

Abstract
Aims
High levels of glucose and reactive carbonyl intermediates of its degradation pathway such as methylglyoxal (MG) may contribute to diabetic complications partly via increased generation of reactive oxygen species (ROS). This study focused on glutathione peroxidase-1 (GPx1) expression and the impact of carbonylation as an oxidative protein modification on GPx1 abundance and activity in human umbilical vein endothelial cells (HUVEC) under conditions of mild to moderate oxidative stress.
Results
High extracellular glucose and MG enhanced intracellular ROS formation in HUVECs. Protein carbonylation was only transiently augmented pointing to an effective antioxidant defense in these cells. Nitric oxide synthase expression was decreased under hyperglycemic conditions but increased upon exposure to MG, whereas superoxide dismutase expression was not significantly affected. Increased glutathione peroxidase (GPx) activity seemed to compensate for a decrease in GPx1 protein due to enhanced degradation via the proteasome. Mass spectrometry analysis identified Lys-114 as a possible carbonylation target which provides a vestibule for the substrate H2O2 and thus enhances the enzymatic reaction.
Innovation
Oxidative protein carbonylation has so far been associated with functional inactivation of modified target proteins mainly contributing to aging and age-related diseases. Here, we demonstrate that mild oxidative stress and subsequent carbonylation seem to activate protective cellular redox signaling pathways whereas severe oxidative stress overwhelms the cellular antioxidant defense leading to cell damage.
Conclusions
This study may contribute to a better understanding of redox homeostasis and its role in the development of diabetes and related vascular complications.
Abbreviations: AV, adenovirus; NOS3, endothelial nitric oxide synthase; GPx, Glutathione peroxidase; HUVEC, human umbilical vein endothelial cells; PTM, post-translational modification; ROS, reactive oxygen species; SOD, superoxide dismutase
Keywords: Protein carbonylation, Reactive oxygen species, Hyperglycemia, Glutathione peroxidase-1, Endothelial cells
Graphical abstract
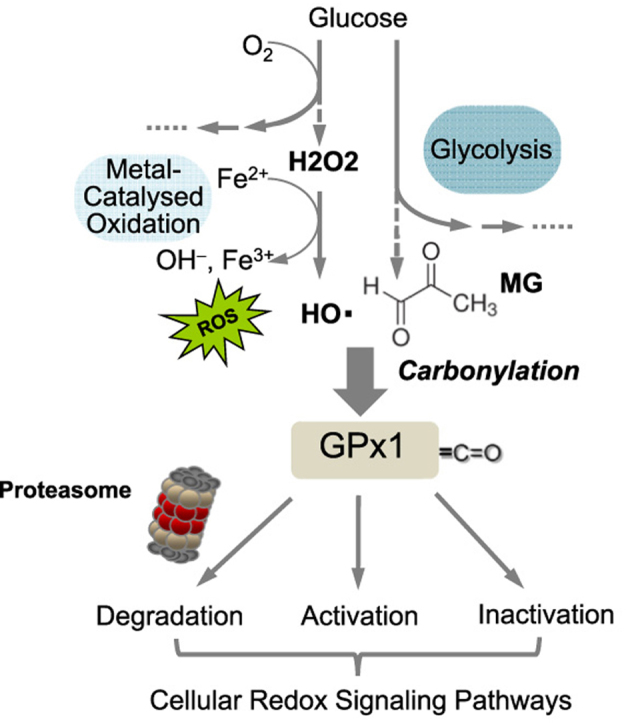
1. Introduction
Endothelial cells are generally considered to be chief targets of hyperglycemic damage [14]. However, it is very likely that they play a role in the initiation and progression of this complication of diabetes as they integrate metabolic, biochemical and hemodynamic signals [22]. There are three seemingly independent biochemical pathways involved in the mechanism of hyperglycemia-induced cellular damage: glucose-induced activation of protein kinase C (PKC) isoforms, increased formation of glucose-derived advanced glycation end products (AGEs) and increased glucose flux through the aldose reductase pathway [39]. Increased formation of reactive oxygen species (ROS) has been shown to serve as causal link between elevated glucose and each of these major pathways responsible for diabetic damage [39]. Thus, diabetes has been characterized as an oxidative stress disorder [60] which arises from an imbalance between the generation of reactive oxygen species (ROS) and their removal by antioxidants. In addition, ROS generated by AGE binding to endothelial cells itself can modulate the expression of the receptor for AGEs which is mediated through the formation of the glycolytic intermediate methylglyoxal [4], [59], thus establishing a self-amplifying cycle [38].
ROS derived from oxygen are radicals, such as the superoxide or hydroxyl radical, but they do not necessarily contain an unpaired electron like hydrogen peroxide (H2O2). An important mechanism by which ROS can alter macromolecular and hence cellular, function is through reversible or irreversible post-translational modification (PTM) that may lead to inactivation of critical proteins. It is thus well known that oxidative stress-related PTMs accumulate in the aging proteome and may become harmful due to unspecific cell damage. However, over recent years it has been shown that such oxidative protein modifications and ROS themselves play a pivotal role in cellular signaling processes, for example in the promotion of cell growth or insulin signaling [29], [54]. Conversely, they have been implicated in pathophysiological processes, such as hypertension, cancer or diabetes [54].
Protein carbonylation represents the most common type of PTM triggered by oxidative stress [6], and selectively occurs at the surface of proteins [33]. Thus, carbonyl groups are introduced into proteins by direct metal-catalyzed oxidation of certain amino acids or indirectly by reaction with reactive carbonyl species derived from the oxidation of sugars like, e.g. methylglyoxal (MG) [19]. Carbonylation-prone sites are chiefly located in regions enriched for the susceptible amino acids arginine, lysine, proline or threonine, probably due to the fact that carbonylation increases the reactivity of neighboring carbonylatable sites [33]. Methylglyoxal, on the other hand, modifies the side chains of arginine, lysine and cysteine, and thus can affect, e.g. enzyme activity [36]. Moreover, methylglyoxal can glycate proteins leading to the formation of AGEs which show a disrupted secondary structure that might explain their altered functions as compared to unmodified proteins [48].
Cells have developed complex antioxidant defense mechanisms for the protection against oxidative stress and maintaining the redox equilibrium. Amongst others, these comprise various antioxidant enzymes [11] such as superoxide dismutase (SOD), which converts superoxide radicals into hydrogen peroxide that is further detoxified to water and molecular oxygen through catalase, peroxiredoxins or glutathione peroxidases (GPx) [29]. However, in chronic hyperglycemia, this compensatory response seems insufficient, leading to both ROS and reactive nitrogen species formation and induction of stress- and redox-sensitive gene expression e.g., via the redox-sensitive transcription factor NF-κB [11]. Furthermore, the redox-sensitive transcription factor nuclear factor E2-related factor 2 (Nrf2), which regulates antioxidant response element-mediated expression of detoxifying and antioxidant enzymes is activated by carbonylation of the cytoplasmic Kelch-like ECH associated protein 1 (Keap1), a known negative regulator of Nrf2 [7]. Nrf2 mediates the expression of enzymes in endothelial cells that contribute to maintaining redox homeostasis, like e.g. SOD-1, GPx, catalase and peroxiredoxin 1, which directly metabolize H2O2 [31]. Moreover, Nrf2 also induces expression of cytoprotective enzymes such as heme oxygenase-1 or glutathione reductase. Nevertheless, aberrant activation of Nrf2 signaling has been shown to be detrimental [49] and to upregulate NADPH oxidase (NOX4) expression, thus increasing ROS production [24].
There are five different GPx isoenzymes that differ in their tissue and subcellular distribution. GPx1 is a cytosolic and mitochondrial isoenzyme that is expressed almost ubiquitously [45]. Its main substrate is H2O2; however, GPx1 can also degrade fatty acid hydroperoxides. This study aims at elucidating the role of protein carbonylation in GPx1 enzymatic activity in endothelial cells in the context of hyperglycemia and elevated levels of methylglyoxal.
