruses fail to spread in SPCA1 KO cells. In contrast, lymphocytic choriomeningitis virus (LCMV), which utilizes the early Golgi protease SKI-1/S1P (Beyer et al., 2003), is unaffected (Figure S4B). This is consistent with our results that show LCMV spread is unimpaired in furin-deficient Lovo cells (Figure S3F).
Viral glycoproteins are improperly cleaved in SPCA1-deficient cells
The decreased furin levels in SPCA1 KO cells suggested that they might produce virus particles with uncleaved glycoproteins that are fusion impaired. To test this, we infected WT and SPCA1 KO cells with hPIV-3 and RSV, collected the supernatants, and quantified viral RNA by qRT-PCR and infectivity of progeny virus by plaque assay. Supernatants from both cell types had a similar number of viral RNA copies, but supernatants from SPCA1 KO cells were significantly less infectious than those from WT cells (Figures 4A–4D).
Next, we evaluated the glycoprotein conformation of particles released from hPIV-3-infected WT and KO cells. HPIV-3 infectivity requires cleavage of the precursor F0 viral glycoprotein to mediate virus-cell membrane fusion and syncytia. For most paramyxoviruses this cleavage is performed by the cellular TGN protease, furin (Chang and Dutch, 2012). Notably, we found that the F proteins in viral particles released from SPCA1 KO cells were mostly uncleaved (Figure 4H). The defect in viral spread was fully rescued when we added trypsin to cell supernatants during viral infection (Figure 4E). These results support a model that when SPCA1 is absent or unable to transport Ca2+ into the TGN, furin is less active and glycoprotein processing and viral infectivity are reduced.
Selection of hPIV-3 variant capable of spread in SPCA1 deficient cells
Viruses readily adapt to environmental changes and arising escape mutations help to understand which viral proteins are targeted. We passaged RSV and hPIV-3 on SPCA1 KO cells to select a virus capable of spread. We were unable to select a resistant RSV variant; however, after five passages, two mutations appeared in the hPIV-3 F protein: D104G and G398D. The G398D mutation is located within the F protein’s antigenic site A and has been shown to confer resistance to neutralizing monoclonal antibodies (Coelingh and Tierney, 1989). It was identified in clinical isolates in conjunction with changes in the proteolytic cleavage site (Coelingh and Winter, 1990). Interestingly, the D104G mutation is located within the furin-binding pocket at the P6 position directly adjacent to the consensus furin cleavage site (Figure 5A). For other respiratory viruses, it has been shown that mutations in the F protein cleavage site can alter recognition by cellular proteases (Bando et al., 1991; Collins et al., 1993; Glickman et al., 1988; Tashiro et al., 1988). We therefore hypothesize that the D104G mutation facilitates hPIV-3 spread in SPCA1 deficient cells either by increasing the F protein’s susceptibility for furin cleavage or by creating a better substrate for a different protease (Remacle et al., 2008). Additional work is required to determine if D104G alone is sufficient for viral spread in SPCA1-deficient cells or if G398D also plays a role.
Figure 5. A passaged hPIV-3 isolate in SPCA1 KO cells and primary HAE cultures.
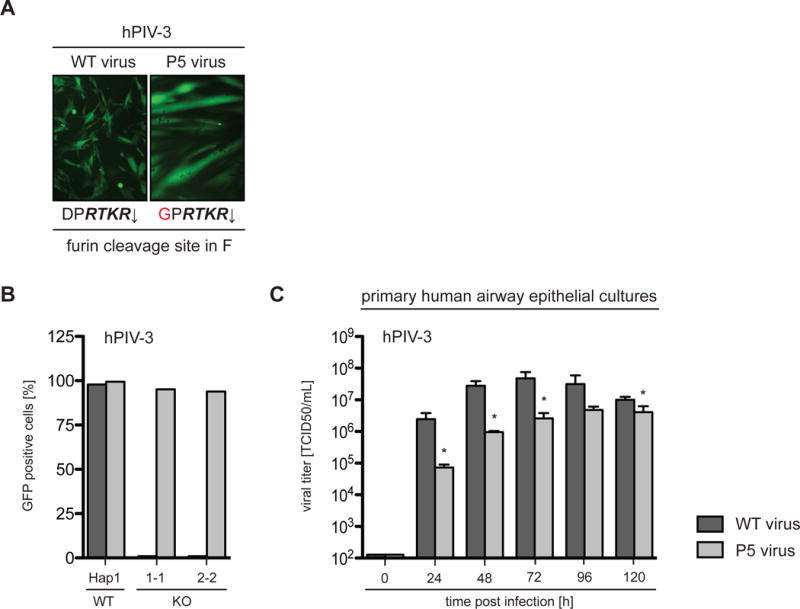
(A) The passaged hPIV-3-GFP isolate (P5) bears a mutation (D104G indicated in red) within the furin cleavage site of the fusion protein F. The sequence in italics represents the minimal furin consensus sequence with the arrow indicating the cleavage site. The microscopy pictures display infections of BHK-21 cells with both WT and P5 virus at a MOI of 1 at 24 hpi, indicating altered fusion properties. (B) Hap1 cells and SPCA1 KO clones were infected with both WT and passaged hPIV-3-GFP viruses at a MOI of 0.0001, harvested at 72 hpi, analyzed by flow cytometry and plotted as a percentage of GFP positive cells. (C) Primary human airway epithelial (HAE) cultures were infected with both WT and passaged hPIV-3-GFP viruses at a MOI of 0.1. Cultures were washed daily with PBS to harvest the released virus, which was subsequently titered by TCID50 assay. Data represent the mean and SD of 3 independent experiments. Statistical analysis was performed with a 2-tailed Student t-test and significance is stated with * p≤0.05. See also Figure S3.
The selected virus was able to efficiently spread in SPCA1 Hap1 KO cells, however it appeared to be more cytotoxic than the WT virus. Interestingly, when BHK-21 cells were infected with both viruses, we observed altered fusion properties with the selected variant being hyperfusogenic (Figure 5A). Furthermore, while the hPIV-3 variant spread efficiently in SPCA1 KO cells, it was less fit than WT virus in primary human airway epithelial (HAE) cell cultures. Together, this indicates there is a high fitness cost for this adaptation (Figures 5B and 5C).
