

Abstract
Background
Acute neurological insults caused by infection, systemic inflammation, ischemia, or traumatic injury are often associated with breakdown of the blood-brain barrier (BBB) followed by infiltration of peripheral immune cells, cytotoxic proteins, and water. BBB breakdown and extravasation of these peripheral components into the brain parenchyma result in inflammation, oxidative stress, edema, excitotoxicity, and neurodegeneration. These downstream consequences of BBB dysfunction can drive pathophysiological processes and play a substantial role in the morbidity and mortality of acute and chronic neurological insults, and contribute to long-term sequelae. Preserving or rescuing BBB integrity and homeostasis therefore represents a translational research area of high therapeutic potential.
Methods
Induction of general and localized BBB disruption in mice was carried out using systemic administration of LPS and focal photothrombotic ischemic insult, respectively, in the presence and absence of the monoacylglycerol lipase (MAGL) inhibitor, CPD-4645. The effects of CPD-4645 treatment were assessed by gene expression analysis performed on neurovascular-enriched brain fractions, cytokine and inflammatory mediator measurement, and functional assessment of BBB permeability. The mechanism of action of CPD-4645 was studied pharmacologically using inverse agonists/antagonists of the cannabinoid receptors CB1 and CB2.
Results
Here, we demonstrate that the neurovasculature exhibits a unique transcriptional signature following inflammatory insults, and pharmacological inhibition of MAGL using a newly characterized inhibitor rescues the transcriptional profile of brain vasculature and restores its functional homeostasis. This pronounced effect of MAGL inhibition on blood-brain barrier permeability is evident following both systemic inflammatory and localized ischemic insults. Mechanistically, the protective effects of the MAGL inhibitor are partially mediated by cannabinoid receptor signaling in the ischemic brain insult.
Conclusions
Our results support considering MAGL inhibitors as potential therapeutics for BBB dysfunction and cerebral edema associated with inflammatory brain insults.
Electronic supplementary material
The online version of this article (10.1186/s12974-018-1166-9) contains supplementary material, which is available to authorized users.
Keywords: Monoacylglycerol lipase, 2-arachidonoylglycerol, Neuroinflammation, Blood-brain barrier, Neurovasculature
Background
The blood-brain barrier (BBB) is a selectively permeable barrier that regulates protein, metabolite, and ion transport to and from the central nervous system (CNS). It is composed of endothelial cells connected by tight junctions, pericytes, and astrocyte end-feet [1]. Acute neurological insults caused by infection, ischemia, or traumatic insults are often associated with breakdown of the BBB followed by infiltration of circulating immune cells and extravasation of plasma components. BBB breakdown and extravasation of these peripheral components into the brain parenchyma result in inflammation, oxidative stress, edema, excitotoxicity, and neurodegeneration [2]. Each of these insults can drive pathophysiological processes and play a substantial role in the morbidity and mortality of acute and chronic neurological diseases. Preserving or rescuing BBB integrity and function is therefore a research area with high therapeutic potential.
Systemic inflammatory challenges such as lipopolysaccharide (LPS), an immunogenic component of Gram-negative bacteria, promote BBB dysfunction [3]. Several inflammatory pathways have been proposed to contribute to the disruptive effects of LPS on BBB. Central to these mechanisms is the activation of the cerebrovascular endothelium and surrounding cells within the neurovascular unit by pro-inflammatory cytokines (IL-1β and TNFα) and eicosanoids induced by LPS [4].
Although the triggering mechanism of BBB disruption in ischemic stroke is relatively distinct from other systemic or central inflammatory stimuli [5], the neurovascular unit is dramatically sensitized to further disruptive changes by peripheral or central inflammation [6]. As a consequence, brain damage and mortality are exacerbated by systemic and central inflammation in clinical outcome as well as in experimental animal models [6, 7].
The interplay between brain injury and the subsequent inflammatory cascade as it pertains to BBB integrity and properties is not completely understood. Moreover, it is still unclear whether cerebrovascular changes and BBB breakdown can initiate pathogenic events that lead directly to neuronal injury, impaired functional activity, and early neurological symptoms in humans [2]. Therefore, a better mechanistic understanding of the molecular events leading to BBB dysfunction is necessary to address the unmet medical needs in acute brain injury and perhaps other chronic neurodegenerative conditions.
We and others have recently shown that monoacylglycerol lipase (MAGL), a serine hydrolase which modulates levels of the abundant endocannabinoid 2-arachidonoylglycerol (2-AG) [8, 9], also regulates neuroinflammation [10, 11]. Through hydrolysis of 2-AG, MAGL produces a pool of arachidonic acid (AA) which contributes to the inflammatory cascade in the CNS. Inhibition of MAGL enhances 2-AG-mediated cannabinoid receptor signaling while lowering arachidonate levels in the brain.
Interestingly, both pathways triggered by MAGL inhibition, elevation of 2-AG and reduction of AA, have been proposed to be implicated in modulation of BBB properties. After neurological insults, endocannabinoids are elevated and are hypothesized to protect the CNS through enhanced cannabinoid receptor signaling [12]. Indeed, administration of 2-AG was shown to be neuroprotective and restored BBB function after traumatic brain injury (TBI) [13]. In contrast, elevation of AA has long been known to induce BBB dysfunction and cerebral edema [14]. Due to the bidirectional effects on brain inflammation, MAGL inhibition has demonstrated a variety of beneficial therapeutic effects including improvement of BBB function in a TBI model [15]. However, the specific mechanism(s) of action of MAGL in BBB function and dysfunction has yet to be elucidated.
In this study, we assessed the effects of systemic inflammation on BBB function and the contribution of the inflammatory cascade to BBB dysfunction subsequent to an ischemic insult. We have characterized and used a selective inhibitor of 2-AG hydrolysis to modulate the central arachidonate inflammatory cascade and to enhance endocannabinoid tone. Transcriptomic analysis coupled with functional measurement of BBB integrity demonstrated that MAGL inhibition promotes preservation of BBB integrity in both inflammatory and ischemic conditions. In addition, we characterized the contribution of central arachidonate lowering effects relative to cannabinoid receptor agonism and demonstrated a dynamic and differential MAGL inhibition-mediated mechanism of BBB protection, which is dependent on the nature (inflammatory vs. ischemic) of the initial insult.
Methods
Animals
Animals were purchased from Charles River (CD1) or Jackson Labs (C57Bl/6). All animals were housed in groups in a temperature-controlled environment, kept on 12 h light/dark cycle, and allowed food and water ad libitum. All procedures were conducted under the approval of the Institutional Animals Care and Use Committee (IACUC) at Pfizer Inc. (Cambridge, MA).
Pharmacokinetic, pharmacodynamic, cytokine, and activity-based protein profiling measurements
CPD-4645 was dissolved in a vehicle of 5:5:90 DMSO:Cremophor:Saline and subcutaneously administered to CD1 mice at a dose of 10 mg/kg. Plasma and brain samples of CPD-4645-treated mice were collected at 0.5, 1, 2, 4, 8, 12, and 24 h post-dose (three mice per time point). Plasma and brain samples of vehicle-treated mice were collected at 1 h post-dose.
Plasma samples were prepared for CPD-4645 measurement on wet ice. Briefly, samples were spiked with IS (internal standard) solution (100 ng/mL Labtalol, 400 ng/mL diclofenac, and 200 ng/mL tolbtamide in CAN/MeOH (v:v 50:50) with 0.1% formic acid) followed by extraction in water/MeOH (v:v 75:25) with 0.1% formic acid. Samples were then centrifuged at 4000 rcf for 10 min at 4 °C, and supernatant was directly injected for LC-MS/MS analysis. Cerebellums were homogenized with three volumes (w:v) of PBS. The homogenates were then spiked with IS solution followed by centrifugation at 15,700 rcf for 15 min at 4 °C. Supernatant was removed and directly injected for LC-MS/MS analysis.