2. Material and methods
2.1. Animals
The aorta of 1, 3, and 6 months old male Ins2Akita and control mice on a C57/Bl6 background were kindly provided by Hans-Peter Hammes (V. Medical Clinic, University Hospital Mannheim, Germany) in accordance with local animal welfare regulations and with permission of the Regional Council Karlsruhe, Germany, and conformed to the Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85-23, revised 2011).
2.2. Cell culture and stimulation conditions
Human umbilical vein endothelial cells (HUVEC) were freshly isolated from umbilical cords, which were not older than 24 h (reference number of the approval by the local ethical review committee: S-130/2009) and were cultured as published previously [37]. HUVECs were exposed to experimental stimulation at passage one. The endothelial cell basal medium contained 5.55 mmol/L D-glucose. 16.45 mmol/L D-mannitol served as an osmotic control for 22 mmol/L D-glucose stimulation. Aminoguanidine (500 µmol/L, # 396494) and methylglyoxal (1–10 µmol/L, #M0252) were from Sigma-Aldrich, Steinheim Germany.
2.3. Cell culture and adenoviral transduction
The human breast adenocarcinoma cell line MCF-7 (ATCC HTB-22™) was chosen due to low levels of endogenous GPx1 and purchased from the American Type Culture Collection (ATCC) VA, USA. Cells were maintained in minimum essential medium (MEM) with Eagle's salts and L-glutamine, 1% MEM nonessential amino acids, 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All adenoviruses were purchased from the Viral Vector Core Facility, Carver College of Medicine, University of Iowa, IA, USA. Adenoviral infections and cell culture following infection was performed in medium supplemented with 35 nM sodium selenite. Varying multiplicity of infection (MOI) was tested and transduction efficiency was maximal (> 80%) at an MOI of 500 viral particles/cell as indicated by efficient transduction of the EGFP gene (Ad.eGFP reporter gene expression). Adenoviral infections were carried out in serum-free medium for 2 h, followed by the addition of equal amount of fresh medium supplemented with 20% FCS. The medium was replaced 24 h after infection, and cells were analyzed 72 h after infection. Three different types of recombinant GPx1 adenoviruses were used: Ad.GPx1, Ad.Mut (GPx1 with glutamic acid at position 113, lysine at position 114, cysteine at position 115 and glutamic acid at position 116 replaced by serine, alanine, isoleucine and serine, respectively), Ad.K114 (GPx-1 with lysine at position 114 replaced by alanine) and Ad.E116 (GPx-1 with glutamic acid at position 116 replaced by serine).
2.4. Protein detection and analysis of carbonylated proteins
Protein detection by Western Blot and immunohistochemistry was done according to standard protocols. In brief, for Western Blot analysis protein extracts (10–20 μg protein per lane) were mixed with 4× sample buffer (Carl Roth GmbH, Karlsruhe, Germany) and boiled at 95 °C for 5 min. The samples were then separated by denaturing 10 or 12% SDS-polyacrylamide gel electrophoresis according to standard protocols and subsequently transferred to a polyvinylidene fluoride transfer membrane (Immobilon-PSQ Membran, 0.2 µm, #ISEQ. 00010, MERCK Millipore, Darmstadt, Germany). The membrane was blocked in 5% (w/v) BSA or powdered milk (Carl Roth, Karlsruhe, Germany) in TBS-T for 1 h and then probed with antibodies against endogenous GPx1 (GeneTex, Irvine, CA, USA, GTX116040, 1:1000 dilution), MnSOD (Enzo Life Sciences, Lörrach, Germany, ADI-SOD-110, 1:2500 dilution), NOS3/NOS3 (BD Transduction Laboratories, Franklin Lakes, NJ, USA, #610297, 1:2500 dilution), or β-actin (abcam, Cambridge, UK, ab6276, 1:5000 dilution) at 4 °C over night. An anti-GPx1 (Clone: GPX-347, Biozol, Eching, Germany, MBL-M015-3) reacting with the N-terminal epitope of human GPX1 was used for detection of the adenoviral expressed protein in MCF-7 cells. The secondary, horse radish peroxidase (HRP) -labeled antibody (Sigma-Aldrich) was added for 1 h at room temperature. After washing, proteins were detected using Luminata Western HRP substrate (Millipore) and a chemiluminescence system (ImageQuant LAS 4000 mini, GE Healthcare, Freiburg, Germany). A stripping protocol was used to investigate more than one protein on the same membrane by washing the membrane after the first staining once in distilled H2O for 5 min followed by washing in 0.2 M NaOH (5 min) and washing again in dH2O (5 min). Thereafter the protocol started again with the blocking step. For the detection of carbonylated proteins (OxyBlot), 10 µg whole cells lysates were treated with 2,4-dinitrophenolhydrazine (DNPH) solution (OxyBlot protein detection kit, Merck, S7150) according to manufacturer's instructions. The reaction was stopped by addition of neutralization solution (OxyBlot protein detection kit). To neutralize the pH of the samples for loading on an SDS gel, the proteins were precipitated by adding four times the volume of ice-cold ethanol. After incubation on ice for 15 min the samples were centrifuged at 20.800 ×g at 4 °C for 15 min. The supernatants were removed and the protein pellet was air-dried before resuspended in 10 μl of 2× SDS sample buffer. Detection of carbonylated proteins by Western Blot was done using an anti-DNP-antibody (invitrogen, Darmstadt, Germany, A6430). The specific quantification of GPx1 carbonylation was achieved by immobilizing GPx1 from the DNPH-derivatized samples to a 96-well-plate (Corning, Kaiserslautern, Germany, #3591) using a GPx1/2 antibody (Santa Cruz Biotechnology, Heidelberg, Germany, sc-133152) or a control-IgG-antibody (Cell Signaling Technology, Danvers, MA, USA, #5415S)) and subsequent quantification of the amount of DNP using the anti-DNP-antibody.
For localization of carbonylated proteins in aortic tissue sections, the tissue had been fixed and was subsequently incubated with DNPH solution diluted 1:100 in PBS. Afterwards unspecific antibody binding sides were blocked and the tissue stained with an anti-DNP-antibody (1:100 dilution) according to standard procedures. Moreover, CD31 staining (Clone JC70A, #M0823; Dako, Hamburg Germany) revealed the presence of an intact endothelial cell layer.
2.5. LC-MS/MS of tryptic peptides and database search
Mass spectrometry of in vitro modified recombinant GPx1 and whole cell lysates of HUVEC made hyperglycemic was performed with two independent replicates with at least three technical replicates for each analysis. Proteins (15 µg) were separated by SDS-PAGE and stained with Coomassie Brilliant Blue G-250. Protein lanes were cut, dehydrated and dried by vacuum concentration as published previously [15]. After digested with trypsin peptides were extracted, combined, vacuum concentrated and stored at − 20 °C. Before MS analysis peptides were dissolved in 10 μl of 0.5% formic acid in 60% aqueous acetonitrile and further diluted 1:20 with 0.1% formic acid in 3% aqueous acetonitrile.
A nano-Acquity UPLC (Waters GmbH, Eschborn, Germany) was coupled online to an LTQ Orbitrap XL ETD mass spectrometer equipped with a nano-ESI source (Thermo Fischer Scientific, Bremen, Germany) and used as described previously [15]. The acquired tandem mass spectra were searched against the Uniprot Homo sapiens database using the Sequest search engine (Proteome Discoverer 1.4, Thermo Scientific), allowing up to two missed cleavages and a mass tolerance of 10 ppm for precursor ions and 0.8 Da for product ions. A list of variable modifications used for database search always included oxidation of Met, Cys, Trp, Phe, and Pro, direct carbonylation on Lys (aminoadipic semialdehyde), Arg and Pro (glutamic semialdehyde), Thr (amino-ketobutyric acid), MGO derived modifications of Lys, Arg and Cys, and propionamide on Cys (in-gel digestion dataset).