Partial reduction of SPCA1 decreases viral burden in primary cells
SPCA1’s role in promoting virus spread was not limited to Hap1 cells as we observed similar defects in virus spread when SPCA1 was knocked out in A549 and HeLa cells. Furthermore, the defect was dose-dependent and apparent even in cells with reduced SPCA1 expression (Figure S5). Encouraged by these results, we obtained primary fibroblasts from patients with Hailey-Hailey disease (HHD) - a haplo-insufficiency disorder that occurs when only one allele of ATP2C1 is functional (Hu et al., 2000; Sudbrak et al., 2000). As expected, SPCA1 protein levels were lower in fibroblasts from HHD patients compared to those from WT controls, and though furin protein levels were similar (Figure S6A), hPIV-3 produced in patient-derived fibroblasts was less infectious than virus produced in WT cells. Further, we were able to fully restore hPIV-3 infectivity by adding trypsin (Figure 6E).
Figure 6. Decreased levels of SPCA1 impair viral spread and production of infectious particles.
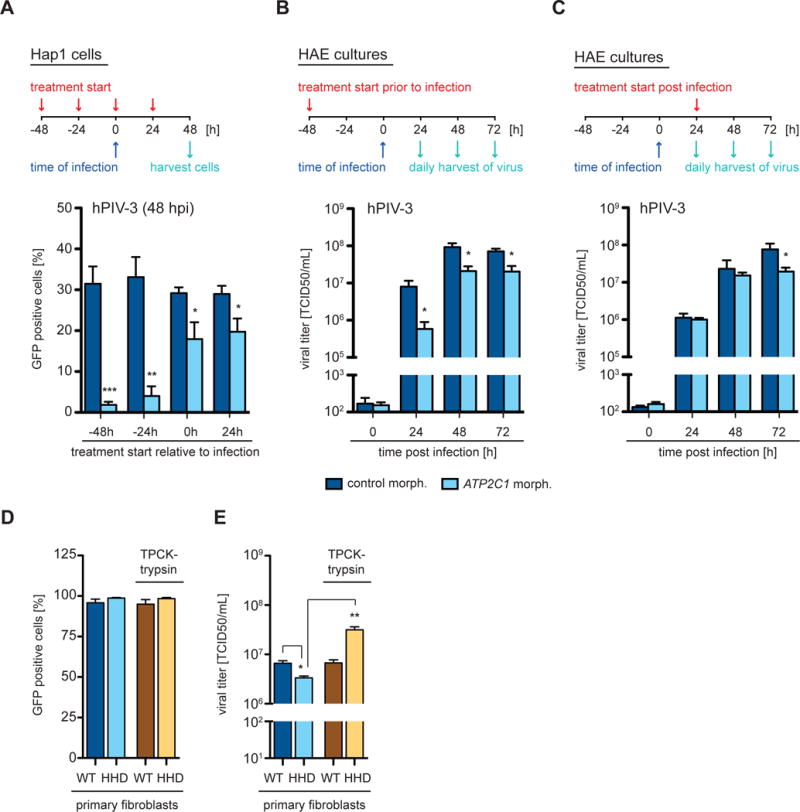
(A) Hap1 cells and (B and C) primary human airway epithelial (HAE) cultures were treated with a control Morpholino or a Morpholino targeting ATP2C1 at a concentration of (A) 5 μM and (B and C) 20 μM. The experiments are outlined schematically above graphs indicating treatment start relative to infection - (A) pre-treatment and post-treatment; (B) pre-treatment; and (C) post-treatment. Infections were performed with hPIV-3-GFP viruses at a MOI of (A) 0.01 and (B and C) 0.1. (A) Cells were harvested at 48 hpi, analyzed by flow cytometry and plotted as a percentage of GFP positive cells. Data represent the mean and SD of 3 independent experiments. (B and C) The cultures were washed daily with PBS to harvest the released virus, which was subsequently titered by TCID50 assay. Data represent the mean and SD of 3 independent experiments. (D and E) Primary fibroblasts derived from healthy control individuals (C1, C2, C3, C4) and HHD patients (H1, H2, H3, H4) were infected with hPIV-3-GFP at a MOI of 4 in the presence or absence of TPCK-trypsin. Cells and supernatants were harvested at 24 hpi. (D) Cells were analyzed by flow cytometry and the average of control fibroblasts (WT) and HHD fibroblasts (HHD) plotted as a percentage of GFP positive cells. (E) Supernatants were titered by TCID50 assay and the average of control fibroblasts (WT) and HHD fibroblasts (HHD) plotted as indicated. Data represent the mean and SD of 3 independent experiments of 4 biological replicates. Statistical analysis was performed with a 2-tailed Student t-test and significance is stated with: * p≤0.05, ** p≤0.005 and *** p≤0.0005. See also Figure S6.
In order to test whether transient suppression of SPCA1 in WT cells could also impair virus spread, we treated Hap1 cells and HAE cell cultures with Morpholino oligomers. Morpholinos are synthetic molecules that can modify gene expression by blocking translation or splicing, and their efficacy in decreasing protein abundance has been demonstrated in vitro and in vivo (Bottcher-Friebertshauser et al., 2011; Krahling et al., 2009; Moerdyk-Schauwecker et al., 2009; Moulton and Moulton, 2010). The Morpholino we designed binds specifically to an intron-exon junction of ATP2C1, interferes with its splicing, and therefore reduces its abundance (Figure S6B). We observed that hPIV-3 spread and titers were significantly decreased in both culture systems (Figures 6A–6C). This was true regardless of whether cultures were treated prior to or after infection and this may be important with respect to antiviral therapies (Figures 6A–6C).
SPCA1 deficiency inhibits multiple viruses from different families
In addition to paramyxoviruses, many viruses require furin and other TGN proteases for glycoprotein processing and infectivity. We therefore set out to determine whether the loss of SPCA1 impacted diverse viral pathogens.
All of the flaviviruses we tested—yellow fever virus (YFV), DENV, WNV, and ZIKV—required SPCA1 to spread efficiently (Figure 7). Similarly, though they were less affected than the flaviviruses, all of the togaviruses we tested—Venezuelan equine encephalitis virus (VEEV), Ross River virus (RRV), and chikungunya virus (CHIKV)—also required SPCA1 for efficient spread (Figure S7). Interestingly, VEEV and CHIKV spread to a minor extent in furin-deficient Lovo cells (Figures S3G and S3H). This is consistent with the finding that shows, along with furin, CHIKV uses additional proprotein convertases for glycoprotein processing (Ozden et al., 2008). The ability to use additional proprotein convertases may explain why CHIKV and other togaviruses are less sensitive to SPCA1 deficiency than paramyxo- and flaviviruses.
Figure 7. Impaired spread of flaviviruses in SPCA1 KO cells.