Levels of brain 2-AG and AA were measured by homogenizing brain with three volumes (w:v) of homogenization buffer (1% PMSF and 5% 100 mM NH4OAc in water pH 2.0, adjusted with formic acid). The brain homogenates were then spiked with IS2 [1 μg/mL d5-2-AG or d8-AA (deuterated 2-arachidonoylglycerol or deuterated arachidonic acid) (Cayman Chemical Ann Arbor, Michigan) in ACN] solution followed by precipitation and organic extraction in ACN. Samples were then centrifuged at 15,700 rcf for 15 min at 4 °C and supernatant directly injected onto a C-18 UPLC column held at 50 °C at a flow rate of 0.5 mL/min. For 2-AG measurement, mass spectrometry was run in positive ESI mode with SRM detection. The 2-AG m/z was 379.4/287.3. The d5-2-AG m/z was 384.4/287.4. For AA measurement, mass spectrometry was run in negative ESI mode with SRM detection. The AA m/z was 303.1/205.2. The d5-2-AG m/z was 311.2/267.0. Calibration standards and quality control samples were prepared using the same methodology. The 2-AG calibration curve consisted of 50–50,000 ng/mL 2-AG in homogenization buffer. The AA calibration curve consisted of 50–50,000 ng/mL AA in homogenization buffer.
Brain cytokine levels were measured by preparing soluble proteomes as previously described [11] followed by ELISA measurement using V-Plex Proinflammatory Panel 1 (Meso Scale Diagnostics, Rockville MD) with detection antibodies for IL1β and IL6. Cytokine levels were normalized for total protein as determined by bicinchoninic acid (BCA) protein assay (Thermo Fisher, Waltham, MA).
Activity-based protein profiling was performed as previously described [9] with minor modifications. Briefly, membrane proteomes were isolated by homogenizing brain tissue in PBS buffer and centrifuged at 145,000×g for 45 min at 4 °C. Pellets were washed three times in PBS. Samples were diluted to 1 mg/mL total protein and incubated with 2 μM final fluorophosphonate-rhodamine. Reactions were incubated for 30 min at room temperature and quenched with 4× SDS loading buffer and boiled for 10 min at 95 °C. Samples were run on 12% SDS mini-gels and visualized using a fluorescent scanner (GE ImageQuant Las4000). Densitometry analysis was performed on the in-gel fluorescence images using Image Studio version 4 software (LI-COR, Lincoln, Nebraska).
Induction of BBB disruption by lipopolysaccharide
Male CD1 mice aged 8–10 weeks were intraperitoneally injected with 3 mg/kg of salmonella enterica typhimurium (Sigma L2262) at 0, 6, and 24 h as previously described [3]. For the pharmacology studies, mice were dosed with 10 mg/kg subcutaneous CPD-4645 in a vehicle of 5:5:90 DMSO:Cremophor:Saline with and without combination of 3 mg/kg rimonabant and AM630 in vehicle (5:5:90; DMSO:Cremophor:Saline) 30 min post each LPS dose. For assessment of BBB function, mice were euthanized at 28 h after the first LPS injection. Animals were perfused with heparinized PBS, and brains were collected and frozen on dry ice for fluorescent immunostaining or ELISA. For the RNA-seq studies, the brains were not perfused as above and frozen brains were transferred into RNA-later-ICE Frozen Tissue Transition Solution (Life Technologies AM7030) in stored for 24 h at − 20 °C. Brain vasculature was then isolated as previously described [16] with the addition of two extra washes in sucrose buffer to remove remaining traces of myelin. Total RNA was purified using Qiagen RNeasy kits. RT-qPCR was performed to determine which cell types are present in the preparations.
| Gene symbol | Protein name | Taqman assay ID |
|---|---|---|
| Gfap | Glial Fibrillary Acidic Protein | Mm01253033_m1 |
| Pdgfrb | Platelet-Derived Growth Factor Receptor Beta | Mm00435546_m1 |
| Pecam1 | Platelet Endothelial Cell Adhesion Molecule | Mm01242576_m1 |
| Sypl | Synaptophysin-like | Mm01289818_g1 |
| Aif1 (Iba1) | Allograft Inflammatory Factor 1 | Mm00520165_m1 |
Transcriptomics
Library preparation
Stranded cDNA libraries were prepared from 50 ng RNA using TruSeq Stranded mRNA NeoPrep kits (Illumina) and sequenced on a NextSeq 500 (Illumina) at a read depth of 10–20 million reads per sample (75 base pair single-end reads). FASTQ files were assembled using bcl2fastq.
Sequence data processing
Sequence reads were aligned to mouse genome mm10/GRCm38 assembly using STAR 2.5.2a (http://github.com/alexdobin/STAR; parameters: --outFilterMultimapNmax 20 --outFilterType BySJout --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --outFilterMismatchNoverLmax 0.1 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000). Read counts for gene expression quantification were calculated using STAR --quantMode based on GENCODE release M9 basic annotation.
Differential gene expression analysis
The read count data were normalized by the trimmed mean of the M-values method [17] using the calcNormFactors() function from the edgeR package [18]. The mean-variance relationship of the counts was estimated using the voom() function [19] from the limma package [20]. To identify differentially expressed genes, the log2 fold differences and p values were estimated by fitting a linear model for each gene and applying empirical Bayes to moderate residual variances, using lmFit() and eBayes() functions from the limma package. Benjamini-Hochberg procedure for multiple hypothesis testing was applied to adjust p values. Differentially expressed genes were selected at twofold change (FC) cutoff and false discovery rate (FDR) of 0.05.
Gene ontology analysis
DAVID 6.7 bioinformatics tools (http://david-d.ncifcrf.gov) were applied for gene ontology (GO) analysis. The enriched GO categories were identified using the functional annotation clustering tool.
Assessment of extravascular fibrinogen via fluorescent immunostaining
Fresh frozen brains were stored at − 20 °C until sectioning. Twelve-micrometer sections were cut using a cryostat and were heat mounted to slides and stored at − 20 °C until use. Sections were incubated in Zinc fix (BD Pharmingen #51-7538KZ) for 3 h prior to permeabilization (0.1% citrate, 0.1% Triton-X100 in PBS) for 10 min at 4 °C. Next, sections were blocked in 3% bovine serum albumin (BSA) for 1 h. Sections were incubated overnight at 4 °C in sheep anti-Fibrinogen (1:500; Serotec #4440-8004) and rat anti-CD31 (1:250; Serotec MCA2388T) made in 0.1% BSA. The next day, sections were washed in PBS-T and incubated in Alexa Fluor anti-sheep-555 (Invitrogen #A21436) and Alexa Fluor anti-rat-488 (Life Technologies #A21208) made in 0.1% BSA for 1 h at 37 °C. Slides were imaged on a Zeiss 710 confocal microscope. Images were taken with 20× objective and analyzed using ImageJ software. All images and subsequent analysis were performed blinded. To analyze extravascular fibrinogen, images were thresholded to remove signal where CD31 staining (vascular endothelial marker) and fibrinogen staining were co-localized. The areas of exclusively fibrinogen signal in the brain were quantified using the ImageJ integrity density function.
Measurement of brain and plasma fibrinogen levels
Brain homogenates were made by sonicating one cerebral hemisphere (without cerebellum) in radioimmunoprecipitation assay buffer (Sigma #R0278) containing protease and phosphatase inhibitors (Pierce #88669). Lysates were centrifuged twice at 13,300 rpm for 15 min. Pellets were discarded and supernatants were kept for analysis. For plasma sample collection, blood samples were collected in heparin-sodium (1:10) via cardiac puncture and centrifuged at 6000 rpm for 5 min. Plasma was collected and spun again at 13,300 rpm for 5 min to remove any residual blood cells. Brain homogenates (1:30 dilution) and plasma samples (1:20,000 dilution) were analyzed for fibrinogen levels by ELISA (Genway; cat #GWBBB0BA2) following the manufacturer’s protocol.
Measurement of FITC dextran permeability in LPS model
At 1 h prior to tissue collection, 200 μL of 25 mg/mL solution of fluorescein isothiocyanate (FITC) conjugated dextran (70 kDa, Sigma Aldrich, St Louis, MO) in sterile saline was injected retro-orbitally. At 1-h post-fluorescent tracer administration, mice were anesthetized with isoflurane and transcardially perfused with approximately 20 mL of phosphate buffered saline (PBS) with heparin (5 U/ml). The brain was collected, frozen in liquid nitrogen, and stored at − 80 °C until use.