2.6. Visualizing GPx1 sequence modifications on 3D structure
For structural analysis of the human GPx1 protein, the crystal structure of the selenocysteine to glycine mutant was used (resolution: 1.5 Å, PDB ID: 2f8a). To enable the representation of the selenocysteine in the human Gpx1 structure in a more appropriate way, the crystal structure of the selenoenzyme glutathione peroxidase from Bos taurus (resolution: 2.0 Å, PDB ID: 1gp1) that includes a selenic acid (residue name: SE7) was superimposed on the human structure with the PyMOL tool (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC) (RMSD: 0.29 Å). Afterwards, the glycine in the structure of human Gpx1 (2f8a) was replaced by the aligned selenic acid. The distances between the C-alpha atoms of the backbone structure for the respective amino acid residues were calculated using the Visual molecular dynamics (VMD) software support ([21] http://www.ks.uiuc.edu/Research/vmd/), which was also used for the visualization. The labeled residues K114 and E116 are numbered according to the residue numbering in the UniProt entry with the accession number P07203, and correspond to K112 and E114 in the PDB structure, 2f8a. The inserted selenic acid is at position 49 in the UniProt sequence and position 47 in the PDB file, 2f8a.
2.7. GPx activity assay
GPx activity was determined indirectly by a coupled reaction with glutathione reductase and the oxidation of NADPH to NADP+ (Glutathione Peroxidase Assay Kit, No. 703102, Cayman Chemical, Ann Arbour, MI, USA). A negative control without protein samples was used for background subtraction whereas a positive control consisting of recombinant GPx1 assured proper working of the assay. In brief, 10–20 μg of total protein lysate in a volume of 20 μl was added to 50 μl of a master mix composed of NADPH (0.25 mM), glutathione reductase (1 U/ml), reduced glutathione (1 mM). Directly before measurement, cumene hydroperoxide (0.05% w/w) in a volume of 10 μl was added and the absorbance at 340 nm was measured after 30 s intervals for 10 min at room temperature with the microplate spectrophotometer PowerWave XS and analyzed by the program KCJunior.
For some approaches in vitro metal catalyzed oxidation of recombinant GPx1 protein was performed as previously published elsewhere [33] followed by activity measurements or mass spectrometry analysis. For this purpose recombinant GPx1 (Creative BioMart, #GPX1-1273H) was dissolved at 0.5 mg/ml in sample buffer (50 mM Tris-HCl, pH 7.6, containing 5 mM EDTA). Oxidation was accomplished by supplementing 10–20 μl of protein solution (5 µg) with a freshly prepared FeSO4 with a final concentration of 1.2 mmol/L and incubating for 10–30 min at room temperature after addition of 0.05% (w/w) cumene hydroperoxide (Sigma-Aldrich, #247502) or 0.3% (w/w) hydrogen peroxide (Merck Millipore, #107209). Modification of recombinant GPx1 using the reactive dicarbonyl compound methylglyoxal (10 µmol/L final concentration) was performed under comparable conditions as described above.
2.8. Real-time polymerase chain reaction
Relative mRNA expression was determined by quantitative real-time PCR analysis as published previously [50], using appropriate forward and reverse primer for the amplification of the target mRNA (Table 1). The expression levels were normalized to ribosomal protein RPL32 that served as a housekeeping gene and loading control. Relative expression ratio was calculated using the ΔCP quantification method [44].
Table 1.
Primers used for qPCR.
| Gene | Primer | Sequence (5′→3′) | Annealing temperature | Cycles |
|---|---|---|---|---|
| GPx1 | Forward | TTC ATG CTC TTC GAG AAG TGC GAG G | 58 °C | 40 |
| Reverse | ACA TCG TTG CGA CAC ACC GGA GAC C | 58 °C | 40 | |
| MnSOD | Forward | CAG ATA GCT CTT CAG CCT GC | 62 °C | 40 |
| Reverse | CAG TGG ATC CTG ATT TGG AC | 62 °C | 40 | |
| eNOS | Forward | GGA TGT GGC TGT CTG CAT GGA C | 58 °C | 35 |
| Reverse | TGG TCC ACG ATG GTG ACT TTG | 58 °C | 35 | |
| RPL32 | Forward | AGG CAT TGA CAA CAG GGT TC | 61 °C | 40 |
| Reverse | GTT GCA CAT CAG CAG CAC TT | 61 °C | 40 |
2.9. Detection of intracellular reactive oxygen species
The ROS indicator dichlorodihydrofluorescein diacetate (DCFH-DA, Thermo Fisher Scientific, D-399, dissolved in DMSO) was used for the measurement of oxygen radicals. DCFH fluorescence results from oxidation by potent oxidants, such as those produced from metal ion-catalyzed reactions, like hydroxyl radicals [13] produced via the Fenton reaction, which oxidize protein amino acids to carbonyls [19]. Moreover, redox-active metals (e.g., Fe(II)) promote DCFH oxidation in the presence of oxygen or H2O2 [23].
Cells were cultured in a 12-well-plate prior to the assay. A pre-incubation with 5 µmol/l DCFH-DA for 30 min was sufficient for cellular uptake. The endothelial cell basal medium contained 5.55 mmol/L D-glucose (control). Glucose (11 and 20 mmol/L) or MG (3 and 10 µmol/L) was added for 1 h. As a positive control H2O2 was added as a bolus to the cells (100 µmol/L final concentration). Transduced MCF-7 cells were stimulated with 40 µmol/L H2O2 for 30 min. Cells were then carefully washed with PBS and light emission at 538 nm after excitation with 485 nm was measured (Fluoroskan Ascent, Thermo Fisher Scientific).
2.10. Measurement of intracellular glucose and methylglyoxal levels
Measurements of intracellular glucose and methylglyoxal levels were performed using high-performance liquid chromatography as previously published [35]. HUVECs grown on 60 mm cell culture dishes were detached by trypsin. After centrifugation they were resuspended in 1 ml Hank's and adjusted to 1–2 × 106 cells per sample. Then they were centrifuged again, resuspended in 100 mmol/L sodium acetate (pH 4.0, 0.1% TritonX100) and stored at − 20 °C until analysis.
2.11. Statistical analysis
Statistical analysis was performed for experiments with at least three independent replicates using the GraphPad Prism 6.0 software (GraphPad, USA). The n-number reflects biological replicates and parallel measurements of biologically distinct samples. Results are expressed as mean ± standard error of the mean (SEM) of n independent experiments. Unpaired student's t-test was used for two-group comparisons and one sample t-test for values expressed as relative to a control sample. For data encompassing three groups or more, one-way ANOVA with Tukey's multiple comparisons test was employed. P-values < 0.05 were considered statistically significant.
3. Results
3.1. Protein carbonylation in aortic endothelial cells in hyperglycemic mice
Immunofluorescence analysis of tissue sections from the thoracic aorta of diabetic Ins2Akita mice (1–6 months old), which develop significant hyperglycemia as early as 4 weeks of age [2], revealed an age-dependent increase in carbonylated proteins in the endothelial cell monolayer (Fig. 1). As oxidative stress is induced by excess superoxide due to chronic hyperglycemia in these mice [57], we investigated the effect of high levels of glucose and methylglyoxal, a reactive glucose metabolite, on protein carbonylation and expression of antioxidant enzymes in primary cultured human umbilical vein endothelial cells (HUVEC). Because even short-term culture of endothelial cells from venous or arterial origins erases their differential arteriovenous gene expression pattern [8], HUVECs can be taken as a general model for macrovascular endothelial cells.
Fig. 1.

