

Abstract
Assessing the impact of kinase in gene fusion is essential for both identifying driver fusion genes (FGs) and developing molecular targeted therapies. Kinase domain retention is a crucial factor in kinase fusion genes (KFGs), but such a systematic investigation has not been done yet. To this end, we analyzed kinase domain retention (KDR) status in chimeric protein sequences of 914 KFGs covering 312 kinases across 13 major cancer types. Based on 171 kinase domain-retained KFGs including 101 kinases, we studied their recurrence, kinase groups, fusion partners, exon-based expression depth, short DNA motifs around the break points and networks. Our results, such as more KDR than 5′-kinase fusion genes, combinatorial effects between 3′-KDR kinases and their 5′-partners and a signal transduction-specific DNA sequence motif in the break point intronic sequences, supported positive selection on 3′-kinase fusion genes in cancer. We introduced a degree-of-frequency (DoF) score to measure the possible number of KFGs of a kinase. Interestingly, kinases with high DoF scores tended to undergo strong gene expression alteration at the break points. Furthermore, our KDR gene fusion network analysis revealed six of the seven kinases with the highest DoF scores (ALK, BRAF, MET, NTRK1, NTRK3 and RET) were all observed in thyroid carcinoma. Finally, we summarized common features of ‘effective’ (highly recurrent) kinases in gene fusions such as expression alteration at break point, redundant usage in multiple cancer types and 3′-location tendency. Collectively, our findings are useful for prioritizing driver kinases and FGs and provided insights into KFGs’ clinical implications.
Keywords: kinase, gene fusion, kinase domain retention, gene fusion network, precision medicine
Introduction
Gene fusion event frequently occurs in cancer cells by chromosomal rearrangements such as translocations, deletions, duplications, insertions, transcription read-through of neighbor genes or trans-splicing of pre-mRNAs [1]. A growing understanding of the clinical importance of fusion genes (FGs) has led to an increasing emphasis on genetic features. The World Health Organization classifications set the translocation and/or gene fusion status as mandatory for the diagnosis of some types of tumors such as ‘acute myeloid leukemia (AML) with t(8;21)(q22;q22), RUNX1-RUNX1T1’ and ‘B lymphoblastic leukemia/lymphoma with t(5;14)(q31;q32), IL3-IGH’ [2]. Most of all, FGs involving oncogenic kinases are promising therapeutic targets in cancer. As a result, kinase inhibitors have been well studied in molecularly targeted therapies to treat patients carrying FGs [3]. The first anti-cancer drug for fusion is imatinib, a tyrosine kinase (TK) inhibitor for ABL proto-oncogene 1 (ABL1) in FG BCR-ABL1 in leukemia, which was approved by the US Food and Drug Administration in May 2001 [4, 5]. In up to 95% of the chronic myeloid leukemia (CML) patients, the strong promoter of the gene BCR fuses with the TK gene ABL1, which constitutively drives an activated expression of ABL1 that led to uncontrolled cell proliferation [6]. Kinase fusion genes (KFGs) are also critical drivers in solid tumors, for example EML4-ALK in lung adenocarcinoma [7], TMPRSS2-ETS in prostate cancer [8] and FRFG3-TACC3 in glioblastoma [9].
Discovery of KFGs in various cancer types has been greatly accelerated, thanks to the rapid advances in next-generation sequencing (NGS) technologies. Many gene fusion events have been reported and are available in public resources [10–12]. To distinguish bona fide driver FGs from random chimeras, the recurrence of FGs and/or retention of functional domains provide the most compelling rationale for functional characterization [13]. Driver FGs involving oncogenic kinases are typically marked by a continuous open reading frame (ORF) that retains kinase domain in gene fusion. In other words, break points tend to maintain reading frames and protein globularity [1]. However, thus far, no studies have reported FGs with functional domain retentions. On the other hand, many studies have been conducted to infer unique features of FGs, aiming to facilitate the discovery of driver gene fusion events using multiple methods such as network [14–16], consensus sequence [17, 18] and enriched functional domain-based approaches [19]. Such enriched features in FGs, especially those including kinase genes, motived many investigators to build pipelines [20] or develop machine learning methods for prioritizing driver fusion candidates [21, 22]. However, there are no studies that have systematically explored the features of human kinase’s FGs regarding the kinase domain retention (KDR) in large-scale cancer data.