To measure FITC-Dextran fluorescence, a hemi-brain was homogenized using a TissueLyser II (Qiagen, Germantown, MD) set at 25 Hz for 2 min at 4 °C in 600 μL of ice-cold radioimmunoprecipitation (RIPA) assay buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) (Sigma Aldrich, St Louis, MO) with protease inhibitors (cOmplete EDTA-free, Roche, Indianapolis, IN). Each sample was then centrifuged at 10,000 rcf for 10 min at 4 °C to pellet large debris.
Supernatant was collected and diluted 1:1 with PBS placed into a black 96-well plate. Fluorescence was measured in a SpectraMax M5 (Molecular Devices, Sunnyvale, CA) with excitation at 488 nm and emission at 525 nm. After the initial reading, samples were further diluted (1:4 and 1:8) and fluorescence intensity re-measured. Each reading was multiplied by the corresponding dilution factor, and an average fluorescence reading was determined.
Induction of focal photothrombotic ischemia
To alleviate any potential pain or discomfort, mice were treated with 0.05 mg/kg of buprenorphine prophylactically. Mice were anesthetized with isoflurane for the duration of the procedure. Mice (C57BL/6) received a retro-orbital injection of 30 μL of a 15 mg/mL solution of rose bengal dissolved in sterile saline. The head of the mice was shaved and a small incision aseptically made along the midline of the skull. The mice were then placed into a stereotaxic frame, and a green laser held approximately 4 cm from the surface of the skull was positioned over the somatosensory cortex. Mice were subjected to the laser exposure for 10 min. After green laser illumination, the skin incision was closed with wound clips and animals monitored as they woke from the anesthesia. Mice were subcutaneously administered vehicle or 10 mg/kg CPD-4645 30 min post-photothrombosis. Mice received a daily injection of vehicle of CPD-4645 for 3 days.
On day 3, each mouse was retro-orbitally injected with Alexa Fluor® 555-conjugated Cadaverine (Thermo A30677) at 500 μg/20 g body weight as previously described [21] or 200 μL of 25 mg/mL solution of fluorescein isothiocyanate (FITC)- conjugated dextran (70 kDa, Sigma Aldrich, St Louis, MO) in sterile saline. At 1 h post-fluorescent tracer administration, mice were anesthetized with isoflurane and transcardially perfused with approximately 20 mL of PBS with heparin (5 U/ml). The brain was collected and the cortex dissected. A 5-mm tissue punch was taken from the core of the thrombotic infarct and from the identical region of contralateral cortex. Tissues were frozen in liquid nitrogen and stored at − 80 °C until use.
To measure FITC-Dextran fluorescence, a 5-mm stainless steel bead was added to each tube along with 300 μL of ice-cold radioimmunoprecipitation (RIPA) assay buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) (Sigma Aldrich, St Louis, MO) with protease inhibitors (cOmplete EDTA-free, Roche, Indianapolis, IN). Samples were then homogenized using a TissueLyser II (Qiagen, Germantown, MD) set at 25 Hz for 2 min at 4 °C followed by incubation on ice for 30 min. Each sample was then centrifuged at 10,000 rcf for 10 min at 4 °C to pellet large debris.
Supernatant was collected and diluted 1:1 with PBS placed into a black 96-well plate. Fluorescence was measured in a SpectraMax M5 (Molecular Devices, Sunnyvale, CA) with excitation at 488 nm and emission at 525 nm. After the initial reading, samples were further diluted (1:4 and 1:8) and fluorescence intensity re-measured. Each reading was multiplied by the corresponding dilution factor, and an average fluorescence reading was determined.
Results
Profiling of inflammatory gene expression in neurovascular-enriched brain fraction following systemic insult
Brain fraction enriched for cerebrovascular tissue was isolated as previously described [16]. To confirm enrichment of the neurovasculature, we assessed expression of cell type-specific genes characteristic of the different cell types that compose the blood-brain barrier. Gene expression analysis by RT-qPCR demonstrated enrichment in the expression of Gfap, Pdgfrb, and Pecam1 and low abundance of Syp and Aif1 (Fig. 1a) demonstrating prevalence of astrocytes, pericytes, and endothelial cells over neurons and microglia which is consistent with the cell types that comprise the neurovascular unit.
Fig. 1.
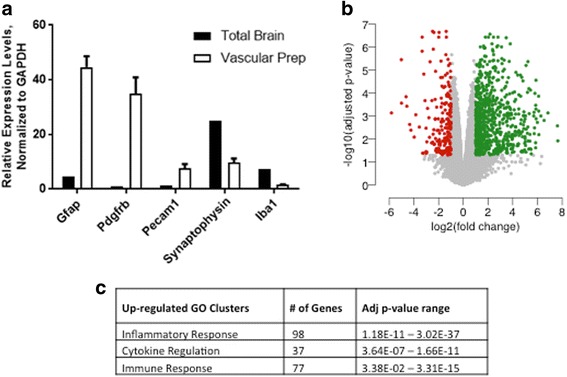
Transcriptomic profiling of neurovascular unit after LPS challenge. a Expression profile of cell type-specific makers in neurovasculature preparation. b Volcano plot of statistically significant upregulated (green) and downregulated (red) genes in the neurovascular unit after LPS challenge. c Gene ontology clusters for up- and downregulated genes identified using DAVID bioinformatics resource. Neurovasculature was isolated 4 h post last LPS dose. Data are means ± SEM, n = 4/5 mice per group
Activation of the neurovasculature through inflammatory challenge has previously been shown to disrupt the integrity of the BBB and substantially induce its permeability [3, 22]. After systemic LPS administration, brain vasculature was isolated and extracted RNA was analyzed by next generation sequencing for gene expression quantification. The RNA-seq analysis identified 949 differentially regulated genes (false discovery rate ≤ 0.05, increased or decreased by at least twofold) by LPS challenge compared to vehicle controls (Fig. 1b and Additional file 1: Table S1). Among those, 745 were statistically significantly upregulated, and 204 were significantly downregulated. The GO analysis of the upregulated genes using DAVID bioinformatics tools identified the top three enriched functional annotation clusters as inflammatory response, regulation of cytokine production, and immune response (Fig. 1c and Additional file 2: Table S2). The inflammatory response cluster contained genes pertaining to TLR (Toll-like receptor) signaling and inflammasome activation as expected in an LPS challenge model. The cytokine regulation and immune response clusters contained a number of genes coding for cytokines, chemokines, and complement system proteins. Of the LPS downregulated genes, there were no statistically significant clusters identified (Additional file 2: Table S2), and therefore in this study, we only focus on the upregulated genes.
Inhibition of 2-AG hydrolysis rescues altered transcriptional profile of the neurovascular unit after LPS challenge
Previous studies have highlighted the anti-inflammatory effects of MAGL inhibition in acute and chronic brain insults and transgenic mouse models [10, 11, 23, 24]. Additionally, the protective role of 2-AG in preserving BBB integrity after acute brain injury has been described [13]. Therefore, we sought to determine whether inhibition of MAGL activity in vivo can reverse BBB pathogenesis and rescue neurovascular integrity and function after insults.
CPD-4645 (Fig. 2a) is a hexafluoroisopropyl carbamate covalent MAGL inhibitor [25]. CPD-4645 is brain penetrant (Fig. 2b), and administration of subcutaneous CPD-4645 at 10 mg/kg to CD1 mice resulted in ~ 3-fold increase in brain 2-AG (Fig. 2c) which persisted for 8 h. Levels of brain AA were reduced by 50% over the same time period (Fig. 2d). CPD-4645 inhibited MAGL (~ 99% max), alpha-beta-hydrolase domain containing 6 (ABHD6) (~ 70% max), and fatty acid amide hydrolase (FAAH) (~ 50% max) in the brain as determined by activity-based protein profiling (Additional file 3: Figure S1). No inhibition of additional serine hydrolases was detected (data not shown). ABHD6 and FAAH are serine hydrolases responsible for the degradation of 2-AG and anandamide (AEA), respectively. ABHD6 is closely related to MAGL and is responsible for ~ 15% of the hydrolysis of 2-AG while MAGL is responsible for the remaining ~ 85% [26]. Although the majority of the pharmacodynamic effects seen with CPD-4645 may be attributed to inhibition of MAGL, the partial inhibition of ABHD6 may also be contributing to the 2-AG elevation and AA lowering seen in the brain. It has previously been shown that > 85% inhibition of FAAH is required to maintain significant elevations in brain AEA levels [27], as such the 50% inhibition of FAAH seen in these experiments is below the threshold level needed to elicit a pharmacodynamic effect.