Detection of carbonylated proteins in aortic endothelial cells of diabetic Ins2Akitamice. (A) Representative immunofluorescence images with anti-DNP (Cy3, red) detection of carbonylated proteins and endothelial cell marker CD31 (Cy2, green) in 1 month (1 M) and 6 months (6 M) old mice. DAPI (blue) served as a nuclear counterstain. Scale bar represents 100 µm. (B) Fluorescence intensity of carbonylated proteins (mean pixel brightness) in individual images was measured to quantify the amount of carbonylated proteins in the aortic endothelial cell layer of diabetic mice (statistical summary, n = 3, *p < 0.05 versus non-diabetic control).
3.2. High glucose and MG-induced oxidative stress in HUVEC
Both, intracellular glucose and MG concentrations rose in a linear manner upon raising the extracellular concentration of glucose in the medium from 5 to 33 mmol/L (Suppl. Fig. 1A, C; note that the endothelial cell basal medium itself used as control contained 5.55 mmol/L D-glucose). As expected, raising the extracellular concentration of MG (1–30 µmol/L) did not affect the intracellular level of glucose (Suppl. Fig. 1B) but caused a significant rise in intracellular MG (Suppl. Fig. 1D). Under these conditions, aminoguanidine, a potent MG scavenger, decreased the intracellular concentration of MG to near control levels. Both, diabetes-relevant concentrations of glucose (22 mmol/L) and MG (3 µmol/L) [26] significantly increased ROS formation by up to 6- and 3-fold, respectively, in the endothelial cells after one hour of exposure (Suppl. Fig. 1E).
3.3. Protein carbonylation is transiently augmented in HUVEC following exposure to high glucose or MG
Neither high extracellular glucose nor MG significantly affected protein carbonylation over 24 h (Fig. 2A). Time course experiments with MG (3 µmol/L) up to 24 h revealed no significant changes (Fig. 2B). In contrast, a transient increase in protein carbonylation (maximum at 16–20 h, Fig. 2C) was observed, which returned to near baseline after 24 h (Fig. 2C).
Fig. 2.

Effect of high glucose and MG treatment on protein carbonylation. (A) HUVECs were exposed to either D-glucose (Glc, n = 5) or methylglyoxal (MG, n = 3) at the indicated concentrations for 24 h. D-Mannitol (D-Man) was used as osmotic control for 22 mmol/L D-glucose; MG was scavenged by aminoguanidine (AG, 500 µmol/L). Time course of carbonylation using (B) MG (3 µmol/L) and (C) hyperglycemia (22 mmol/L D-glucose), as indicated. Statistical summaries (n = 5 each) and (D) exemplary immunoblot showing anti-DNP staining in HUVECs made hyperglycemic with β-actin as a loading control.
3.4. Effects of high glucose and MG on redox enzyme expression, protein abundance and activity
In HUVECs exposed to hyperglycemia (22 mmol/L glucose) or high extracellular MG (10 µmol/L) for 0–24 h, there was no effect on NOS3 or MnSOD mRNA expression (n = 3, data not shown). Under these culture conditions, GPX1 mRNA also did not change significantly within a period of up to 3 days (Suppl. Fig. 2A). In contrast, GPx1 protein abundance significantly decreased in HUVECs exposed to high glucose or MG while GPx activity was significantly upregulated after 1 day (Fig. 3A,B). This opposite effect was still detectable after two days, albeit somewhat weaker, but no longer after three days (Suppl. Fig. 2B,C). The decline in GPx1 abundance was prevented by inhibiting the 20S proteasome with low concentrations of bortezomib (5 nmol/L, Fig. 3C), pointing to an activation of the proteasome and enhanced degradation under conditions of hyperglycemia.
Fig. 3.

Enhanced GPx activity compensates for the decreased GPx1 expression in HUVECs treated with high glucose or MG. HUVECs were exposed to D-glucose (Glc, n = 5) or MG (n = 4) for 24 h. D-Mannitol (D-Man) was used as osmotic control for 22 mM D-glucose, MG was scavenged by aminoguanidine (AG, 500 µM). GPx1 protein levels were determined by (A) Western blot analysis (the insert shows exemplary Western blots). (B) GPx enzymatic activity was measured as indicated in the Material and Methods section, and normalized to the GPx1 protein content. (C) For inhibition of the proteasome (exemplary Western blot analysis) cells were pre-incubated for 1 h with 5 nmol/L bortezomib (0.01% in DMSO); therefore a DMSO control was added. *p < 0.05 vs control. Note that the endothelial cell basal medium used as control (contr) itself contained 5.55 mmol/L D-glucose.
Oxidatively modified proteins have also been shown to be prone to form aggregates [16]. Increased aggregation accounting for the loss of GPx1 in HUVECs following exposure to hyperglycemia or high extracellular MG could be excluded though as there was no discernible change in GPx1 aggregate content in whole cell lysates (data not shown).
NOS3 abundance was slightly decreased on the protein level following exposure to hyperglycemic conditions (Suppl. Fig. 3A). D-mannitol, used as a control for the effects of glucose on osmotic pressure, had the same inhibiting effect as D-glucose, suggesting that NOS3 protein abundance is affected by hyperosmotic stress. In contrast, extracellular MG (3 or 10 µmol/l) increased NOS3 protein abundance (Suppl. Fig. 3A). Moreover, the abundance of other antioxidant enzymes such as MnSOD did not change significantly under conditions of hyperglycemia s (Suppl. Fig. 3B) whereas extracellular MG had a tendency to decrease MnSOD protein abundance (Suppl. Fig. 3B).
3.5. Posttranslational oxidative modification of GPx1
The gain in enzymatic activity may compensate for the loss of GPx1 protein in the presence of hyperglycemia or high extracellular MG that is preceded or paralleled by a significant rise in oxidative stress in these cells. Main protein modifications originating from increased oxidative stress comprise direct oxidation, oxidative glycation and carbonylation [6]. In fact, carbonylation of GPx1 was confirmed using an ELISA for detection of protein carbonyl groups (Fig. 4A). Using a recombinant human GPx1 protein, we detected an up to 13-fold increase in GPx activity under conditions of mild oxidative stress hence metal-catalyzed carbonylation (0.05% (w/w) cumene hydroperoxide, 1.2 mmol/L Fe(II); Fig. 4B) whereas conditions favouring strong carbonylation (0.3% (w/w) hydrogen peroxide, 1.2 mmol/L Fe(II)) resulted in a complete loss of enzymatic activity (not shown) and aggregate formation (Fig. 4B inset). The molecular mass of the aggregated GPx1 protein seemed to correspond to that of three or four GPx1 subunits. It has been reported that oxidative stress can induce multimerization by forming complexes via oxidative linkage between subunits [40]. Interestingly, treatment with MG did not affect activity of the recombinant enzyme. Specificity of the metal-catalyzed carbonylation of GPx1 leading to the subsequent increase in activity was demonstrated using divalent Fe(II) ions whereas Fe(III) and Cu(II) had not such an effect (Fig. 4B).
Fig. 4.