To explore the signatures of driver kinase fusion genes (KFGs), we performed systematic annotation of 914 KFGs and found multiple features in 171 KFG’s retaining kinase domains. Our results revealed multiple lines of evidence supporting a positive selection on 3′-kinase fusion genes (3′-KFGs) rather than 5′-kinase fusion genes (5′-KFGs) such as more KDR, combinatorial effect between 3′-KDR kinases and their 5′-partners and a signal transduction-specific DNA sequence motif in the break point intron sequences. We also found common features of ‘effective’ kinases involved in gene fusion; here, effectiveness denotes those KFGs with high recurrence. These features include expression alteration at break points, redundant usage in multiple cancer types and 3′-location tendency. Through these analyses, we pinpointed several effective but understudied kinase candidates in FGs, such as PRKCB, SGK2, WNK1, PRKCH, MELK and CDK12, for future investigation.
Results
Overview of the KDR in pan-cancer FGs
Figure 1 illustrates the definition of kinase domain-retained fusion genes (KDR FGs) and the pipeline used in this study. Starting with 7993 FGs from TCGA Fusion Gene Data Portal [23], we identified 525 FGs whose 5′-partner gene was a kinase gene (5′-KFGs) and 389 FGs whose 3′-partner gene was a kinase gene (3′-KFGs). These KFGs were then filtered for those with intact ORFs in both of their partner genes. As a result, 357 5′-KFGs and 256 3′-KFGs remained. The rationale of this filtering step is that with intact reading frames, the resultant KFGs would have comparable sequences with their wild-type counterparts. Next, we investigated whether the kinase domains were retained in the fusion amino acid sequence (see Materials and Methods section). A total of 35 5′-KFGs involving 28 kinases and 136 3′-KFGs involving 76 kinases were identified as kinase domain-retained events (Supplementary Table S1). The observation of nearly four times of KDRs in 3′-FGs versus that in 5′-FGs suggested that kinases tended to occur at the 3′-end than in the 5′-end in the formation of KFGs, or 3′-end KDRs might have undergone adaptive selection during tumorigenesis (Figures 1B and 2A). This result is consistent with the study on TK gene fusions involved in cancer [24]. In most TK proteins, the TK domain is located at the C-terminus, whereas inhibitory domains are at the N-terminus. In TK FGs, the partner gene always replaced the N-terminus, whereas the C-terminal TK domain was retained. Therefore, most TK FGs lost the entire extracellular ligand-binding domain of receptor tyrosine kinase (RTK), and the expression level of fusion product was driven by the promoter of the partner genes. As explained above, we identified a positive selection on 3′-KFGs, where they are more likely to fuse with a stronger 5′-partner. This selection on 3′-KFGs results in the continuous activation of kinase function and can eventually contribute to cancer development [25].
Figure 1.

Illustration of KDR in FGs and flow chart of KDR annotation. (A) Illustration of the KDR in the 5′- and 3′-KFGs. (B) Flow chart of annotation of KDR.
Figure 2.

KDR ratios and DNA motif sequences in the break point introns in 5′- and 3′-KFGs. (A) The relative percentage of KDR in 5′- and 3′-KFGs. (B) KDR kinases in classical kinase group. (C) Short DNA motif sequences in the break point introns of the 5′- and 3′-KDR FGs.
Next, we asked whether each KDR FG preferred specific kinase groups when forming KFGs. The Human Kinome database [26] defined 10 groups of kinases: containing protein kinase A, G, and C families (AGC); atypical; calcium/calmodulin-dependent protein kinase (CAMK); casein kinase 1 (CK1); containing cyclin-dependent kinase (CDK), mitogen-activated protein kinase (MAPK), glycogen synthase kinase 3 (GSK3), CDC-like kinase (CLK) families (CMGC); other; receptor guanylate cyclases (RGC); homologs of yeast sterile 7 (STE7), sterile 11 (STE11), sterile 20 (STE20) genes (STE); tyrosine kinase (TK); and tyrosine kinase-like (TKL). Among the 43 kinases from the KDR FGs that have been assigned kinase groups, 23 (53.5%) kinases were TK group or TKL group (Figure 2B). Surprisingly, 81% kinases from the TK or TKL kinases (17 of 23) belonged to 3′-KDR FGs. Furthermore, the information of kinase activity that was predicted by searching for the presence of kinase catalytic motifs was compiled from the Human Kinome data (Supplementary Table S2). The kinase catalytic motifs were kept intact in all of the kinase sequences in 3′-KDR FGs, but 3′-non-KDR FGs had three inactive kinases. This fact could also support the importance of KDR in KFGs.