Fig. 2.
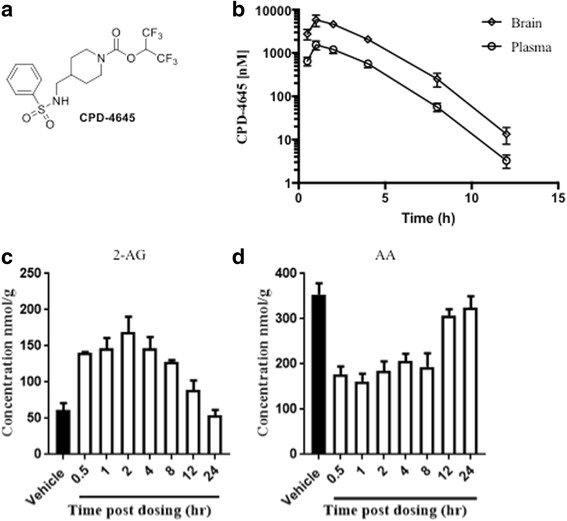
Pharmacokinetic and pharmacodynamic profiling of CPD-4645 in naïve mouse brain. a Structure of the 2-AG hydrolysis inhibitor CPD-4645. Total CPD-4645 concentrations in brain and plasma (b) and bulk levels of brain 2-AG (c) and AA (d) at given time points following single 10 mg/kg subcutaneous dose of CPD-4645 in CD1 mice. Data are means ± SEM, n = 3/5 mice per group
Mice were subjected to the LPS challenge as described above, in the presence and absence of CPD-4645 which was administered 45 min post each dose of LPS. Assessment of brain 2-AG levels 3.5 h post last dose of CPD-4645 revealed no effect of LPS while CPD-4645 treatment significantly elevated levels of the endocannabinoid (Fig. 3a). There was a modest but significant elevation in brain AA levels due to LPS challenge, which was significantly attenuated by CPD-4645 treatment (Fig. 3b). Given the potent anti-inflammatory effects of MAGL inhibition, we next measured levels of the proinflammatory cytokines IL1β and IL6 in the brains of mice challenged with LPS. The LPS challenge significantly elevated levels of both cytokines above baseline levels while treatment with CPD-4645 significantly reduced the inflammatory mediators (Fig. 3c, d). We next profiled the effects of LPS and CPD-4645 on the transcriptional profile of the neurovasculature. As expected, transcriptional signatures showed a clear distinction between the sham and LPS group (separated by the first principal component as shown in Additional file 4: Figure S2). However, the CPD-4645 treatment exhibited a striking reversal of the transcriptional induction observed in the LPS group (Fig. 4a). Among the 745 statistically significantly upregulated genes in response to LPS challenge, the expression of 550 (74%) showed reduction in gene expression with CPD-4645 treatment (95% confidence interval 0.70–0.77, proportion test). The majority of the LPS upregulated genes in the annotation clusters related to inflammation, cytokine regulation, and immune response (Fig. 1c and Additional file 2: Table S2) were attenuated by CPD-4645 treatment (Fig. 4b). The top enriched GO clusters identified for the significantly downregulated genes (Additional file 1: Table S1) in the CPD-4645-treated animals included leukocyte migration, inflammatory response, and chemotaxis (Additional file 2: Table S2). These results suggest a role of MAGL inhibition in reducing mediators of leukocyte-endothelial cell interaction and inflammation.
Fig. 3.
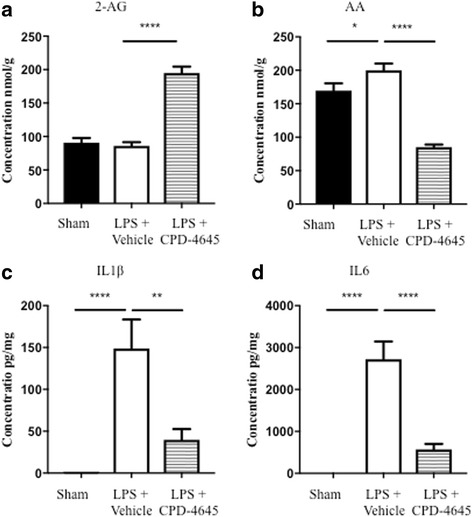
Pharmacodynamic and anti-inflammatory activity of CPD-4645 in LPS-challenged mouse brain. Inhibition of MAGL by CPD-4645 resulted in significant elevation of brain 2-AG (a) and concomitant reduction in brain AA (b). Levels of the proinflammatory cytokines, IL1β (c) and IL6 (d), were significantly modulated in brain tissue following LPS challenge and CPD-4645 treatment. Bar graphs were plotted with mean ± SEM and data analyzed using one-way analysis of variance (ANOVA) with Bonferroni post-hoc comparisons. n = 10 mice per group. Significance is shown as *p < 0.05; **p < 0.01; ****p < 0.0001
Fig. 4.
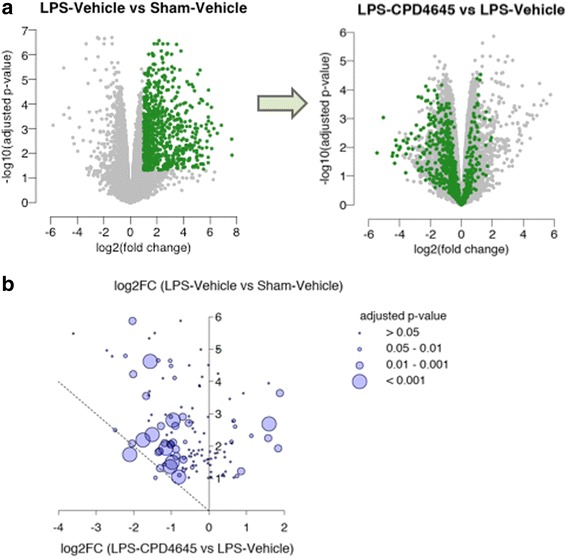
CPD-4645 alters LPS-induced gene expression profiles. a Volcano plots showing expression changes of the LPS upregulated genes (left, green points) after treatment with CPD-4645 (right). b Bubble plot showing changes in expression of genes that are induced by LPS and related to cytokines and inflammation. The y-axis represents the log2-fold change due to LPS challenge while the x-axis represents the log2-fold change due to CPD-4645 treatment. Dotted line represents a return to basal expression level. Size of the bubbles represents the multiple comparison adjusted p values for vehicle versus CPD-4645 treatment. Neurovasculature was isolated 4 h post last LPS dose. n = 4/5 mice per group
In addition to the notable changes in genes directly involved in immune and inflammatory responses, several genes associated with endothelial activation and loss of BBB integrity were significantly altered by LPS challenge and attenuated in animals treated with CPD-4656 (Fig. 5a). We found that in mice treated with CPD-4645, the transcript levels of Vwf, Selp, Sele, Vcam1, Itga5, and Tgfb1, markers of endothelial cell activation and inflammatory responses, were significantly reduced compared to vehicle-treated animals following the LPS challenge (Fig. 5b). In addition, transcript levels of the extracellular proteases Adamts9, Adamts4, and Mmp8 were also upregulated by LPS challenge and significantly reduced following CPD-4645 treatment (Fig. 5c).
Fig. 5.