Carbonylation-associated increase in GPx activity. (A) Specific quantification of GPx1 carbonylation in HUVECs incubated with 22 mmol/L D-glucose (Glc) for 24 h using a sandwich GPx1 ELISA and DNPH-derivatized cell lysates. IgG-antibody (IgG) and a derivatization control solution (contr) were used to demonstrate overall specificity. n = 3, *p < 0.05 vs IgG control. (B) Measuring enzymatic activity of a recombinant GPx1 protein (contr) under mild oxidative conditions in the presence of iron (Fe(II)) and MG. Statistical summary, n = 7–13, *p < 0.05 vs control. Fe (II) metal specificity of hydroperoxide-dependent GPx1 carbonylation was demonstrated using comparable concentrations of Fe(III) and Cu(II) (n = 3). The insert shows recombinant GPx1 in vitro carbonylated under mild (0.05% (w/w) cumene hydroperoxide) or strong (0.3% (w/w) hydrogen peroxide) oxidative conditions and detected by Western blot analysis (exemplary images). Note the aggregated GPx1 protein marked by arrows, which might reflect an oxidative linkage between GPx1 monomers. (C, D) Location of the mutated residues in the 3D structure of human GPx1. (C) Side view of GPx1 homodimers showing selenic acid at the selenocysteine site (yellow, van der Waals spheres representation), the four EKCE amino acids (stick representation), and the intermolecular distances between the C-alpha atoms (dashed lines). The surface representation of the left monomer in the dimeric structure (white) shows the active site located in a shallow depression around the selenic acid. (D) GPx1 dimer interface. The right monomer (gray, cartoon representation) of the dimer structure shown in (C) was rotated by 90° to show the dimer interface. Additionally, the selenic acid is shown in yellow spheres on the monomer along with the intramolecular distances between the corresponding C-alpha atoms for the mutated residues. (E) Effects of different mutations on GPx1 enzymatic activity. GPx activity assay with MCF-7 lysates and adenovirally mediated overexpression of wild-type and mutant GPx1 proteins (Ad.GPx1, wild-type protein; Ad.Mut, EKCE replaced by SAIS; Ad.K114, Ad.E116). Ad.eGFP reporter gene expression and NTC (non-transduced control) were used as controls (statistical summary, n = 4, *p < 0.05 vs NTC). The insert shows an exemplary Western blot of recombinant wild-type and mutant GPx1 proteins, demonstrating the presence of roughly equal quantities of overexpressed wild-type and mutant proteins in MCF-7 cells. β-Actin served as a loading control.
Mass spectrometry analyses of tryptically digested recombinant GPx1 before and after exposure to cumene hydroperoxide plus Fe(II) or MG identified both common and differently carbonylated amino acids (Table 2). Analysis of the protein structure shows that nearly two thirds of all modified amino acid residues were found in structurally undefined regions, about one third was located in helical regions and only one modification was localized to a beta-strand region. Comparison of these in vitro induced modifications in the recombinant protein with mass spectrometry analysis of tryptic digests from whole cell lysates of HUVECs made hyperglycemic identified Lys-114 (K114) as a possible target amino acid responsible for the observed increase in activity under these conditions. According to the published environmental requirements for carbonylation sites [32], this lysine residue may be highly prone to carbonylation because of its close proximity to the iron binding site Glu-116 (E116) [33]. Fig. 4C,D shows the location of K114 in the three-dimensional structure of the GPx1 homodimer and its distance from the selenocysteine in the active site of the opposite subunit. We hypothesized therefore that the K114 side-chain and its surroundings provide a vestibule for the substrate H2O2 that enhances the enzymatic reaction.
Table 2.
GPx1 carbonylation sites differ between metal-catalyzed oxidation and MG treatment. Recombinant human GPx1 was exposed to cumene hydroperoxide (0.05% (w/w)), hydrogen peroxide (0.3% (w/w)) or MG plus 1.2 mmol/L Fe(II) ions (Lys: adipic semialdehyde, Arg/Pro: glutamic semialdehyde, Thr: ketobutyric acid, Cys: cysteine, *: MG adduct). Mass spectrometry of in vitro modified recombinant GPx1 was performed with two independent replicates with at least three technical replicates for each analysis.
| Position | Carbonylation sites |
Secondary structure | |||
|---|---|---|---|---|---|
| Control | Cumene | H2O2 | Methylglyoxal | ||
| 37 | Lys | Lys | |||
| 50 | Thr | ||||
| 53 | Arg | Arg | Arg | Arg /Arg* | helical |
| 63 | Arg | Arg | Arg | Arg Arg* | helical |
| 64 | Arg | Arg | helical | ||
| 68 | Arg | helical | |||
| 87 | Lys | ||||
| 107 | Pro | Pro | |||
| 114 | Lys | Lys | |||
| 115 | Cys* | ||||
| 133 | Pro | Pro | |||
| 135 | Pro | Pro | |||
| 144 | Thr | Thr | |||
| 147 | Lys | Lys | helical | ||
| 150 | Thr | Thr | |||
| 153 | Pro | Pro | beta-strand | ||
| 165 | Lys | Lys | |||
| 170 | Pro | ||||
| 181 | Arg | Arg | Arg | ||
| 185 | Thr | Thr | helical | ||
| 189 | Pro | helical | |||
| 199 | Pro | Pro | Pro | Pro | |
| 201 | Cys* | ||||
3.6. Adenovirus-mediated overexpression of wild-type and mutant GPx1 in MCF-7 cells
The potential importance of the two amino acids, K114 and E116, for the catalytic activity of GPx1 was evaluated further by adenoviral overexpression of human recombinant GPx1 and mutated forms in the human breast cancer cell line MCF-7. This cell line was chosen due to its low intrinsic GPx activity and the fact that adenoviral transduction of HUVEC is quite difficult to achieve. Somewhat unprecedented, overexpression of the GPx1 EKCE-containing mutant (Ad.mut) where the amino acid quartet at position 113–116 was substituted by the amino acid sequence SAIS resulted in a complete loss of GPx activity despite detection of reasonable amounts of protein in the transduced cells (Fig. 4E). The single K114A mutation (Ad.K114) also resulted in a complete loss of GPx activity whereas the single E116S mutation (Ad.E116) retained approximately 30% of the activity of the overexpressed wild-type GPx1 protein. Activity of all three mutant GPx1 proteins was not significantly recovered under mild carbonylating conditions (not shown). According to Western blot analysis, comparable levels of wild-type and mutant GPx1 protein were attained in the MCF-7 cells through adenoviral transduction (Fig. 4E).
4. Discussion
Diabetes mellitus is a metabolic disease of increasing worldwide importance and prevalence. Typically, hyperglycemia as well as increased levels of reactive metabolites such as MG cause formation of ROS [14], [36], which modify macromolecules leading to lipid peroxidation and DNA damage but also to metal-catalyzed carbonylation of selected proteinogenic amino acids [14], [20], [33]. Excessive cellular ROS formation, i.e. oxidative stress, arises as a result of an imbalance between the generation of these reactive intermediates and the ability to detoxify them [60]. This study investigated the influence of hyperglycemia and high extracellular MG on protein carbonylation as well as the expression and activity of certain antioxidant enzymes in macrovascular endothelial cells (HUVECs). The underlying working hypothesis was that severe oxidative stress leads to cell damage whereas mild oxidative stress can have signaling function and reinforce antioxidant defense mechanisms, thus being cell protective. Signaling through mild oxidative stress probably comprises moderate carbonylation of antioxidant enzymes such as GPx1, reinforcing their activity and maintaining their physiological function.
Besides large amounts of Fe(II) in the blood that are mainly complexed by hemoglobin small amounts of Fe(II) in the plasma are found to be redox-active. This so-called labile plasma iron can cross plasma membranes leading to cellular and tissue damage [5]. Labile plasma iron increases in patients with diabetes as compared to healthy subjects [47] and possibly promotes metal-catalyzed oxidation of cellular proteins. It has been described that this posttranslational oxidative modification inter alia causes protein carbonylation [52], leading to the assumption that metal-catalyzed oxidation-dependent protein carbonylation is increased under pro-diabetic conditions. Despite increased oxidative stress caused by hyperglycemia or MG in the cultured HUVECs, overall protein carbonylation was only transiently increased.
It is still a matter of debate whether carbonylation of proteins is reversible or not. So far, carbonylation is considered to reflect an irreversible modification of a protein [33] which from a chemical point of view is rather likely since carbonylated amino acid side chains probably cannot be completely restored [6]. On the contrary, there may be a non-enzymatic or enzymatic decarbonylation involving thiols and thioredoxins [53]. In this study, time course experiments showing a transient carbonylation under conditions of hyperglycemia implying that HUVEC are indeed able to decarbonylate carbonylated proteins (see Fig. 2C). Nevertheless, it remains to be elucidated whether decarbonylation contributes to intracellular signaling through carbonylated proteins and which proteins are involved therein.