Enriched pathways of partner genes suggest different regulation mechanisms between four KDR FG groups
Early studies suggested that the role of the partner genes of KFGs was limited to oligomerization, but increasing evidence highlighted additional roles such as the recruitment of proteins involved in signaling or protein stabilization [24]. Specific partner genes can also serve to regulate the functions of the FGs. A 3′-kinase fused with broadly expressed partners like transcription factors and housekeeping genes would lead to continuous expression of the kinase domains. In addition, 5′-kinases can also be impacted by their 3′-partners through mechanisms like microRNA regulation of the 3′-untranslated region (UTR) regions. To this end, we analyzed the features of partner genes of four KDR FG groups (5′-KDR FGs, 5′-non-KDR FGs, 3′-KDR FGs and 3′-non-KDR FGs). We found 13 3′-KDR kinases having more than three partner genes: BRAF (number of partner genes: 12), RET (10), NTRK1 (6), ALK (6), MET (6), NTRK3 (6), PRKCB (5), FGFR1 (3), MERTK (3), ROS1 (3), FYN (3), NTRK2 (3) and RAF1 (3). In contrast, only two 5′-KDR kinases had more than three partner genes: FGFR3 and FGFR2 (Supplementary Table S3). To determine the functions that the partner genes may be involved in, we conducted gene set enrichment tests for the partner genes in each of four KDR FG groups. We found the partner genes of 3′-KDR FGs were enriched in the ‘dephosphorylation’- and ‘receptor protein signaling’-related pathways (WebGestalt, adjusted P-value (i.e. q-value) <0.05, hypergeometric test followed by multiple test correction using Benjamini–Hochberg’s method, Supplementary Table S4) [27, 28]. Abnormal phosphorylation [29, 30] and receptor protein signaling had long been implicated in cancer. This is consistent with several examples of evidences that these partner genes found to frequently fuse with kinase genes [24]. On the other hand, the partner genes for 3′-non-KDR FGs were plentiful in ‘catabolism’- and ‘negative regulation of cell cycle’-related pathways. Catabolic wasting or cachexia was often seen in the end stage of cancer, and about 50% of all cancer patients suffered from cachexia [3, 31, 32]. If a cell carried a FG for negative cell cycle regulatory proteins like tumor suppressors, then the cell might become carcinogenic [33]. In the 3′-non-KDR FGs, the kinase domain is not preserved and may lead to cancer through ‘catabolism’ and ‘negative regulation of cell cycle’ processes. On the other hand, for the partner genes of 5′-KFGs, there were no significantly enriched pathways, but only ‘mitochondrial part’ was determined to be significant in the cellular component of gene ontology (GO) analysis. Taken together, our pathway enrichment analysis showed the combinatorial effect between 3′-KDR kinases and their 5′-partner genes.
Short DNA motif sequence in the break point introns and break point usage in FGs
To provide mechanistic insights, many studies have attempted to find motifs near the break points in translocation events. We previously scanned the TK domain-specific motif ‘GXGXXG’ in the TK FGs and found that the fusion break points were located within a three-exon range from this motif [17]. Another study suggested that there is a strong association between the short homologous sequence at the break point and the generation of chimeric transcript in eukaryotes with the transcriptional slippage model [18]. With this specific aim, we sought for the break point motifs in KFGs. However, break points mainly occurred in introns, and the exact location of break points was not always available, especially in FG events detected by using RNA sequencing (RNA-seq) data. To overcome this challenge, we searched for DNA motifs using the intron sequences located next to the exon junction break point because these intron sequences were the regions where breakage of genome was expected to occur. Based on this assumption, we scanned sequences of 106 intron sequences that were located on the 5′-end of the exon junction break point of the 70 3′-KDR kinase genes. We also searched for motifs using the 31 intron sequences located on 3′-end of the exon junction break point of the 28 5′-KDR kinase genes. These analyses were performed using the Discriminative Regular Expression Motif Elicitation (DREAM) function in motif-based sequence analysis tools (MEME suite) [34]. For the 3′-KDR kinases, 43 significant sequence motifs were predicted (P-value <0.05; Supplementary Table S5). The most significant motif was ‘CCKCGGCC’ where K represents nucleotide G or T. This motif was present in 77 of the 106 introns, corresponding to 58 of the 70 kinases (Figure 2C). Subsequent gene set enrichment analysis by Gene Ontology for Motifs (GOMO) showed that the most significantly enriched biological processes for the genes that have this motif in their promoter regions were ‘negative regulation of signal transduction (GO ID: 0009968)’ with a q-value 4.48 × 10−6 and ‘Wnt receptor signaling pathway, calcium modulating pathway (GO ID: 0007223)’ with a q-value 4.481 × 10−6 [35, 36]. As shown in Supplementary Table S6, this motif was mainly enriched in the ‘regulation of transcription’- and ‘signal transduction’-related pathways. These results showed the evidence of enrichment of this motif in the kinase genes. We applied the similar analyses to the 5′-KDR kinases. We found only one motif ‘CGGGAGGC’, and this motif was present in 18 of the 31 introns, corresponding to 17 of the 28 kinases. Interestingly, this sequence was found in the predicted viral microRNA candidate hairpin structure sequence of the viral genome of bovine herpesvirus 1 (complete genome: NC_001847.1) with a minimum free energy (MFE) of −57.6 kcal/mol in the Vir-Mir database [37]. Recently, this motif was also identified in microRNA-like molecules derived from the anti-genome RNA of hepatitis C virus with −22.3 kcal/mol of MFE [38]. These findings might be related with the recent reports that a number of viruses known to target TK function during infection displayed obvious structure modifications and cell growth regulation that are extremely unusual [39, 40]. To show the information with more details of these two motifs, we created a graph of the distance distribution of each motif in their intron sequences (Supplementary Figure S1).