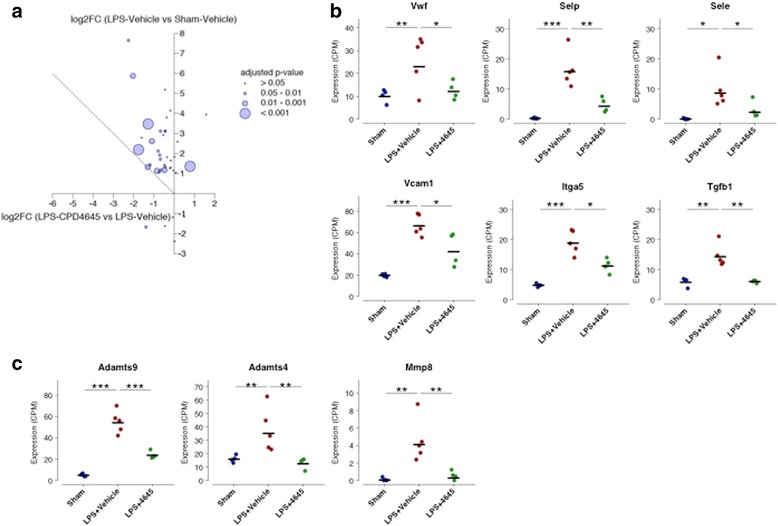
Genes related to BBB (dys) function and proteases are differentially expressed after LPS challenge and treatment with CPD-4645. a Changes in the expression of 40 genes related to BBB function and dysfunction. The y-axis represents the log2-fold change due to LPS challenge while the x-axis represents the log2-fold change due to CPD-4645 treatment. Dotted line represents a return to basal expression level. Size of the bubbles represents the multiple comparison adjusted p values for vehicle vs. CPD-4645 treatment. b Expression levels of selected BBB genes. c Expression levels of selected extracellular proteases. n = 4–5 mice/group. Significance is shown as adjusted p values *p ≤ 0.05, **p < 0.01, *** p < 0.001. Differential gene expression was analyzed using the limma package in R/Bioconductor (see Methods)
Inhibition of 2-AG hydrolysis reduces BBB permeability after LPS challenge
In order to assess the physiological consequences of reversing the inflammatory signature of brain endothelial activation and dysfunction, we evaluated BBB integrity by measuring extravasation of an abundant plasma protein, fibrinogen, into the brain via ELISA analysis and immunofluorescent staining in LPS-challenged animals treated with either vehicle or CPD-4546. We measured the ratio of the brain to plasma fibrinogen using ELISA. As fibrinogen is an acute phase protein upregulated under inflammatory conditions, to account for potential differences in plasma fibrinogen, we normalized the brain fibrinogen to the amount of fibrinogen present in the plasma. We showed that the ratio of the brain to plasma fibrinogen was significantly decreased in animals treated with CPD-4645 following LPS challenge (Fig. 6a). Of note, LPS challenge resulted in elevated levels of circulating fibrinogen in both vehicle- and MAGL inhibitor-treated mice to the same degree, suggesting that reduction of brain fibrinogen in the LPS/CPD-4645 group relative to LPS/veh is not due to reduction in systemic fibrinogen (Fig. 6b). In addition, we stained for CD31+ endothelial cells and fibrinogen in the striatum. Images were merged to determine the degree of extravascular fibrinogen staining as previously reported [28]. Vehicle-treated mice show significant leakage of fibrinogen into the brain parenchyma (Fig. 6c, d, and g) a phenotype that was significantly reversed when LPS-challenged animals were treated with CPD-4546 (Fig. 6e–g).
Fig. 6.
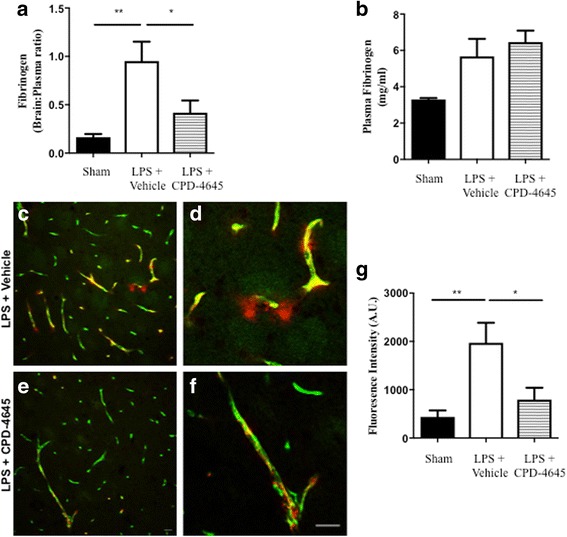
Inhibition of 2-AG hydrolysis reduces LPS-induced BBB permeability. a, b Fibrinogen levels in b plasma and the a ratio of brain to plasma fibrinogen were assessed by ELISA. n = 5/7 mice per group. c, d Fluorescent immunostaining in the striatum for fibrinogen (red) and vascular marker (CD31; green) demonstrated leakage of fibrinogen into the brain with vehicle treatment, whereas vascular integrity was preserved when (e, f) MAGL was inhibited. g Extravascular fibrinogen was semi-quantitated in fluorescently labeled sections of the striatum. Bar graphs were plotted with mean ± SEM and data analyzed using one-way analysis of variance (ANOVA) with Tukey post-hoc comparisons. n = 5/7 mice per group. Significance is shown as *p < 0.05, **p < 0.01. Scale bar = 20 μm
Inhibition of 2-AG hydrolysis reduces blood-brain barrier permeability following photothrombosis
Ischemic injury results in a brain inflammatory cascade that also alters BBB properties and permeability. To assess efficacy of MAGL inhibition at reducing BBB permeability following ischemic inflammatory injury, the photosensitizing dye, rose bengal, was combined with exposure to green laser light to induce focal ischemic injury in the somatosensory cortex which results in BBB breakdown [29, 30]. The extravasation of high molecular weight fluorescent tracers, which are typically excluded from the brain parenchyma, was utilized as a surrogate marker of altered BBB permeability. Treatment with CPD-4645 30 min after inducing the lesion resulted in significant attenuation of BBB damage (Fig. 7a, b). To further test the clinical utility of a MAGL inhibitor, animals were treated 6 h after the onset of the ischemic lesion. Interestingly, leakage of the fluorescent tracer was significantly attenuated in CPD-4645-treated animals compared to vehicle (Fig. 7c, d). These results indicate that inhibition of MAGL, even when withheld for 6 h, could significantly improve BBB function after ischemic injury.
Fig. 7.
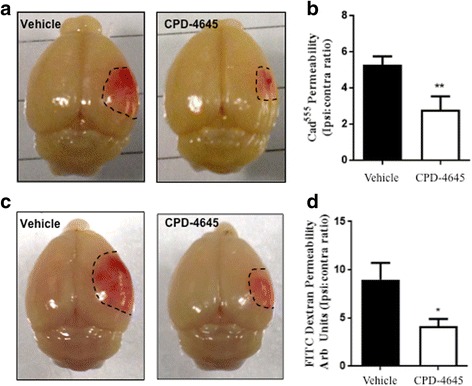
CPD-4645 rescues BBB integrity after ischemic challenge. The rose bengal photothrombosis model induces a focal ischemic injury that leads to BBB dysfunction. CPD-4645 administered 30 min post lesion (a, b) reduced the penumbra size (a) and BBB permeability (b) as assessed by extravasation of Cadavarin555 into the brain parenchyma. CPD-4645 administered 6 h post ischemic lesion (c, d) reduces penumbra size (c) and BBB permeability (d) as assessed by extravasation of 70 kDa FITC conjugated dextran in the brain parenchyma. Data were plotted with means ± SEM and data analyzed using unpaired t test (n = 5 mice/group). Significance is shown as *p ≤ 0.05 and **p < 0.01 compared to vehicle group
Differential mechanisms of action of 2-AG hydrolysis inhibition in models of BBB damage
To assess the contribution of cannabinoid signaling to the mechanism of action of MAGL inhibition in preservation of blood-brain barrier integrity, we tested if the efficacy of MAGL (and possibly ABHD6) inhibition was reversed by blockade of cannabinoid type 1 and 2 receptor signaling. A combination of CB1 and CB2 receptor antagonists, Rimonabant and AM630 (3 mg/kg s.c.), were used in the LPS- and focal ischemia-induced BBB disruption models. Co-treatment of LPS-challenged mice with CB1 and CB2 antagonists did not significantly reverse the effect of MAGL inhibition on BBB breakdown (Fig. 8a). In contrast, co-treatment of photothrombotic mice with MAGL inhibitor and CB1 and CB2 antagonists partially reversed the protective effect of MAGL inhibition on BBB dysfunction (Fig. 8b). These results suggest that 2-AG hydrolysis differentially regulates BBB permeability after systemic or ischemic inflammatory injury, and that enhanced cannabinoid signaling, through CB1 and/or CB2, contributes to the rescue of BBB permeability following ischemic injury. The contribution of cannabinoid signaling to BBB rescue in the LPS model still cannot be ruled out based on these results alone and will require additional molecular and pharmacological characterizations.