Carbonylated proteins like GPx1 seemed to be rapidly degraded as demonstrated through inhibiting the 26S proteasome by bortezomib [30]. The 26S proteasome contains the 20S proteasome that serves as the catalytic core and two 19S regulatory subunits. Increased proteasomal degradation by the 20S proteasome [16] is the most important mechanism for elimination of oxidized proteins in cells [25]. Primary activation of the proteasome under pro-diabetic conditions has been described in retinal endothelial cells [1]. Both hyperglycemia and MG decreased the amount of GPx1 protein and hyperglycemia carbonylation-dependently increased overall GPx activity in the cultured HUVECs. This carbonylation-dependent increase in GPx activity may stand for a novel mechanism emphasizing the protective effect of mild oxidative stress. There are already some reports on enhanced GPx1 activity in endothelial cells. However, this was always associated with an increased GPx1 expression so that the overall rise in activity rather seemed to correlate with the higher protein level [12], [61].
In rat kidney epithelial cells, decreased GPx1 activity due to uncontrolled nitric oxide production leads to a compensatory rise in GPx1 protein [10]. In contrast, MG has been reported to decrease GPx1 activity in a cell-free system under non-physiological conditions (20 mmol/L for five days) [48] but also in rat aortic smooth muscle cells which had been exposed to 100 µmol/L MG [55]. Moreover, it is known that GPx1 activity is increased by other posttranslational modifications, particularly phosphorylation [45] and O-GlcNAcylation [58]. Such protein modifications affect the conformation and subsequently substrate affinity or binding and thus might also explain the initially increased enzymatic activity as observed by us. Further propagation of protein modifications may then lead to an increase of accessible and modifiable neighboring sites and thus cause a drop of activity due to increasing detrimental changes to the conformation of the protein. Mass spectrometry analysis revealed that metal-catalyzed oxidation and MG treatment lead to rather different carbonylation-dependent modifications of GPx1. Moreover, it could be demonstrated that higher peroxide concentrations hence more severe metal-catalyzed oxidation modifies further amino acid residues when compared to milder conditions as proposed previously [33].
The selenocysteine at position 49 in the active site of a GPx1 monomer forms a "catalytic triad" with tryptophan and glutamine, which activates the selenium moiety for an efficient reduction of peroxides [29]. The catalytic triad itself is not affected by oxidative modification. However, three amino acids in close proximity to the active site harboring the selenocysteine undergo carbonylation under different conditions. Thr-50, closest to the selenocysteine at position 49, is already carbonylated under control conditions. This modification gets lost upon metal-catalyzed oxidation but not MG treatment. In contrast, metal-catalyzed oxidation causes carbonylation of Arg-165 located next to the tryptophan residue of the catalytic triad. MG treatment modifies Lys-87 which is not only close to the catalytically active glutamine but also part of the glutathione tripeptide binding site within GPx1. MG has been shown to inactivate bovine GPx in a dose- and time-dependent manner by modifying Arg-184 and Arg-185 located near the glutathione binding site [41]. All of the aforementioned modifications may affect the affinity of GPx1 for its substrate H2O2 by conformational changes thus augmenting enzyme activity. In line with our results, it has been shown recently that oxidative carbonylation of carnosinase-1 increases its dipeptidase activity [43], which might play a role in the pathogenesis of diabetic nephropathy by degrading the endogenous antioxidant L-carnosine.
Moreover, it seems possible that carbonylation-induced structural changes further open the catalytic pocket or provide a vestibule for the substrate H2O2, thereby enhancing the probability or rate of substrate binding. In fact, by mutating both Lys-114 and Glu-116, we caused a total loss of enzymatic activity which emphasizes the functional or structural relevance of these residues and the potential consequences of their oxidative modification. Active GPx1 normally is a homotetramer. K-114 within one GPx1 subunit is located quite close to the active site of the opposite subunit (see Fig. 4c). In addition to the reactive selenocysteine and the surrounding four arginine residues (R57, R103, R184, R185; numbered according to Uniprot accession number P00435) a lysine of the adjacent subunit (K-91') is considered to bind glutathione [3].
We may have thus uncovered a novel adaptive mechanism of increasing transiently the antioxidant defense of, e.g. endothelial cells, triggered by a posttranslational oxidative modification of a pivotal antioxidant enzyme. In this context, low levels of ROS probably lead to cell protection whereas excessive oxidative stress may exaggerate the degree of posttranslational oxidative modification of such antioxidant enzymes. After an initial period of increased activity, accumulating further carbonylation might then lead to inactivation, degradation or aggregation of the protective antioxidant enzymes.
Is high GPx1 activity always protective and a main goal in therapy? Interestingly, GPx1 deficiency protects from high-fat induced insulin resistance in mice [28] and does not deteriorate diabetic nephropathy as assessed by glomerular damage and oxidative renal injury [9]. On the contrary, general GPx1 overexpression leads to insulin resistance, hyperglycemia and obesity [34]. This might be due to the fact that insulin signaling is influenced by ROS and thus insulin sensitizing is diminished by excess antioxidant capacity [34]. However, specific GPx1 overexpression in pancreatic β-cells is protective resulting even in a clear amelioration of hyperglycemia in db/db mice because pancreatic islets have low levels of antioxidant enzymes [17]. Moreover, GPx1 overexpression and depletion of H2O2 has been shown to down-regulate DNA binding of the redox-sensitive and inflammatory transcription factor NF-κB, preferentially by inhibiting IκB kinase α activity [27]. In contrast, in primary aortic endothelial cells isolated from GPx1 knockout mice, IκB degradation was prolonged suggesting an augmentation of NF-κB activity leading to an increased expression of pro-inflammatory genes [46]. Therefore, a compensatory increase in GPx1, as observed in this study, is presumably cell type specific, must maintain baseline activity and should not overcompensate.
Apart from GPx1, additional cellular enzymes involved in redox homeostasis in endothelial cells were investigated in our study. Mitochondrial MnSOD protein abundance was found to be decreased by MG but unaffected by hyperglycemia. These findings are in accordance with the literature where MnSOD protein levels in HUVECs have been reported to remain constant under hyperglycemic conditions [12] whereas MnSOD in rat aortic smooth muscle cells is inactivated in the presence of MG [51]. Moreover, anti-atherosclerotic NOS3 protein expression was increased by MG but decreased by hyperglycemia in the cultured HUVECs, thus confirming previous reports [14], [42], [56]. Taken together, the hyperglycemic abnormality in the course of diabetes may cause mitochondrial superoxide overproduction, which activates several pathways involved in the pathogenesis of diabetic complications and is the central and major mediator of cell and tissue damage in diabetes [14].
Since oxidative stress plays an important role in the development of diabetic complications, there have been several attempts to pharmaceutically affect the cellular redox balance. So far, pharmacologically reinforcing antioxidant defense mechanisms hardly produced any beneficial effect but contrariwise even deteriorated diabetes and its sequelae [18]. Our study emphasizes the hypothesis that low levels of ROS are pivotal for cellular signaling whereas excessive levels of oxidative stress lead to cell damage. Future research will have to address this differential redox balance in more detail to understand the pathogenesis of vascular complications in patients with diabetes and to develop effective therapeutic intervention strategies.
5. Conclusions
Molecular mechanisms through which reactive oxygen species or carbonyls contribute to the development and progression of diabetic complications are only partly known. A pre-diabetic mild hyperglycemic state seems to activate antioxidant defense systems in endothelial cells by augmenting glutathione peroxidase-1 activity through metal-catalyzed carbonylation providing a vestibule for the substrate hydrogen peroxide thus reinforcing the enzymatic reaction. The results further indicate that there might be a threshold for beneficial carbonylation-dependent redox signaling. This might explain why clinical trials with high-dose antioxidant supplementation abolishing this protective redox regulation mostly produced negative results in the setting of preventative therapeutic strategies.