Next, we searched the break point usage for each kinase (Supplementary Table S7). Twenty-six kinases (34%) in the 3′-KDR FGs had more than two break points. Among these, six kinases (BRAF, RET, ALK, MET, NTRK3 and NTRK1) had at least five break points. Here, we identified that the break point usage had cancer-type specificity. For example, ALK had five break points in the data from TCGA Fusion Gene Data Portal, but only one was used in the five EML4-ALK positive samples of lung adenocarcinoma (LUAD). On the other hand, BRAF had eight break points in thyroid cancer (THCA), and all the break points were used to form eight fusion isoforms in 10 samples. Such patterns of diverse break point usage can provide insights into designing pan-cancer FG detection kits in the clinical application of NGS technology. Most of the commercial kits that are widely used to detect gene fusions only target one major break point for each gene and often fail in detecting fusions with rare break points.
Classification of kinases based on gene expression alteration at the break point
Fusion genes in cancer not only result in constitutive kinase activity but also in aberrant overexpression, which can be monitored as a clue of gene fusion [24]. To describe such FG expression, we explored the gene expression alteration of KDR kinases at the break point using reads per kilobase per million (RPKM) value per exon from TCGA data [41]. As shown in Figure 3A, we found three patterns of expressional alterations at break points, which are clearly distinguishable by simple criteria (see Materials and Methods section; Figure 3B and Supplementary Table S8). We named these three patterns as ‘Class A’, ‘Class B’ and ‘Class C’, and gave them weights of ‘3’, ‘2’ and ‘1’, respectively. The weights only aimed to provide a relative ranking of the three classes of patterns. We calculated the average weight for each kinase for all KDR kinases. Kinases with an average weight >2.5 were ALK, DYRK1A, EPHA6, KSR2, MAP3K15, NTRK1, RET and ROS1. We were able to identify that 3′-KDR kinases had changed their expression levels dramatically compared with 5′-KDR kinases (Supplementary Table S9). These altered fusion transcript expressions of 3′-KDR kinases might have been caused by the promoters of partner genes. Interestingly, kinases with a weight >1.5 were enriched in ‘regulation of phospholipase C activity’ and ‘activation of MAPKK activity’ pathways (ClueGO app in CytoScape, adjusted P-value <0.05, hypergeometric test followed by multiple test correction using the Bonferroni method, Supplementary Figure S2). The MAPK signaling pathways are key mediators of eukaryotic transcriptional responses to extracellular signals. These pathways control gene expression in a number of ways including the phosphorylation and regulation of transcription factors [42]. This enriched pathway also suggested the important function, i.e. why these kinases had a major gene expression alteration at the break point.
Figure 3.

KDR kinase classification based on the pattern of gene expression alteration at break points. (A) Gene expression plots of KDR kinases. Each dot presents RPKM value of an exon in each sample. Blue vertical line indicates the break point. X axis: exon of the kinase. Y axis: RPKM value. (B) Three kinase groups classified by the expression alteration at break points based on Figure 3A. We marked each kinase group by different color. The value in each cell is the number of samples having the corresponding class of the KDR kinase.