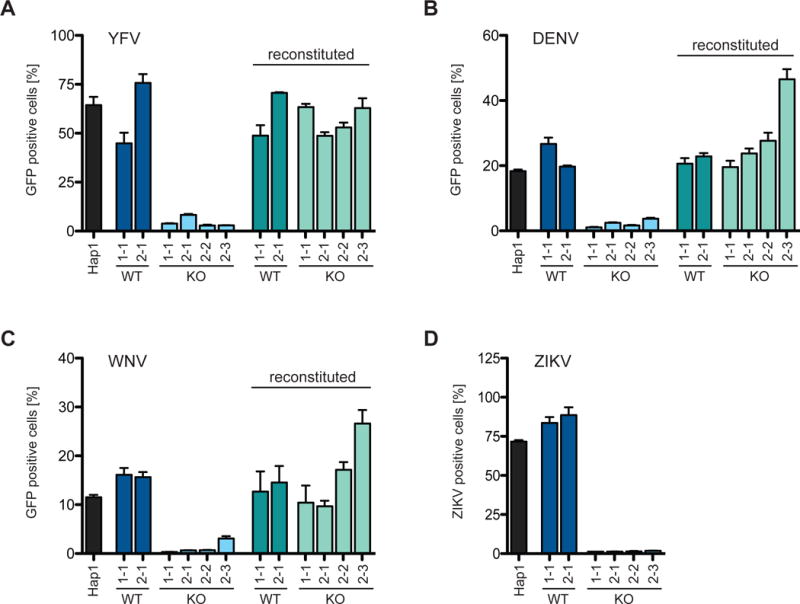
Hap1 cells, CRISPR-generated Hap1 clones and SPCA1-reconstituted clones infected with (A) YFV-venus at a MOI of 0.01, (B) DENV-GFP at a MOI of 0.1, (C) WNV-GFP at a MOI of 10 and (D) ZIKV at a MOI of 0.25. Cells were harvested at (A and D) 48 hpi and (B and C) 72 hpi, and analyzed by flow cytometry and plotted as a percentage of GFP positive cells. Data represent the mean and SD of 3 independent experiments. Cells are colored differently to indicate: parental WT = black; CRISPR WT clones = dark blue and green; and CRISPR KO clones = light blue and green. See also Figures S3, S4 and S7.
Not all of the viruses we tested required SPCA1 to spread. Viruses that were unaffected by loss of SPCA1 included herpes simplex 1 (HSV-1), vesicular stomatitis (VSV), bunyamwera (BUNV), and LCMV (Figure S4). HSV-1’s glycoprotein responsible for fusion possesses features of the VSV glycoprotein (Heldwein et al., 2006), which does not require proteolytic processing (Roche et al., 2006), and BUNV and LCMV glycoproteins are cleaved by SKI-1/S1P (Beyer et al., 2003; Vincent et al., 2003), a protease within the early Golgi.
DISCUSSION
Here we present results from a genome-wide knockout screen where we found that SPCA1, a Ca2+ pump in the TGN, is critical for RSV infection. Besides affecting viral entry, SPCA1 deficiency had a major impact on viral spread. Upon further study we found that like RSV, other viruses from the Paramyxoviridae, Flaviviridae and Togaviridae families also failed to spread in SPCA1-deficient cells. We then showed that when SPCA1 was absent or unable to transport Ca2+ into the TGN, furin protease levels were reduced, viral glycoprotein processing was impaired, and newly generated virus particles were less infectious.
It is likely that other proteases in the TGN, besides furin, are also negatively affected by SPCA1 deficiency. Indeed, we found that while hPIV-3 spread was completely inhibited in SPCA1-deficient cells, it spread to low levels in furin-deficient Lovo cells. In the absence of furin, hPIV-3 might utilize other proteases for glycoprotein cleavage, which are also impaired in SPCA1-deficient cells.
Whereas furin-dependent viruses were highly impaired in SPCA1-deficient cells, VSV and HSV-1, which do not rely on host proteases for glycoprotein processing, were unaffected. BUNV and LCMV were also unaffected by SPCA1-deficiency. This is likely because BUNV and LCMV glycoproteins are processed by the SKI-1/S1P protease located in the early Golgi. In the early Golgi, SERCA1-3, not SPCA1, maintains proper Ca2+ levels We next showed that even reduced levels of SPCA1 decreased viral spread. Primary fibroblasts from patients with Hailey-Hailey disease, a skin disorder caused by ATP2C1 haplo-insufficiency, were less susceptible to hPIV-3 infections. Similarly, primary lung cell culture systems and Hap1 cells treated with Morpholinos targeting ATP2C1 were also less susceptible to hPIV-3.
Along with gaining insight into virus-host biology, a goal of this work was to identify potential targets for antiviral therapies. In this regard, SPCA1 has several attractive qualities: it affects viruses from multiple families; it has an enzymatic active site that can be targeted by small molecule compounds; reduced SPCA1 is tolerated in humans; and even partial loss of SPCA1 activity is sufficient to decrease viral burden. Further, the mechanism of action of SPCA1 and its impact on TGN proteases suggests that filoviruses like Ebola virus and highly pathogenic avian influenza viruses might also be sensitive to SPCA1 deficiency. For acute viral infections like these, decreasing viral spread could significantly improve disease outcomes.
Another attractive quality of targeting SPCA1 may be its impact on viral entry. We found that SPCA1 deficiency not only impaired viral spread but also reduced susceptibility to RSV infection. Besides proteases, other Golgi-residing enzymes such as glycosyl- and sulfotransferases may be negatively impacted by the loss of SPCA1. The absence of SPCA1 may therefore decrease glycosylation or maturation of cell surface proteins thereby affecting the ability of viral particles to attach to and engage specific receptors. More work is needed to elucidate the role of SPCA1 in shaping the plasma membrane proteome but targeting SPCA1 may interfere with two critical steps of the viral life cycle: viral glycoprotein maturation required for release of infectious particles, and the optimal constellation of cell surface determinants for efficient virus binding and entry.
Aside from viral infections, SPCA1 is likely to be important in a variety of biological settings. Since SPCA1 controls furin activity, it may also have roles in other diseases where furin is important. Bacterial toxins, for example, require cleavage by cell surface furin (e.g. anthrax toxin and Clostridium septicum α-toxin) or endosomal furin (e.g. Pseudomonas exotoxin A, shiga toxin, and diphtheria toxins) for their activation and pathogenicity (Gordon et al., 1997; Gordon and Leppla, 1994; Molloy et al., 1992). Modulation of SPCA1 may also benefic patients with other diseases caused or exacerbated by furin, such as tumor metastasis (Bassi et al., 2001) and Alzheimer’s disease (Bennett et al., 2000).