Acknowledgments
We are indebted to Franziska Mohr and Nadine Scholz for expert technical assistance.
Acknowledgments
Funding
This work was supported by a grant to M.H. by the German Research Foundation (IGK 1874 “Diabetic Microvascular Complications”, SP 10) and lump sum funds provided by the CRC 1118 "Reactive metabolites as a cause of diabetic complications.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.02.018.
Appendix A. Supplementary material
Supplementary material
References
- 1.Aghdam S.Y., Gurel Z., Ghaffarieh A., Sorenson C.M., Sheibani N. High glucose and diabetes modulate cellular proteasome function: implications in the pathogenesis of diabetes complications. Biochem. Biophys. Res. Commun. 2013;432:339–344. doi: 10.1016/j.bbrc.2013.01.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barber A.J., Antonetti D.A., Kern T.S., Reiter C.E., Soans R.S., Krady J.K., Levison S.W., Gardner T.W., Bronson S.K. The Ins2Akita mouse as a model of early retinal complications in diabetes. Investig. Ophthalmol. Vis. Sci. 2005;46:2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 3.Brigelius-Flohe R., Maiorino M. Glutathione peroxidases. Biochim. Biophys. Acta. 2013;1830:3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 4.Brownlee M. Negative consequences of glycation. Metabolism. 2000;49:9–13. doi: 10.1016/s0026-0495(00)80078-5. [DOI] [PubMed] [Google Scholar]
- 5.Cabantchik Z.I., Breuer W., Zanninelli G., Cianciulli P. LPI-labile plasma iron in iron overload. Best Pract. Res. Clin. Haematol. 2005;18:277–287. doi: 10.1016/j.beha.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Cattaruzza M., Hecker M. Protein carbonylation and decarboylation: a new twist to the complex response of vascular cells to oxidative stress. Circ. Res. 2008;102:273–274. doi: 10.1161/CIRCRESAHA.108.172148. [DOI] [PubMed] [Google Scholar]
- 7.Cheng X., Siow R.C., Mann G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox Signal. 2011;14:469–487. doi: 10.1089/ars.2010.3283. [DOI] [PubMed] [Google Scholar]
- 8.Conway E.M. New specs for arteriovenous identity. Blood. 2013;122:3857–3858. doi: 10.1182/blood-2013-10-533141. [DOI] [PubMed] [Google Scholar]
- 9.de Haan J.B., Stefanovic N., Nikolic-Paterson D., Scurr L.L., Croft K.D., Mori T.A., Hertzog P., Kola I., Atkins R.C., Tesch G.H. Kidney expression of glutathione peroxidase-1 is not protective against streptozotocin-induced diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2005;289:F544–F551. doi: 10.1152/ajprenal.00088.2005. [DOI] [PubMed] [Google Scholar]
- 10.Dobashi K., Asayama K., Nakane T., Kodera K., Hayashibe H., Nakazawa S. Induction of glutathione peroxidase in response to inactivation by nitric oxide. Free Radic. Res. 2001;35:319–327. doi: 10.1080/10715760100300851. [DOI] [PubMed] [Google Scholar]
- 11.Evans J.L., Goldfine I.D., Maddux B.A., Grodsky G.M. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 12.Felice F., Lucchesi D., di Stefano R., Barsotti M.C., Storti E., Penno G., Balbarini A., Del Prato S., Pucci L. Oxidative stress in response to high glucose levels in endothelial cells and in endothelial progenitor cells: evidence for differential glutathione peroxidase-1 expression. Microvasc. Res. 2010;80:332–338. doi: 10.1016/j.mvr.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Forman H.J., Augusto O., Brigelius-Flohe R., Dennery P.A., Kalyanaraman B., Ischiropoulos H., Mann G.E., Radi R., Roberts L.J., 2nd, Vina J., Davies K.J. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic. Biol. Med. 2015;78:233–235. doi: 10.1016/j.freeradbiomed.2014.10.504. [DOI] [PubMed] [Google Scholar]
- 14.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circ. Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griesser E., Vemula V., Raulien N., Wagner U., Reeg S., Grune T., Fedorova M. Cross-talk between lipid and protein carbonylation in a dynamic cardiomyocyte model of mild nitroxidative stress. Redox Biol. 2017;11:438–455. doi: 10.1016/j.redox.2016.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grune T., Jung T., Merker K., Davies K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and 'aggresomes' during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 17.Harmon J.S., Bogdani M., Parazzoli S.D., Mak S.S., Oseid E.A., Berghmans M., Leboeuf R.C., Robertson R.P. Beta-cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology. 2009;150:4855–4862. doi: 10.1210/en.2009-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasanain B., Mooradian A.D. Antioxidant vitamins and their influence in diabetes mellitus. Curr. Diabetes Rep. 2002;2:448–456. doi: 10.1007/s11892-002-0110-6. [DOI] [PubMed] [Google Scholar]
- 19.Hecker M., Wagner A.H. Role of protein carbonylation in diabetes. J. Inherit. Metab. Dis. 2018;41:29–38. doi: 10.1007/s10545-017-0104-9. [DOI] [PubMed] [Google Scholar]
- 20.Higdon A., Diers A.R., Oh J.Y., Landar A., Darley-Usmar V.M. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem. J. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(33–8):27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 22.Jaimes E.A., Hua P., Tian R.X., Raij L. Human glomerular endothelium: interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am. J. Physiol. Ren. Physiol. 2010;298:F125–F132. doi: 10.1152/ajprenal.00248.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalyanaraman B., Darley-Usmar V., Davies K.J., Dennery P.A., Forman H.J., Grisham M.B., Mann G.E., Moore K., Roberts L.J., 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovac S., Angelova P.R., Holmstrom K.M., Zhang Y., Dinkova-Kostova A.T., Abramov A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta. 1850;794–801:2015. doi: 10.1016/j.bbagen.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kretz-Remy C., Arrigo A.P. Modulation of the chymotrypsin-like activity of the 20S proteasome by intracellular redox status: effects of glutathione peroxidase-1 overexpression and antioxidant drugs. Biol. Chem. 2003;384:589–595. doi: 10.1515/BC.2003.066. [DOI] [PubMed] [Google Scholar]
- 26.Lapolla A., Flamini R., Dalla Vedova A., Senesi A., Reitano R., Fedele D., Basso E., Seraglia R., Traldi P. Glyoxal and methylglyoxal levels in diabetic patients: quantitative determination by a new GC/MS method. Clin. Chem. Lab Med. 2003;41:1166–1173. doi: 10.1515/CCLM.2003.180. [DOI] [PubMed] [Google Scholar]
- 27.Li Q., Sanlioglu S., Li S., Ritchie T., Oberley L., Engelhardt J.F. GPx-1 gene delivery modulates NFkappaB activation following diverse environmental injuries through a specific subunit of the IKK complex. Antioxid. Redox Signal. 2001;3:415–432. doi: 10.1089/15230860152409068. [DOI] [PubMed] [Google Scholar]