Fig. 8.
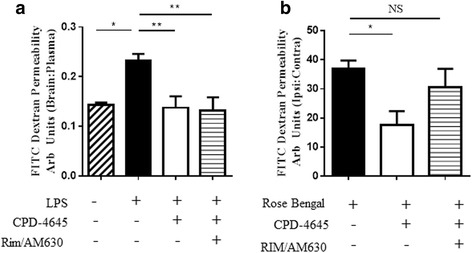
CPD-4645 rescues BBB integrity via endocannabinoid dependent and independent mechanisms. Blockade of endocannabinoid signaling with rimonabant and AM630 (3 mg/kg) does not reverse the BBB protective effects of MAGL inhibition in the inflammation-driven LPS model (a), whereas blockade of endocannabinoid signaling partially reverses the protective effects of MAGL inhibition (administered 30 min post lesion) in the photothrombotic ischemia-driven model (b) as measured by extravasation of 70 kDa FITC-dextran into the brain parenchyma. Data were plotted with means ± SEM (n = 6–9 mice/group) and analyzed with one-way ANOVA with Bonferroni’s post-hoc test. Significance is shown as *p ≤ 0.05, **p ≤ 0.01
Discussion
The integrity of the BBB is often compromised following acute insults or in cases of chronic neuroinflammation leading to infiltration of circulating immune cells and extravasation of blood components into the brain parenchyma. The entrance of these restricted components into the CNS can lead to a multitude of pathophysiological responses and further exacerbate the initial trauma or disease. In this study, we demonstrate that inhibition of 2-AG hydrolysis (inhibition of MAGL and partial inhibition of ABHD6) by CPD-4645 is sufficient to reduce BBB damage in both inflammatory- and ischemic-driven models of BBB dysfunction.
We show that CPD-4645 is a potent and brain penetrant covalent MAGL inhibitor. Treatment with CPD-4645 was sufficient to elevate brain levels of the major endocannabinoid, 2-AG, while reducing levels of brain AA in naïve and LPS-challenged mice. In addition to modulating AA levels, inhibition of 2-AG hydroylsis resulted in significant attenuation of the proinflammatory cytokines IL1β and IL6 in the brains of LPS-challenged mice. These results highlight the potent anti-inflammatory mechanism of MAGL inhibition.
RNA-seq analysis of isolated brain vasculature after LPS challenge showed a significant elevation of genes related to the immune and inflammatory responses as well as endothelial cell activation. Treatment with CPD-4645 was able to restore these gene clusters closer to sham-treated levels. The results of this particular experiment are confounded by the fact that peripheral immune cells like macrophage and neutrophils may be attaching to the cerebral endothelium and/or entering the brain after LPS challenge. The transcriptomic signature shown in this study is a combination of all cells present in the isolated neurovascular unit preparation including those peripheral cells. The changes in gene expression are therefore a snapshot of the overall state of health of the BBB and do not necessarily imply that MAGL inhibition is directly responsible for altering transcription.
Further analysis into the gene expression dataset revealed a number of interesting findings related to endothelial activation, leukocyte-endothelial interaction, and BBB integrity-related genes. Although assumed based on previous reports, our RNA-seq results confirmed that endothelial activation occurs in this LPS model as is evidenced by the upregulation of von Willebrand factor (Vwf), P-Selectin (Selp), and E-Selectin (Sele). Vwf and Selp are two key components of Weibel-Palade bodies which are released from endothelial cells in response to cytokines, playing dual roles in regulating hemostasis and additional inflammatory processes [31]. Interestingly, Selp knockout mice show reduced BBB breakdown following transient ischemic stroke, likely due to preventing leukocyte-endothelial cell interaction and downstream inflammatory process damaging the endothelial cell integrity [32]. In addition, mice genetically engineered to produce high levels of soluble Selp in circulation showed an increase in BBB permeability [33]. We also demonstrated CPD4-656 treatment reduced the upregulation of vascular cell adhesion molecule 1 (Vcam1), which is known to occur in response to cytokine release and plays an additional roll in leukocyte-endothelial interactions. Similar to P-selectin, soluble Vcam1 in CSF and plasma correlates with BBB lesion severity, in multiple sclerosis (MS) for example [34]. Moreover, the mechanism of action of the approved MS therapy Natalizumab involves prevention of the α4β1-integrin receptor molecules on immune cells from interacting with Vcam1 on endothelial cells to reduce disease-related lesions and leukocyte trafficking into the brain.
The interaction of endothelial cells with the extracellular matrix protein fibrinonectin is mediated through integrin alpha-5 (Itga5). It was recently found that endothelial Itga5 protein levels are increased in the inflammatory EAE model of multiple sclerosis and coincide with fibrinogen leakage into the spinal cord [35]; thus, our findings that Itga5 mRNA is upregulated with LPS treatment is not surprising. Moreover, deletion of Itga5 specifically in endothelial cells strengthens BBB integrity and increases tight junction protein expression following acute brain injury [36]. Interestingly, MAGL inhibition following LPS challenge also reduced the levels of Tgfb1, a known upstream regulator of Itga5 and other integrins [37]. The action of Tgfb1 on the vasculature is complex and highly dependent on the cellular and environmental context [38]. Whether the effects of CPD-4645 on Itga5 expression are dependent or independent on Tgfb1 signaling remains elusive and should be addressed in future experiments.
Although interesting, it is currently unknown whether the attenuation of upregulation of these genes in LPS-challenged animals with CPD-4645 treatment is a direct result of inhibiting the MAGL pathway in endothelial cells or is due to an indirect effect of reducing neuroinflammation. Regardless, maintenance of these genes at a baseline level after exposure to LPS most likely plays a role in preservation of BBB as these proteins have been implicated in modulation of BBB integrity [5, 39].
Gene expression analysis also revealed changes in the matrix metalloproteinases (MMPs) and the disintegrin and metalloproteinases with thrombospondin motifs (ADAMTSs). MMPs and ADAMTs are classes of extracellular proteases which cleave a wide variety of extracellular matrix, tight junction, and basement membrane proteins leading to the hypothesis that their activities contribute to the dysfunction of the BBB in inflammatory and ischemic injuries [40–43]. In this study, we found LPS challenge to induce the expression of several MMPs including Mmp-3, 8, 9, and 19. Of these, Mmp8 was significantly reduced by treatment with CPD-4645. MMP-8 is an inducible MMP that has been shown to be upregulated in experimental models of multiple sclerosis [44] and elevated in the CSF of children with bacterial meningitis [45]. Consistent with our findings in the LPS model, which mimics a bacterial challenge, infection of human brain microvascular endothelial cells with Neisseria meningitides resulted in an increase in MMP-8 activity and was shown to be directly involved in the cleavage of the occludin and subsequent BBB dysfunction [46].
The role of ADAMTS family of proteins has been extensively studied for their ability to remodel extracellular matrix (ECM) in the context of osteoarthritis. Recent emerging data suggest that astrocytic ADAMTSs may play a variety of roles within the CNS including modulation of neuronal signaling, inflammation, and permeability of the BBB [42]. We found that LPS challenge stimulated the expression of Adamts-1, 4, 9, and 14 in the neurovasculature. Treatment with CPD-4645 significantly downregulated expression of Adamts4 and 9. It has been shown that IL1β and TNFα, cytokines which are elevated in our LPS model, can induce the expression of Adamts4 and 9 [47, 48] in astrocytes and macrophage. Versican is the major chondroitin sulfate proteoglycan within the vasculature and is a cleavage substrate for ADAMTS-4 [49] and ADAMTS-9 [50] leading to the hypothesis that ADAMTS activity may contribute to BBB dysfunction via degradation of ECM [42, 51].
Taken together, our data suggest that inflammation induced by LPS challenge can serve to upregulate expression of Mmp8 and Adamts4 and 9 which may contribute to subsequent damage of the BBB through degradation of occludin and versican. Treatment with a MAGL inhibitor elicits an anti-inflammatory response which in turn serves to reduce levels of the neurovascular damaging proteases. Regulation of proteases through reduction in inflammation is one possible explanation of many for the BBB protective effects observed in this study. A deeper understanding of the complex interplay between neuroinflammation, protease activity, and BBB function will be required to determine if MMP-8, ADAMTS-4, or ADAMTS-9 are solely responsible or act in concert with each other or with other pathophysiological processes leading to BBB dysfunction.