In conclusion, our results demonstrate that SPCA1 affects cellular proteases in the TGN and likely other Golgi-residing enzymes such as glycosyl- and sulfotransferases as well. More work is needed, but understanding how these enzymes influence viral entry, replication and pathogenicity is an exciting area for future research.
METHODS AND MATERIALS
Contact for Reagent and Resource Sharing
Further information and requests for resources, reagents, and data should be directed to and will be fulfilled by the Lead Contact, Charles M Rice (ricec@rockefeller.edu).
Experimental Model and Subject Details
Cells
A549 (human; sex: male), HeLa (human; sex: female), Vero (African green monkey; sex: female), Huh-7.5 (human; sex: male) and Lenti-X 293T (human; sex: female) cells were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS). Lovo (human; sex: male) cells were maintained in F12K medium (Invitrogen) supplemented with 10% FBS. BHK-21 (Syrian golden hamster; sex: male) cells were grown in MEM (Invitrogen) supplemented with 7.5% FBS. Hap1 (human; sex: male) cells were maintained in IMDM supplemented with 10% FBS. Primary human dermal fibroblasts (sex: male and female) (INSERM) were maintained in DMEM supplemented with 10% FBS and 1× penicillin/streptomycin. Informed consent was obtained from human subjects according to local regulations. Samples were subsequently transferred for experimental testing under respective ethics approvals at INSERM in France and at the Rockefeller University in the United States. All protocols involving the use of human tissue were reviewed and exempted by The Rockefeller University Institutional Review Board. Primary human normal airway tracheobronchial epithelial cells (NHBE) (sex: male and female) provided by Lonza, Inc. (Walkersville, MD) were grown on collagen-coated transwell membrane supports at air-liquid interface for 6 weeks to yield well-differentiated polarized human airway epithelial (HAE) cultures. Hap1 cells were used for the haploid screen and together with A549, HeLa, Lovo cells, primary fibroblasts, and HAE cultures for hit validation and functional studies. Huh-7.5, Vero, and BHK-21 cells were used for virus production and virus titration. All cell lines have been tested negative for contamination with mycoplasma and were obtained from the ATCC (with exceptions for Huh-7.5, Hap1 and Lenti-X 293T cells). Cells have not been further authenticated.
Method Details
Viruses
The generation of viral stocks for the following viruses has been previously described: hPIV3-GFP (Zhang et al., 2005) (based on strain JS), RSV-GFP (Biacchesi et al., 2004) (based on strain A2), MeV-GFP (del Valle et al., 2007) (MVvac2-GFP, based on vaccine strain, Edmonston lineage), BUNV-GFP (Shi et al., 2010) (based on rBUN-del7GFP), YFV-Venus (Jones et al., 2010) (derived from YF17D-5′C25Venus2AUbi), WNV-GFP (McGee et al., 2010) (derived from pBELO-WNV-GFP-RZ ic), DENV-GFP (Schoggins et al., 2012) (derived from IC30P-A, a full-length infectious clone of strain 16681), RRV-GFP (Shabman et al., 2007) (derived from the T48 strain of RRV), CHIKV-GFP (Tsetsarkin et al., 2006) (derived from pCHIKV-LR 5′GFP), VEEV-GFP (Petrakova et al., 2005) (derived from pTC83-GFP infectious clone), HSV-1 (generously provided by Margaret MacDonald, The Rockefeller University), VSV-GFP (Dalton and Rose, 2001) (generously provided by John Rose, Yale). ZIKV (PRVABC59 obtained from the CDC, Ft. Collins) was amplified in Hap1 cells and titered by plaque assay on Huh-7.5 cells. Experiments with WNV and CHIKV were carried out in biosafety level 3 (BSL3) containment in compliance with institutional and federal guidelines.
Plasmids
A full-length ORF clone encoding SPCA1 isoform 1C (Sino Biological Inc.) was used to generate isoforms 1A-F. Isoforms 1A, 1B and 1D were made by PCR using the Expand High Fidelity PCR System (Roche) with synthesized DNA (G-Blocks, IDT) encoding for the relevant C-terminus. Isoform 1E and 1F were made using isoforms 1B and 1A with an internal primer downstream of the SPCA1 initiation codon. Mutations G309C and D742Y were introduced into isoform 1D using the QuikChange Site-Directed Mutagenesis Kit (Agilent). All constructs were cloned into pDONR221, sequenced and transferred into the pLX304 lentiviral backbone via the Gateway cloning system (Invitrogen). The pLX304 plasmid was then used to for lentivirus production. Small guide RNAs (sgRNA) for CRISPR editing targeting ATP2C1 (sgRNA #1: 5′-GCATACACTTGCCCGAGACT-3′ and sgRNA #2: 5′-TCAAACAAGCGTAAGTCAGC-3′) were designed using http://crispr.mit.edu. They were cloned into pX330 for subsequent transfection or into lentiCRISPRv2 for subsequent lentivirus production as described previously (Cong et al., 2013; Sanjana et al., 2014).
Lentivirus production and transduction
Lentiviral stocks were generated in Lenti-X 293T (Clontech) cells by co-transfection of plasmids expressing (1) the ORF or sgRNA of interest (2), HIV gag-pol, and (3) the vesicular stomatitis virus glycoprotein (VSV-G) in a ratio of 0.55:0.35:0.1. For blasticidin-selectable lentiviral vectors, we used the pLX304 lentiviral backbone in which the V5 tag was removed, and for puromycin-selectable lentiviral vectors, we used the lentiCRISPRv2 lentiviral backbone (gift from Feng Zheng). For each transfection, 6 μl Lipofectamine 2000 (Invitrogen) was combined with 2.0 μg total DNA in 250 μl Opti-MEM (Gibco). Transfections were carried out for 6 h followed by a media change to DMEM with 3% FBS. Supernatants were collected at 48 h, cleared by centrifugation at 2,850 RCF, filtered using 0.45 micron syringe filters and stored at −80°C. For lenZviral transduction, Hap1, A549, and HeLa cells were seeded into 6-well plates at a density of 2 × 105 cells/well and transduced with lentiviral pseudoparticles by spinoculation at 930 RCF for 60 min at 37°C in medium containing 3% FBS, 20 mM HEPES, and 4 μg/ml polybrene. Cells were selected starting at 24 h post transduction with either 2.5 μg/ml blasticidin or 1 μg/ml puromycin.