- 28.Loh K., Deng H., Fukushima A., Cai X., Boivin B., Galic S., Bruce C., Shields B.J., Skiba B., Ooms L.M., Stepto N., Wu B., Mitchell C.A., Tonks N.K., Watt M.J., Febbraio M.A., Crack P.J., Andrikopoulos S., Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubos E., Loscalzo J., Handy D.E. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011;15:1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luker G.D., Pica C.M., Song J., Luker K.E., Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat. Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 31.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madian A.G., Regnier F.E. Proteomic identification of carbonylated proteins and their oxidation sites. J. Proteome Res. 2010;9:3766–3780. doi: 10.1021/pr1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maisonneuve E., Ducret A., Khoueiry P., Lignon S., Longhi S., Talla E., Dukan S. Rules governing selective protein carbonylation. PLoS One. 2009;4:e7269. doi: 10.1371/journal.pone.0007269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McClung J.P., Roneker C.A., Mu W., Lisk D.J., Langlais P., Liu F., Lei X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA. 2004;101:8852–8857. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLellan A.C., Phillips S.A., Thornalley P.J. The assay of methylglyoxal in biological systems by derivatization with 1,2-diamino-4,5-dimethoxybenzene. Anal. Biochem. 1992;206:17–23. doi: 10.1016/s0003-2697(05)80005-3. [DOI] [PubMed] [Google Scholar]
- 36.Miyamoto Y., Koh Y.H., Park Y.S., Fujiwara N., Sakiyama H., Misonou Y., Ookawara T., Suzuki K., Honke K., Taniguchi N. Oxidative stress caused by inactivation of glutathione peroxidase and adaptive responses. Biol. Chem. 2003;384:567–574. doi: 10.1515/BC.2003.064. [DOI] [PubMed] [Google Scholar]
- 37.Moller K., Adolph O., Grunow J., Elrod J., Popa M., Ghosh S., Schwarz M., Schwale C., Grassle S., Huck V., Bruehl C., Wieland T., Schneider S.W., Nobiling R., Wagner A.H., Hecker M. Mechanism and functional impact of CD40 ligand-induced von Willebrand factor release from endothelial cells. Thromb. Haemost. 2015;113:1095–1108. doi: 10.1160/TH14-04-0336. [DOI] [PubMed] [Google Scholar]
- 38.Mukherjee T.K., Mukhopadhyay S., Hoidal J.R. The role of reactive oxygen species in TNFalpha-dependent expression of the receptor for advanced glycation end products in human umbilical vein endothelial cells. Biochim. Biophys. Acta. 1744;213–23:2005. doi: 10.1016/j.bbamcr.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Nishikawa T., Edelstein D., Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int. 2000;(Suppl. 77):S26–S30. doi: 10.1046/j.1523-1755.2000.07705.x. [DOI] [PubMed] [Google Scholar]
- 40.Park S.J., Ciccone S.L., Beck B.D., Hwang B., Freie B., Clapp D.W., Lee S.H. Oxidative stress/damage induces multimerization and interaction of Fanconi anemia proteins. J. Biol. Chem. 2004;279:30053–30059. doi: 10.1074/jbc.M403527200. [DOI] [PubMed] [Google Scholar]
- 41.Park Y.S., Koh Y.H., Takahashi M., Miyamoto Y., Suzuki K., Dohmae N., Takio K., Honke K., Taniguchi N. Identification of the binding site of methylglyoxal on glutathione peroxidase: methylglyoxal inhibits glutathione peroxidase activity via binding to glutathione binding sites Arg 184 and 185. Free Radic. Res. 2003;37:205–211. doi: 10.1080/1071576021000041005. [DOI] [PubMed] [Google Scholar]
- 42.Patel H., Chen J., Das K.C., Kavdia M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc Diabetol. 2013;12:142. doi: 10.1186/1475-2840-12-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peters V., Lanthaler B., Amberger A., Fleming T., Forsberg E., Hecker M., Wagner A.H., Yue W.W., Hoffmann G.F., Nawroth P., Zschocke J., Schmitt C.P. Carnosine metabolism in diabetes is altered by reactive metabolites. Amino Acids. 2015;47:2367–2376. doi: 10.1007/s00726-015-2024-z. [DOI] [PubMed] [Google Scholar]
- 44.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sadi G., Guray T. Gene expressions of Mn-SOD and GPx-1 in streptozotocin-induced diabetes: effect of antioxidants. Mol. Cell Biochem. 2009;327:127–134. doi: 10.1007/s11010-009-0050-4. [DOI] [PubMed] [Google Scholar]
- 46.Sharma A., Yuen D., Huet O., Pickering R., Stefanovic N., Bernatchez P., de Haan J.B. Lack of glutathione peroxidase-1 facilitates a pro-inflammatory and activated vascular endothelium. Vasc. Pharmacol. 2016;79:32–42. doi: 10.1016/j.vph.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Sulieman M., Asleh R., Cabantchik Z.I., Breuer W., Aronson D., Suleiman A., Miller-Lotan R., Hammerman H., Levy A.P. Serum chelatable redox-active iron is an independent predictor of mortality after myocardial infarction in individuals with diabetes. Diabetes Care. 2004;27:2730–2732. doi: 10.2337/diacare.27.11.2730. [DOI] [PubMed] [Google Scholar]
- 48.Suravajjala S., Cohenford M., Frost L.R., Pampati P.K., Dain J.A. Glycation of human erythrocyte glutathione peroxidase: effect on the physical and kinetic properties. Clin. Chim. Acta. 2013;421:170–176. doi: 10.1016/j.cca.2013.02.032. [DOI] [PubMed] [Google Scholar]
- 49.Sykiotis G.P., Bohmann D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev. Cell. 2008;14:76–85. doi: 10.1016/j.devcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagner A.H., Hildebrandt A., Baumgarten S., Jungmann A., Muller O.J., Sharov V.S., Schoneich C., Hecker M. Tyrosine nitration limits stretch-induced CD40 expression and disconnects CD40 signaling in human endothelial cells. Blood. 2011;118:3734–3742. doi: 10.1182/blood-2010-11-320259. [DOI] [PubMed] [Google Scholar]
- 51.Wang H., Liu J., Wu L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem. Pharmacol. 2009;77:1709–1716. doi: 10.1016/j.bcp.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 52.Wong C.M., Bansal G., Marcocci L., Suzuki Y.J. Proposed role of primary protein carbonylation in cell signaling. Redox Rep. 2012;17:90–94. doi: 10.1179/1351000212Y.0000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong C.M., Cheema A.K., Zhang L., Suzuki Y.J. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 2008;102:310–318. doi: 10.1161/CIRCRESAHA.107.159814. [DOI] [PubMed] [Google Scholar]
- 54.Wong C.M., Marcocci L., Liu L., Suzuki Y.J. Cell signaling by protein carbonylation and decarbonylation. Antioxid. Redox Signal. 2010;12:393–404. doi: 10.1089/ars.2009.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu L., Juurlink B.H. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension. 2002;39:809–814. doi: 10.1161/hy0302.105207. [DOI] [PubMed] [Google Scholar]
- 56.Wu Y., Lee S., Bobadilla S., Duan S.Z., Liu X. High glucose-induced p53 phosphorylation contributes to impairment of endothelial antioxidant system. Biochim. Biophys. Acta. 2017;1863:2355–2362. doi: 10.1016/j.bbadis.2017.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan L.J. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J. Diabetes Res. 2014;2014:137919. doi: 10.1155/2014/137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang W.H., Park S.Y., Ji S., Kang J.G., Kim J.E., Song H., Mook-Jung I., Choe K.M., Cho J.W. O-GlcNAcylation regulates hyperglycemia-induced GPX1 activation. Biochem. Biophys. Res. Commun. 2010;391:756–761. doi: 10.1016/j.bbrc.2009.11.133. [DOI] [PubMed] [Google Scholar]
- 59.Yao D., Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes. 2010;59:249–255. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zatalia S.R., Sanusi H. The role of antioxidants in the pathophysiology, complications, and management of diabetes mellitus. Acta Med. Indones. 2013;45:141–147. [PubMed] [Google Scholar]
- 61.Zhang Y., Handy D.E., Loscalzo J. Adenosine-dependent induction of glutathione peroxidase 1 in human primary endothelial cells and protection against oxidative stress. Circ. Res. 2005;96:831–837. doi: 10.1161/01.RES.0000164401.21929.CF. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material