Several studies have shown that augmentation of 2-AG, either through direct administration or through inhibition of MAGL or ABHD6, can have beneficial effects on neuroinflammation and blood-brain barrier dysfunction in the context of TBI [13, 15, 52]. Rodent TBI models result in damage to the BBB by trauma (cell death), inflammation, or ischemia. In this study, we sought to determine the molecular mechanisms of MAGL-mediated BBB protection using animal models which utilize inflammation or ischemia to drive damage to the BBB. Here, we demonstrate that inhibition of MAGL activity can attenuate BBB breakdown, as measured by the extravasation of brain impermeable endogenous proteins or fluorescently labeled tracers, after either inflammatory (LPS) or ischemic (rose bengal) insults. To pressure test the clinical utility of a MAGL inhibitor, we withheld treatment for 6 h and then administered the compound. This time frame is within the required treatment window (within 3–4.5 h of onset of ischemia) for ischemic stroke patients to qualify for tPA treatment for example. Our results demonstrate that inhibition of 2-AG hydrolysis still exerted significant protection of BBB function in animals with delayed treatment.
Conclusions
Previous work has not looked at the mechanism of the efficacy of endocannabinoid degradation inhibitors [15], or attributed this efficacy in TBI models to enhanced cannabinoid receptor activation (lesion volume and neurodegeneration) through the use of cannabinoid receptor antagonists [52]. Here, we show for the first time that the effect of MAGL inhibition in preserving BBB function is driven through the reduction of arachidonate production and through enhanced cannabinoid signaling depending on the nature of the initial insult. In the inflammation-driven LPS model, co-treatment with CB1 and CB2 antagonists did not significantly reverse the CPD-4645 effect on BBB breakdown, suggesting a less pronounced cannabinoid-dependent mechanism of action in this particular assay. In contrast, in the ischemia-driven photothrombotic model, co-treatment of CPD-4645 with CB1 and CB2 antagonists partially reversed the effect of MAGL inhibition on BBB disruption, suggesting a pronounced, but partial, cannabinoid receptor-dependent mechanism of action. It would therefore be of future interest to analyze expression profiles of the neurovasculature in the presence and absence of cannabinoid receptor antagonists in a more global model of ischemia such as the middle cerebral artery occlusion model, in order to further ascertain whether the BBB pathology in the two models is different or comparable. These results highlight a bi-directional mechanism of action (simultaneous enhancement of cannabinoid signaling through elevation of 2-AG and decrease of AA and downstream eicosanoids) that can achieve therapeutic efficacy through either pathway depending on the nature of the insult.
Additional files
Table S1. Differentially regulated genes. Differentially regulated genes (increased or decreased by at least twofold) and p values for LPS-vehicle vs sham-vehicle and LPS-CPD-4645 vs LPS-vehicle groups. (XLSX 294 kb)
Table S2. Gene ontology analysis. Gene ontology analysis was performed using the DAVID bioinformatics tools. The enriched functional annotation clusters are listed along with p values for LPS vs Sham upregulated genes, LPS vs Sham downregulated genes, and CPD-4645 vs vehicle downregulated genes. (XLSX 157 kb)
Figure S1. In vivo selectivity of CPD-4645. Levels of inhibition of MAGL, ABHD6, and FAAH in the brain after 10 mg/kg subcutaneous dose of CPD-4645 at given tie point as determined by activity-based protein profiling using the pan-serine hydrolase probe FP-rhodamine. Full MAGL inhibition was normalized by effect seen at 2 h post-subcutaneous administration of 16 mg/kg JZL-184. (TIFF 1521 kb)
Figure S2. Principal component analysis. Principal component analysis (PCA) for transcriptional signatures demonstrating a distinction between experimental groups. The first two principal components are plotted with the variance explained by each component shown on the axes. (TIFF 1521 kb)
Acknowledgements
We thank the members of the IMRU at Pfizer Global Research for discussion and critical reading of the manuscript.
Funding
This work was supported by Pfizer Global Research.
Availability of data and materials
The RNA sequencing data has been submitted to the Gene Expression Omnibus data repository and can be accessed through the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113855.
Authors’ contributions
NP isolated the brain vasculature. NP and JRP conducted and analyzed the mRNA expression profiling. JRP, JQ, and TAL conducted the RNAseq. JRP, JQ, HX, RDB, and TAS analyzed the RNAseq. JRP, GLS, YP, SMO, and MI conducted and analyzed the data from the pharmacological studies in the LPS model. JRP, GLS, YP, SMO, and RDB conducted and analyzed data from the pharmacological studies in the rose bengal model. JRP, GLS, RDB, and TAS contributed to study design and manuscript drafting. All authors read and approved the final manuscript.
Ethics approval
All procedures involving animals were conducted with the approval of the Pfizer Institute Animal Care and Use Committee (IACUC) and compliant with the regulations and standards of the Animal Welfare Act.
Competing interests
JRP, GLS, JQ, YP, SMO, MI, NP, TAL, HX, RDB, and TAS are or were current employees of Pfizer Inc. at the time the studies were conducted.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12974-018-1166-9) contains supplementary material, which is available to authorized users.
Contributor Information
Justin R. Piro, Email: JPiro1000@gmail.com
Georgette L. Suidan, Email: georgette.suidan@gmail.com
Jie Quan, Email: jie.quan@pfizer.com.
YeQing Pi, Email: yqpi@hotmail.com.
Sharon M. O’Neill, Email: soneill@alumni.brown.edu
Marissa Ilardi, Email: marissa.ilardi@nyumc.org.
Nikolay Pozdnyakov, Email: pozdnyakovn@yahoo.com.
Thomas A. Lanz, Email: tom_lanz@yahoo.com
Hualin Xi, Email: simonxi111@gmail.com.