Haploid genetic screen
Gene trap virus was produced as previously described in 293T cells by transfection of gene trap plasmid combined with pAdvantage, CMV-VSVG and Gag-pol (Jae et al., 2013). The virus was harvested twice daily for 3 days and concentrated using ultracentrifugation for 2 h at 22,000 RPM in a Beckman SW28 rotor. 1×108 Hap1 cells were infected for 3 consecutive days in the presence of protamine sulfate (8 μg/ml) and passaged for 5 days prior to freezing. For the screen, a library of 1×108 mutagenized cells was exposed to RSV-GFP at a MOI ~1. The resistant colonies were expanded for 11 days and ~1×107 cells were used for genomic DNA isolation.
Sequence analysis of gene trap insertion sites
Insertion sites in RSV-selected cells were identified by sequencing the genomic DNA flanking the gene trap insertion site as previously described (Blomen et al., 2015). Genomic DNA was isolated from ~1×107 cells and subjected to a linear amplification PCR using a biotinylated primer followed by single-stranded DNA linker ligation, PCR and subsequent sequencing using the HiSeq 2000 platform (Illumina). The acquired reads were mapped to the human genome (hg19) using bowtie allowing for a single mismatch, and insertion sites located in Refseq genes were identified. For each Refseq gene, the longest transcript was chosen and overlapping gene regions in opposite orientations were disregarded, since orientation bias cannot be unambiguously assigned in these areas. The number of disruptive mutations (i.e. in sense with transcriptional orientation) and non-disruptive (i.e. in antisense with transcriptional orientation) mutations per individual gene was counted and genes significantly enriched for disruptive mutations following RSV exposure were determined using a binomial test and corrected for multiple testing using a Benjamini-Hochberg false discovery rate (FDR). The significantly enriched genes in the RSV dataset were compared to a reference dataset of wildtype Hap1 cells (NCBI SRA accession no: SRP058962 dataset SRX1045464) that was analyzed similarly. Genes that were already significantly enriched in the wildtype dataset were subtracted from the identified RSV host factor genes.
Identification of CRISPR-induced INDELs
Hap1 cells were cultured for 4 days following transfection with pX330 introducing CRISPR editing of ATP2C1. A549 and HeLa cells were transduced with lentiCRISPRv2 lentiviruses to introduce CRISPR editing of ATP2C1 and selected for 3 days with puromycin (1 μg/ml). Cells were subsequently seeded into 96-well plates at a density of 0.7 cells/well in order to generate single cell clones. Only wells harboring a single clone were tested further for their expression of SPCA1 by western blot as described below. The genomic DNA of Hap1 cell clones was extracted using a DNeasy Blood and Tissue Kit (Qiagen), following the manufacturer’s instructions. The genomic DNA (20 ng) was subjected to PCR amplification using the Expand High Fidelity PCR System (Roche) and a pair of primers that flank the region containing the CRISPR editing site (forward primer 5′-GGGGATAGAGTTCCTGCTGA-3′, reverse primer 5′-ACATTATTTAGCAACTGCAC-3′). The resulting PCR products were resolved on a 1% agarose gel and purified using a Qiaquick Gel Extraction Kit (Qiagen), followed by Sanger sequencing.
Virus replication assays
To test virus infection and virus spread, cells were seeded into 24-well plates at various densities based on their growth rate and size (Lovo − 4×104 cells/well; Hap1 − 3.5×104 cells/well; A549, HeLa and primary fibroblasts − 2.5×104 cells/well). Cells were washed with Opti-MEM prior to infection. Viruses were diluted in Opti-MEM at indicated MOIs and cells were inoculated for 2 h at 37°C. Following incubation, viral inoculum was removed and cultures were washed 3 times with Opti-MEM. In order to remove any residual virus particles from the initial inoculum, cultures were washed additional 3 times at 4 hpi and incubated at 37°C for the duration of the experiment. Supernatant with progeny virus (RSV and hPIV-3) was harvested at 24 hpi to determine viral titers by measuring genome copies with standard qRT-PCR. Briefly, viral RNA was isolated from supernatants using a QiaAmp Viral RNA Mini Kit (Qiagen), followed by cDNA synthesis using SuperScript III First-Strand Synthesis system (Invitrogen). The cDNA products were amplified by qPCR using 2× SYBR Green PCR Master Mix (Applied Biosystems) and primes specific to RSV-GFP (forward: 5′-GTGGTGCCCATCCTGGTCG-3′, reverse: 5′-GGGTAGCGGCTGAAGCACTG-3′) and hPIV-3-GFP (forward: 5′-CTGGATTGCAGGAGATCATGTCG-3′, reverse: 5′-GTGAATGTACGGTTGCCGTATTGG-3′). To determine the infectious titers of harvested supernatants, plaque assays were performed on Vero cells (RSV) or TCID50 assays on BHK-21 cells (hPIV-3-GFP). Cells infected with GFP-tagged viruses and ZIKV were harvested at various time points to assess initial infection and viral spread. Cells were harvested into 300 μl Accumax cell dissociation medium (eBioscience) and transferred to a 96-well block containing 300 μl 4% paraformaldehyde fixation solution. Cells were pelleted at 930 RCF for 5 min at 4°C and resuspended in cold PBS containing 3% FBS, and stored at 4°C until FACS analysis. Cells infected with ZIKV were subsequently stained using a monoclonal antibody anti-flavivirus group antigen 4G2 (Millipore) at 1:500 as a primary antibody and AlexaFluor647 at 1:1000 as secondary antibody. Samples were analyzed using the LSRII flow cytometer (BD Biosciences) equipped with a 488 nm and 561 nm laser for detection of GFP, YFP (venus) and TagRFP. Data were analyzed using FlowJo software (Treestar) with a 0.1% compensation matrix. Cells infected with HSV-1 and BUNV were harvested at 72 hpi to determine cell viability using CellTiterGlo (Promega) according to the manufacturer’s instructions. Luminescence was measured using a plate reader (FLUOstar Omega - BMG labtech). For infections performed with MeV-GFP, we chose a dose, which resulted in 5–10% infected cells (MOI ~0.1) at 20 hpi before syncytia formation started in wildtype cells.
HAE cultures were rinsed with PBS prior to infection to transiently remove apical secretions and supplied with fresh basolateral medium prior to inoculation. We used 3×103 PFU of hPIV-3-GFP per culture which was estimated to be a MOI of 0.1. Virus was diluted in PBS and the inoculum was applied to the apical surface of HAE for 2 h at 37°C. Following incubation, viral inocula were removed, cultures were washed 3 times with PBS and incubated at 37°C for the duration of the experiment. Progeny virus was harvested at indicated times by performing apical washes with 100 μl of PBS for 30 min at 37°C. Washes were collected and stored at −80°C prior to analysis. Viral titers in the apical washes were determined by TCID50 assay on BHK-21 cells.