The expression of 3′-KFGs is regulated by the promoter of partner genes. Therefore, the wild-type kinase gene may not be normally expressed in the cell [24]. From the exon-based expression depth plots for all KDR kinases, we observed that the wild type of ‘Class A’ kinases had zero expression in cells transformed by the fusion product. Generally, wild-type ALK, which belonged to ‘Class A’, is not usually expressed in normal adult tissue except neural tissue [43]. However, when the cancer cells form the ALK FG, the expression of the fusion transcript becomes as high as RPKM 100 in bladder carcinoma (BLCA). On the other hand, ‘Class C’ kinases had no expression change between the wild-type and the chimeric kinases. For example, the wild-type gene of MET proto-oncogene, a RTK, was known to be overexpressed in non-small cell lung cancer (LUAD) [44], so the exon expression depth graph of MET showed an upward trend without a significant alternation at the break point in LUAD (Figure 3A). In addition, FGFR3 had the largest RPKM values in four cancer types [glioblastoma multiforme (GBM), BLCA, lung squamous cell carcinoma (LUSC) and head and neck squamous carcinoma (HNSC)]. EGFR, RPS6KB1 and CDK1 had the largest RPKM value in low grade glioma (LGG), breast carcinoma (BRCA) and LUAD, respectively (Supplementary Figures S3 and S4). As above, there is another explanation about expressional regulation of KFGs, that is several FGs of TKs are known to interact with chaperones and escape the degradation pathways, which results in enhanced protein levels [45–47].
Degree of frequency score reflects the likelihood of recurrence of KFGs
Considering that high-frequency KFGs tend to have major roles in cancer, we hypothesized that they are more likely to be driver events. In this study, we proposed a method to measure quantitatively the possible recurrence of KFGs. We used three characteristics of gene fusions: the number of partner genes of each kinase, the number of break points in each kinase and the number of cancer types related to each kinase. A kinase that had more partner genes, had break points occurring in multiple locations or fused in multiple cancer types was assumed with potentially higher impact because it could fuse with other genes in multiple ways (partner genes or break points) and functions in a wider range of cancer. Using these factors, we defined a degree-of-frequency (DoF) score, which was calculated by multiplying the three numbers above for each kinase (Figure 4A and B and Supplementary Table S7). Assuming that many KFGs occur by chance (e.g. fused with one partner gene in one cancer), we defined a basic threshold where one kinase with two partners, two cancer types and two break points would have a DoF score 8. Thus, we defined kinases with DoF ≥ 9 as high-frequency kinase fusions. Among the 76 kinases involved in 3′-KDR FGs, 13 kinases were assigned in high DoF kinase group. Further, examination revealed that 10 of the 13 kinases belonged to the TK group (ALK, FGFR1, FYN, MERTK, MET, NTRK1, NTRK2, NTRK3, RET and ROS1), two belonged to the TKL group (BRAF and RAF1), but PRKCB was not assigned to any kinase group. These are consistent with the previous study that seven kinases—ALK, BRAF, MET, NTRK1, NTRK2, RAF1 and RET—were described as driver events with their mutual exclusivity in THCA [48]. On the other hand, among the 28 kinases involved in 5′-KDR FGs, only FGFR3 and FGFR2 were high DoF kinases. Moreover, 5′-non-KDR FGs included 17 high DoF kinases. As 5′-non-KDR FGs had no kinase domain, the function of their 3′-partner genes might be related to tumorigenesis through regulations such as blocking microRNA regulation by the truncation in 3′-UTRs.
Figure 4.

The DoF score measures the impact of a kinase in gene fusion event. (A) Kinases of 3′-KDR FGs sorted by DoF score. (B) Kinases of 5′-KDR FGs sorted by DoF score. (C) The average number of gene fusion events for high DoF scored fusions and low DoF scored fusions. Red bar: high DoF scored fusions. Pink bar: low DoF scored fusions. (D) The enriched biological processes of partner genes for the high and low DoF scored cases in 3′-KDR FGs.