Robert D. Bell, Email: Robert.bell@pfizer.com
Tarek A. Samad, Email: samad.tarek@gmail.com
References
- 1.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163(5):1064–1078. doi: 10.1016/j.cell.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav Immun. 2009;23(4):507–517. doi: 10.1016/j.bbi.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017;60:1–12. doi: 10.1016/j.bbi.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87(5):779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denes A, Pinteaux E, Rothwell NJ, Allan SM. Interleukin-1 and stroke: biomarker, harbinger of damage, and therapeutic target. Cerebrovasc Dis. 2011;32(6):517–527. doi: 10.1159/000332205. [DOI] [PubMed] [Google Scholar]
- 7.Palasik W, Fiszer U, Lechowicz W, Czartoryska B, Krzesiewicz M, Lugowska A. Assessment of relations between clinical outcome of ischemic stroke and activity of inflammatory processes in the acute phase based on examination of selected parameters. Eur Neurol. 2005;53(4):188–193. doi: 10.1159/000086355. [DOI] [PubMed] [Google Scholar]
- 8.Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, Piesla MJ, Zhang MY, Bingham B, Uveges A, et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol. 2010;78(6):996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- 9.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5(1):37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334(6057):809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, Schwartz JW, Nomura DK, Samad TA. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012;1(6):617–623. doi: 10.1016/j.celrep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bahr BA, Karanian DA, Makanji SS, Makriyannis A. Targeting the endocannabinoid system in treating brain disorders. Expert Opin Investig Drugs. 2006;15(4):351–365. doi: 10.1517/13543784.15.4.351. [DOI] [PubMed] [Google Scholar]
- 13.Panikashvili D, Shein NA, Mechoulam R, Trembovler V, Kohen R, Alexandrovich A, Shohami E. The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol Dis. 2006;22(2):257–264. doi: 10.1016/j.nbd.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. 2000;20(2):131–147. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz PS, Sulzer JK, Impastato RA, Teng SX, Rogers EK, Molina PE. Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J Neurotrauma. 2015;32(5):297–306. doi: 10.1089/neu.2014.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bales KR, O’Neill SM, Pozdnyakov N, Pan F, Caouette D, Pi Y, Wood KM, Volfson D, Cirrito JR, Han BH, et al. Passive immunotherapy targeting amyloid-beta reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain. 2016;139(Pt 2):563–577. doi: 10.1093/brain/awv313. [DOI] [PubMed] [Google Scholar]
- 17.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid beta efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Teng Z, Song Y, Hu M, Chen C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb Blood Flow Metab. 2015;35(4):706. doi: 10.1038/jcbfm.2014.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pihlaja R, Takkinen J, Eskola O, Vasara J, Lopez-Picon FR, Haaparanta-Solin M, Rinne JO. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J Neuroinflammation. 2015;12:81. doi: 10.1186/s12974-015-0305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terrone G, Pauletti A, Salamone A, Rizzi M, Villa BR, Porcu L, Sheehan MJ, Guilmette E, Butler CR, Piro JR, et al. Inhibition of monoacylglycerol lipase terminates diazepam-resistant status epilepticus in mice and its effects are potentiated by a ketogenic diet. Epilepsia. 2018;59(1):79–91. doi: 10.1111/epi.13950. [DOI] [PubMed] [Google Scholar]
- 26.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14(12):1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3’-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313(1):352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- 28.Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lapilover EG, Lippmann K, Salar S, Maslarova A, Dreier JP, Heinemann U, Friedman A. Peri-infarct blood-brain barrier dysfunction facilitates induction of spreading depolarization associated with epileptiform discharges. Neurobiol Dis. 2012;48(3):495–506. doi: 10.1016/j.nbd.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Underly RG, Levy M, Hartmann DA, Grant RI, Watson AN, Shih AY. Pericytes as inducers of rapid, matrix metalloproteinase-9-dependent capillary damage during ischemia. J Neurosci. 2017;37(1):129–140. doi: 10.1523/JNEUROSCI.2891-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, Goerge T, Schneider SW, Jones C, Nieswandt B, et al. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116(22):4712–4719. doi: 10.1182/blood-2010-03-276311. [DOI] [PubMed] [Google Scholar]
- 32.Jin AY, Tuor UI, Rushforth D, Kaur J, Muller RN, Petterson JL, Boutry S, Barber PA. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neurosci. 2010;11:12. doi: 10.1186/1471-2202-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kisucka J, Chauhan AK, Zhao BQ, Patten IS, Yesilaltay A, Krieger M, Wagner DD. Elevated levels of soluble P-selectin in mice alter blood-brain barrier function, exacerbate stroke, and promote atherosclerosis. Blood. 2009;113(23):6015–6022. doi: 10.1182/blood-2008-10-186650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rieckmann P, Altenhofen B, Riegel A, Baudewig J, Felgenhauer K. Soluble adhesion molecules (sVCAM-1 and sICAM-1) in cerebrospinal fluid and serum correlate with MRI activity in multiple sclerosis. Ann Neurol. 1997;41(3):326–333. doi: 10.1002/ana.410410307. [DOI] [PubMed] [Google Scholar]
- 35.Boroujerdi A, Welser-Alves JV, Milner R. Extensive vascular remodeling in the spinal cord of pre-symptomatic experimental autoimmune encephalomyelitis mice; increased vessel expression of fibronectin and the alpha5beta1 integrin. Exp Neurol. 2013;250:43–51. doi: 10.1016/j.expneurol.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts J, de Hoog L, Bix GJ. Mice deficient in endothelial alpha5 integrin are profoundly resistant to experimental ischemic stroke. J Cereb Blood Flow Metab. 2017;37(1):85–96. doi: 10.1177/0271678X15616979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11(2):97–105. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19(1):116–127. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- 39.Suidan GL, Brill A, De Meyer SF, Voorhees JR, Cifuni SM, Cabral JE, Wagner DD. Endothelial Von Willebrand factor promotes blood-brain barrier flexibility and provides protection from hypoxia and seizures in mice. Arterioscler Thromb Vasc Biol. 2013;33(9):2112–2120. doi: 10.1161/ATVBAHA.113.301362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rempe RG, Hartz AM, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36(9):1481–1507. doi: 10.1177/0271678X16655551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leppert D, Lindberg RL, Kappos L, Leib SL. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res Brain Res Rev. 2001;36(2–3):249–257. doi: 10.1016/S0165-0173(01)00101-1. [DOI] [PubMed] [Google Scholar]
- 42.Lemarchant S, Pruvost M, Montaner J, Emery E, Vivien D, Kanninen K, Koistinaho J. ADAMTS proteoglycanases in the physiological and pathological central nervous system. J Neuroinflammation. 2013;10:133. doi: 10.1186/1742-2094-10-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y, Rosenberg GA. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015;1623:30–38. doi: 10.1016/j.brainres.2015.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nygardas PT, Hinkkanen AE. Up-regulation of MMP-8 and MMP-9 activity in the BALB/c mouse spinal cord correlates with the severity of experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2002;128(2):245–254. doi: 10.1046/j.1365-2249.2002.01855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leppert D, Leib SL, Grygar C, Miller KM, Schaad UB, Hollander GA. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: association with blood-brain barrier damage and neurological sequelae. Clin Infect Dis. 2000;31(1):80–84. doi: 10.1086/313922. [DOI] [PubMed] [Google Scholar]
- 46.Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. 2010;6(4):e1000874. doi: 10.1371/journal.ppat.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cross AK, Haddock G, Stock CJ, Allan S, Surr J, Bunning RA, Buttle DJ, Woodroofe MN. ADAMTS-1 and -4 are up-regulated following transient middle cerebral artery occlusion in the rat and their expression is modulated by TNF in cultured astrocytes. Brain Res. 2006;1088(1):19–30. doi: 10.1016/j.brainres.2006.02.136. [DOI] [PubMed] [Google Scholar]
- 48.Wagsater D, Bjork H, Zhu C, Bjorkegren J, Valen G, Hamsten A, Eriksson P. ADAMTS-4 and -8 are inflammatory regulated enzymes expressed in macrophage-rich areas of human atherosclerotic plaques. Atherosclerosis. 2008;196(2):514–522. doi: 10.1016/j.atherosclerosis.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 49.Westling J, Gottschall PE, Thompson VP, Cockburn A, Perides G, Zimmermann DR, Sandy JD. ADAMTS4 (aggrecanase-1) cleaves human brain versican V2 at Glu405-Gln406 to generate glial hyaluronate binding protein. Biochem J. 2004;377(Pt 3):787–795. doi: 10.1042/bj20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kern CB, Wessels A, McGarity J, Dixon LJ, Alston E, Argraves WS, Geeting D, Nelson CM, Menick DR, Apte SS. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010;29(4):304–316. doi: 10.1016/j.matbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gottschall PE, Howell MD. ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol. 2015;44-46:70–76. doi: 10.1016/j.matbio.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tchantchou F, Zhang Y. Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown, and improves functional recovery in a mouse model of traumatic brain injury. J Neurotrauma. 2013;30(7):565–579. doi: 10.1089/neu.2012.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially regulated genes. Differentially regulated genes (increased or decreased by at least twofold) and p values for LPS-vehicle vs sham-vehicle and LPS-CPD-4645 vs LPS-vehicle groups. (XLSX 294 kb)
Table S2. Gene ontology analysis. Gene ontology analysis was performed using the DAVID bioinformatics tools. The enriched functional annotation clusters are listed along with p values for LPS vs Sham upregulated genes, LPS vs Sham downregulated genes, and CPD-4645 vs vehicle downregulated genes. (XLSX 157 kb)
Figure S1. In vivo selectivity of CPD-4645. Levels of inhibition of MAGL, ABHD6, and FAAH in the brain after 10 mg/kg subcutaneous dose of CPD-4645 at given tie point as determined by activity-based protein profiling using the pan-serine hydrolase probe FP-rhodamine. Full MAGL inhibition was normalized by effect seen at 2 h post-subcutaneous administration of 16 mg/kg JZL-184. (TIFF 1521 kb)
Figure S2. Principal component analysis. Principal component analysis (PCA) for transcriptional signatures demonstrating a distinction between experimental groups. The first two principal components are plotted with the variance explained by each component shown on the axes. (TIFF 1521 kb)
Data Availability Statement
The RNA sequencing data has been submitted to the Gene Expression Omnibus data repository and can be accessed through the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113855.