Antibodies, inhibitors, recombinant proteins
The calcium ionophore calcimycin (A23817) and the furin inhibitor Dec-RVKR-CMK were obtained from Cayman Chemicals. Cells were seeded into 24-well plates for viral infections as described earlier. Compounds A23187 and Dec-RVKR-CMK display cytotoxicity during extended treatments. To minimize adverse effects due to compound treatment, we infected cells with fast replicating viruses such as hPIV-3, and with MeV in order to focus on syncytia formation. After removing and washing off the inoculum, Dec-RVKR-CMK was added at a concentration of 20 μM. The Ca2+ ionophore A23187 was added later during the infection at 12 hpi, at a concentration of 200 nM. Both compounds were present throughout the remainder of the infections. TPCK-trypsin (Sigma) was used to mimic proteolytic cleavage in hPIV-3 infections performed in SPCA1 KO cells and in primary human fibroblasts. Cells were seeded into 24-well plates for viral infections as described earlier. After removing and washing off the inoculum, IMDM containing low serum concentrations (0.1% FBS) was added, supplemented with 1 μg/ml TPCK-trypsin, and kept for the remainder of the infection. For SPCA1 Western Blots we used a monoclonal antibody 2G1 (Abnova) and for furin, the monoclonal antibody MON-152 (Enzo). Western Blots probing for the F protein of hPIV-3 were performed with a monoclonal antibody 176635-7 (generously provided by Anne Moscona, Columbia University). To determine hPIV-3 F protein cleavage, cells were seeded into 6-well plates (5×105 cells/well) and infected the following day with hPIV-3 (MOI=2) for 2 h at 37°C. The virus inoculum was removed and cells were washed 3 times with PBS before adding back medium. In order to remove any residual virus particles from the initial inoculum, cells were washed again 3 times with PBS at 4 hpi before adding back medium. The supernatant was harvested at 24 hpi and virus particles were concentrated by spinning for 2 h at 21,100 RCF at 4°C. The virus pellet was lysed in LDS sample buffer (NuPAGE) containing 5% β-mercaptoethanol, heated for 15 min at 70°C and stored at −80°C. To interfere with transcription of ATP2C1, vivo-Morpholinos were ordered (Gene Tools, LLC) and diluted in distilled water according to the manufacture. The vivo-Morpholino targeting ATP2C1 (5′-TTCACGCACACTGTAACACGAAATA-3′) binds to the intron-exon-junction of exon 7 and the vivo-Morpholino standard control (5′-CCTCTTACCTCAGTTACAATTTATA-3′) targets a human beta-globin intron mutation that causes beta-thalassemia. It has been broadly used as the appropriate negative control for custom vivo-Morpholino oligos. Morpholinos were added to apical site of HAE cultures at different concentrations in a volume of 20 μl (48 h prior to infections or 24 h post infections). The treatment was replenished every 24 h for the duration of the experiment. HAE cultures were harvested at the time of infection (48 h of treatment), to probe for protein levels of SPCA1 by western blot. In comparison, Hap1 cells were seeded into 24-well plates and received a similar treatment. The culture medium was supplemented with Morpholinos (48 h and 24 h prior to infections, at the time of infection or 24 h post infections) and replenished every 24 h for the duration of the experiment. Cells were then harvested at the time of infection (48 h of treatment) for western blot to probe for SPCA1.
Quantification and Statistical Analysis
Statistical analysis was performed in Prism (GraphPad Software, v6.0d, 2013). The statistical tests used and the number of biological replicates is indicated in each figure legend. Unless otherwise stated two conditions were compared using two-tailed Student’s t tests. Statistical significance was defined as a p value of 0.05.
Data and Software Availability
All data are available upon request to the lead contact author.
Supplementary Material
HIGHLIGHTS.
SPCA1 is identified as a host factor required for RSV infection
Lack of SPCA1 impairs viral spread by inhibiting glycoprotein maturation
SPCA1 deficiency reduces the infectivity of viruses requiring the furin protease
Decreased levels of SPCA1 in primary airway epithelial cells lowers viral burden
Acknowledgments
We thank the following investigators for contributing viral molecular clones or viral stocks: R. Cattaneo (MeV), P. Collins (RSV, hPIV-3), I. Frolov (WNV, VEEV), M. Heise (SINV), S. Higgs (CHIKV), J. Rose (VSV), M. MacDonald (HSV-1), R. Elliott (BUNV) and the CDC (ZIKV). We thank A. Moscona for providing the monoclonal hPIV-3 F protein antibody 176635-7. We are thankful for the technical advice of S. Mazel, S. Semova, S. Tadesse and S. Han at The Rockefeller University Flow Cytometry Resource Center. We thank M. Saeed, K. Rozen-Gagnon and J. Luna for critical reading and helpful comments on the manuscript. We also wish to acknowledge Santa Maria Pecoraro Di Vittorio, Joseph Palarca, Glen Santiago, Michael Pearce, Mary Ellen Castillo, Arnella Webson and Sonia Shirley for outstanding administrative and/or technical support. This work was supported in part by a grant from the Greenberg Medical Research Institute, the Robertson Foundation, and several generous donors. W.M.S. was supported by National Institute of Diabetes and Digestive and Kidney Diseases National Research Service Award DK095666 and M.A.S. was supported by the Parker B. Francis Fellowship Program. T.R.B. received funding from the Cancer Genomics Center (CGC.nl<http://cgc.nl>) and the European Research Council (ERC) Starting Grant (ERC-2012-StG 309634).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.-H.H. and C.M.R. designed the project. H.-H.H., M.A.S. and C.M.R. designed the experiments. H.-H.H., M.A.S. and V.A.B. performed the experimental work. H.-H.H., V.A.B., and C.M.R. analyzed the results. W.M.S., A.H. and T.R.B. contributed reagents and expertise. H.-H.H. and W.M.S. wrote the manuscript with input from all coauthors.
BIBLIOGRAPHY
- Anderson ED, Molloy SS, Jean F, Fei H, Shimamura S, Thomas G. The ordered and compartment-specfific autoproteolytic removal of the furin intramolecular chaperone is required for enzyme activation. The Journal of biological chemistry. 2002;277:12879–12890. doi: 10.1074/jbc.M108740200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando H, Kawano M, Kondo K, Tsurudome M, Komada H, Nishio M, Ito Y. Growth properties and F protein cleavage site sequences of naturally occurring human parainfluenza type 2 viruses. Virology. 1991;184:87–92. doi: 10.1016/0042-6822(91)90824-u. [DOI] [PubMed] [Google Scholar]
- Bassi DE, Mahloogi H, Al-Saleem L, Lopez De Cicco R, Ridge JA, Klein-Szanto AJ. Elevated furin expression in aggressive human head and neck tumors and tumor cell lines. Molecular carcinogenesis. 2001;31:224–232. doi: 10.1002/mc.1057. [DOI] [PubMed] [Google Scholar]
- Behne MJ, Tu CL, Aronchik I, Epstein E, Bench G, Bikle DD, Pozzan T, Mauro TM. Human keratinocyte ATP2C1 localizes to the Golgi and controls Golgi Ca2+ stores. The Journal of investigative dermatology. 2003;121:688–694. doi: 10.1046/j.1523-1747.2003.12528.x. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Denis P, Haniu M, Teplow DB, Kahn S, Louis JC, Citron M, Vassar R. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta -secretase. The Journal of biological chemistry. 2000;275:37712–37717. doi: 10.1074/jbc.M005339200. [DOI] [PubMed] [Google Scholar]
- Beyer WR, Popplau D, Garten W, von Laer D, Lenz O. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. Journal of virology. 2003;77:2866–2872. doi: 10.1128/JVI.77.5.2866-2872.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacchesi S, Skiadopoulos MH, Tran KC, Murphy BR, Collins PL, Buchholz UJ. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology. 2004;321:247–259. doi: 10.1016/j.virol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- Bottcher-Friebertshauser E, Stein DA, Klenk HD, Garten W. Inhibition of influenza virus infection in human airway cell cultures by an antisense peptide-conjugated morpholino oligomer targeting the hemagglutinin-activating protease TMPRSS2. Journal of virology. 2011;85:1554–1562. doi: 10.1128/JVI.01294-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Dutch RE. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses. 2012;4:613–636. doi: 10.3390/v4040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelingh KV, Tierney EL. Identification of amino acids recognized by syncytium-inhibiting and neutralizing monoclonal antibodies to the human parainfluenza type 3 virus fusion protein. Journal of virology. 1989;63:3755–3760. doi: 10.1128/jvi.63.9.3755-3760.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelingh KV, Winter CC. Naturally occurring human parainfluenza type 3 viruses exhibit divergence in amino acid sequence of their fusion protein neutralization epitopes and cleavage sites. Journal of virology. 1990;64:1329–1334. doi: 10.1128/jvi.64.3.1329-1334.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MS, Bashiruddin JB, Alexander DJ. Deduced amino acid sequences at the fusion protein cleavage site of Newcastle disease viruses showing variation in antigenicity and pathogenicity. Archives of virology. 1993;128:363–370. doi: 10.1007/BF01309446. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton KP, Rose JK. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology. 2001;279:414–421. doi: 10.1006/viro.2000.0736. [DOI] [PubMed] [Google Scholar]
- del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. Journal of virology. 2007;81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode L, Andersen JP, Raeymaekers L, Missiaen L, Vilsen B, Wuytack F. Functional comparison between secretory pathway Ca2+/Mn2+-ATPase (SPCA) 1 and sarcoplasmic reticulum Ca2+-ATPase (SERCA) 1 isoforms by steady-state and transient kinetic analyses. The Journal of biological chemistry. 2005;280:39124–39134. doi: 10.1074/jbc.M506181200. [DOI] [PubMed] [Google Scholar]
- Fairclough RJ, Dode L, Vanoevelen J, Andersen JP, Missiaen L, Raeymaekers L, Wuytack F, Hovnanian A. Effect of Hailey-Hailey Disease mutations on the function of a new variant of human secretory pathway Ca2+/Mn2+-ATPase (hSPCA1) The Journal of biological chemistry. 2003;278:24721–24730. doi: 10.1074/jbc.M300509200. [DOI] [PubMed] [Google Scholar]
- Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. The New England journal of medicine. 2005;352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- Glickman RL, Syddall RJ, Iorio RM, Sheehan JP, Bratt MA. Quantitative basic residue requirements in the cleavage-activation site of the fusion glycoprotein as a determinant of virulence for Newcastle disease virus. Journal of virology. 1988;62:354–356. doi: 10.1128/jvi.62.1.354-356.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon VM, Benz R, Fujii K, Leppla SH, Tweten RK. Clostridium septicum alpha-toxin is proteolytically activated by furin. Infection and immunity. 1997;65:4130–4134. doi: 10.1128/iai.65.10.4130-4134.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon VM, Leppla SH. Proteolytic activation of bacterial toxins: role of bacterial and host cell proteases. Infection and immunity. 1994;62:333–340. doi: 10.1128/iai.62.2.333-340.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, et al. The burden of respiratory syncytial virus infection in young children. The New England journal of medicine. 2009;360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- Hu Z, Bonifas JM, Beech J, Bench G, Shigihara T, Ogawa H, Ikeda S, Mauro T, Epstein EH., Jr Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nature genetics. 2000;24:61–65. doi: 10.1038/71701. [DOI] [PubMed] [Google Scholar]
- Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, et al. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science. 2014;344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P, et al. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science. 2013;340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN, et al. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nature biotechnology. 2010;28:167–171. doi: 10.1038/nbt.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ, Swaroop M, Murtha-Riel P. Depletion of manganese within the secretory pathway inhibits O-linked glycosylation in mammalian cells. Biochemistry. 1994;33:9813–9819. doi: 10.1021/bi00199a001. [DOI] [PubMed] [Google Scholar]
- Krahling V, Stein DA, Spiegel M, Weber F, Muhlberger E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. Journal of virology. 2009;83:2298–2309. doi: 10.1128/JVI.01245-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissandron V, Podini P, Pizzo P, Pozzan T. Unique characteristics of Ca2+ homeostasis of the trans-Golgi compartment. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9198–9203. doi: 10.1073/pnas.1004702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535:159–163. doi: 10.1038/nature18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee CE, Shustov AV, Tsetsarkin K, Frolov IV, Mason PW, Vanlandingham DL, Higgs S. Infection, dissemination, and transmission of a West Nile virus green fluorescent protein infectious clone by Culex pipiens quinquefasciatus mosquitoes. Vector borne and zoonotic diseases. 2010;10:267–274. doi: 10.1089/vbz.2009.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micaroni M, Perinetti G, Berrie CP, Mironov AA. The SPCA1 Ca2+ pump and intracellular membrane trafficking. Traffic. 2010;11:1315–1333. doi: 10.1111/j.1600-0854.2010.01096.x. [DOI] [PubMed] [Google Scholar]
- Moerdyk-Schauwecker M, Stein DA, Eide K, Blouch RE, Bildfell R, Iversen P, Jin L. Inhibition of HSV-1 ocular infection with morpholino oligomers targeting ICP0 and ICP27. Antiviral research. 2009;84:131–141. doi: 10.1016/j.antiviral.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. The Journal of biological chemistry. 1992;267:16396–16402. [PubMed] [Google Scholar]
- Moulton HM, Moulton JD. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochimica et biophysica acta. 2010;1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O’Brien KL, Roca A, Wright PF, Bruce N, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K. Calcium depletion blocks proteolytic cleavages of plasma protein precursors which occur at the Golgi and/or trans-Golgi network. Possible involvement of Ca(2+)-dependent Golgi endoproteases. The Journal of biological chemistry. 1992;267:17465–17471. [PubMed] [Google Scholar]
- Ozden S, Lucas-Hourani M, Ceccaldi PE, Basak A, Valentine M, Benjannet S, Hamelin J, Jacob Y, Mamchaoui K, Mouly V, et al. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. The Journal of biological chemistry. 2008;283:21899–21908. doi: 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- Petrakova O, Volkova E, Gorchakov R, Paessler S, Kinney RM, Frolov I. Noncytopathic replication of Venezuelan equine encephalitis virus and eastern equine encephalitis virus replicons in Mammalian cells. Journal of virology. 2005;79:7597–7608. doi: 10.1128/JVI.79.12.7597-7608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay S, Carette JE. Hunting Viral Receptors Using Haploid Cells. Annual review of virology. 2015;2:219–239. doi: 10.1146/annurev-virology-100114-055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, Jae LT, Wosen JE, Nagamine CM, Chapman MS, et al. An essential receptor for adeno-associated virus infection. Nature. 2016;530:108–112. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle AG, Shiryaev SA, Oh ES, Cieplak P, Srinivasan A, Wei G, Liddington RC, Ratnikov BI, Parent A, Desjardins R, et al. Substrate cleavage analysis of furin and related proprotein convertases. A comparative study. The Journal of biological chemistry. 2008;283:20897–20906. doi: 10.1074/jbc.M803762200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riblett AM, Blomen VA, Jae LT, Altamura LA, Doms RW, Brummelkamp TR, Wojcechowskyj JA. A Haploid Genetic Screen Identifies Heparan Sulfate Proteoglycans Supporting Rift Valley Fever Virus Infection. Journal of virology. 2016;90:1414–1423. doi: 10.1128/JVI.02055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell reports. 2016;16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Dorner M, Feulner M, Imanaka N, Murphy MY, Ploss A, Rice CM. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14610–14615. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabman RS, Morrison TE, Moore C, White L, Suthar MS, Hueston L, Rulli N, Lidbury B, Ting JP, Mahalingam S, et al. Differential induction of type I interferon responses in myeloid dendritic cells by mosquito and mammalian-cell-derived alphaviruses. Journal of virology. 2007;81:237–247. doi: 10.1128/JVI.01590-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadman KA, Wald ER. A review of palivizumab and emerging therapies for respiratory syncytial virus. Expert opinion on biological therapy. 2011;11:1455–1467. doi: 10.1517/14712598.2011.608062. [DOI] [PubMed] [Google Scholar]
- Shay DK, Holman RC, Newman RD, Liu LL, Stout JW, Anderson LJ. Bronchiolitis-associated hospitalizations among US children, 1980–1996. Jama. 1999;282:1440–1446. doi: 10.1001/jama.282.15.1440. [DOI] [PubMed] [Google Scholar]
- Shi X, van Mierlo JT, French A, Elliott RM. Visualizing the replication cycle of bunyamwera orthobunyavirus expressing fluorescent protein-tagged Gc glycoprotein. Journal of virology. 2010;84:8460–8469. doi: 10.1128/JVI.00902-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DW, Frankel LR, Mathers LH, Tang AT, Ariagno RL, Prober CG. A controlled trial of aerosolized ribavirin in infants receiving mechanical ventilation for severe respiratory syncytial virus infection. The New England journal of medicine. 1991;325:24–29. doi: 10.1056/NEJM199107043250105. [DOI] [PubMed] [Google Scholar]
- Sudbrak R, Brown J, Dobson-Stone C, Carter S, Ramser J, White J, Healy E, Dissanayake M, Larregue M, Perrussel M, et al. Hailey-Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca(2+) pump. Human molecular genetics. 2000;9:1131–1140. doi: 10.1093/hmg/9.7.1131. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Nakagawa T, Kasai K, Banno T, Duguay SJ, Van de Ven WJ, Murakami K, Nakayama K. A second mutant allele of furin in the processing-incompetent cell line, LoVo. Evidence for involvement of the homo B domain in autocatalytic activation. The Journal of biological chemistry. 1995;270:26565–26569. doi: 10.1074/jbc.270.44.26565. [DOI] [PubMed] [Google Scholar]
- Tashiro M, Pritzer E, Khoshnan MA, Yamakawa M, Kuroda K, Klenk HD, Rott R, Seto JT. Characterization of a pantropic variant of Sendai virus derived from a host range mutant. Virology. 1988;165:577–583. doi: 10.1016/0042-6822(88)90601-0. [DOI] [PubMed] [Google Scholar]
- Tsetsarkin K, Higgs S, McGee CE, De Lamballerie X, Charrel RN, Vanlandingham DL. Infectious clones of Chikungunya virus (La Reunion isolate) for vector competence studies. Vector borne and zoonotic diseases. 2006;6:325–337. doi: 10.1089/vbz.2006.6.325. [DOI] [PubMed] [Google Scholar]
- Vanoevelen J, Dode L, Raeymaekers L, Wuytack F, Missiaen L. Diseases involving the Golgi calcium pump. Sub-cellular biochemistry. 2007;45:385–404. doi: 10.1007/978-1-4020-6191-2_14. [DOI] [PubMed] [Google Scholar]
- Vincent MJ, Sanchez AJ, Erickson BR, Basak A, Chretien M, Seidah NG, Nichol ST. Crimean-Congo hemorrhagic fever virus glycoprotein proteolytic processing by subtilase SKI-1. Journal of virology. 2003;77:8640–8649. doi: 10.1128/JVI.77.16.8640-8649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. A public genome-scale lentiviral expression library of human ORFs. Nature methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, Pickles RJ. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. Journal of virology. 2005;79:1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature. 2016;535:164–168. doi: 10.1038/nature18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.