Next, to identify the signatures of driver kinases, we performed three analyses and compared these between high and low DoF kinases in the 3′-KDR FGs: searching the gene expression alteration at the break point, determining the average number of FGs and comparing the enriched GO biological process pathways. First, as shown in Figure 4A and B, the kinases with high DoF scores had an overall weight of the expression pattern >1.0, indicating that the 3′ part of these KFGs after the break points had increased expression, likely because of the fusion with partner genes. The average number of FGs for each of the high and low DoF groups in the 3′-KDR FGs showed a large difference, i.e. 7.5 and 1.2, respectively (Figure 4C). To examine the significance of this difference, we performed a Wilcoxon rank sum test for the number of FG samples between high DoF group (32 values) and low DoF group (250 values). It had P-value 3.099 × 10−30, suggesting significant difference between the two groups. Next, we performed gene set enrichment test of the partner genes for both of the high and low DoF cases in the 3′-KDR FGs [WebGestalt, adjusted P-value (i.e. q-value) <0.05, hypergeometric test followed by multiple test correction using Benjamini–Hochberg’s method, Figure 4D]. Remarkably, the partner genes of high DoF kinases were involved in the ‘signal transduction’- and ‘cellular response to stimulus’-related pathways. These pathways show the typical roles of synergistic combination of the most frequent FGs. On the other hand, the partner genes of the low DoF kinases were involved in ‘dephosphorylation’-related pathways.
KDR gene fusion network highlighted effective kinases in FGs
So far, many studies introduced cancer-type-specific gene fusion networks such as in leukemia [15], neoplasia [14] and ovarian cancer [2]. However, these gene fusion networks just showed the list of FGs with multiple partners in one cancer type without any weighted features. Here, we constructed a pan-cancer KDR gene fusion network by projecting all annotations obtained in this study with visualization of effective kinases in FGs (Figure 5). For each KDR FG, its two partner genes are denoted by the nodes, and the pairing is displayed by the edge. The colors of the edge and background represent cancer types. In this network, there were multiple edges between multiple cancer types. Fusion genes of almost all cancer types shared at least one kinase with the FGs of other cancer types. These frequently observed fusion kinases in the 13 cancer types were ALK, BRAF, CDK1, CDK12, EPHA1, FGFR1, FGFR2, FGFR3, FYN, IKBKB, IRAK4, JAK2, MELK, MERTK, MET, NTRK1, NTRK2, NTRK3, PAK1, PRKCA, PRKCB, PRKCH, RAF1, RET, ROCK1, ROS1, RPS6KB1, SGK2, STRADA, ULK1 and WNK1. Among these recurrent kinases, 54.8% (17 of 31) were in the TK and TKL groups. As shown here, gene fusions found across cancer types are common; therefore, these recurrent kinases would be potential targets for molecular cancer therapy. Specifically, 22 kinases had a DoF score >8. We were able to identify these impacts of kinases at a glance using a gradient color scale of the nodes, which represented the DoF score of each kinase with relevant cancer types. Remarkably, among the top seven kinases (DoF score >60), six were observed in THCA (ALK, BRAF, MET, NTRK1, NTRK3 and RET). The recurrent FGs of THCA come from these high DoF kinases. Interestingly, BRCA had many high-DoF kinases in common with LUAD including ALK, CDK1, FYN, MERTK, MET, RPS6KB1, SGK2 and WNK1. In summary, this network presents an overview of the impact of kinases in pan-cancer FGs.
Figure 5.

KDR gene fusion network. A node represents a kinase and its color reflects the DoF score. The edge color denotes specific cancer type. The node boundary color reflects the kinase group. This gene fusion network of KDRs provides an overview of the impact of kinases in pan-cancer FGs. From this network, we may select kinases with potential clinical implications.
Finally, to investigate the cancer-type-specific functional roles of KDR kinases, we searched the enriched pathways of KDR kinases per cancer type and created a biological process network (ClueGO app in CytoScape, adjusted P-value <0.05, hypergeometric test followed by multiple test correction using the Bonferroni method, Figure 6). Interestingly, the kinases in THCA and prostate adenocarcinoma (PRAD) were enriched only in the ‘activation of MAPKK activity’ pathway, which is the same as ‘activation of MAP kinase kinase activity’. The mutation of MAPK signaling pathway is frequently reported in the thyroid carcinoma for cell proliferation through activating mutations or overexpression [49]. Additionally, the MAPK pathway is also known to be involved in the progress and metastasis of prostate cancer [50]. Therefore, these enriched pathways can be further evidence in that KFGs have deeply involved in tumorigenesis of THCA and PRAD. On the other hand, in LUAD, ‘nuclear envelope organization’, ‘positive regulation of muscle tissue development’ and ‘positive regulation of phosphatidylinositol 3-kinase signaling’ pathways were enriched. However, in BRCA, the kinases were involved in various signaling pathways, likely because of an increased number of kinases and partner genes compared with other cancer types. From these various biological pathways, we may find pivotal roles of KFGs in the tumorigenesis of various cancer types.
Figure 6.
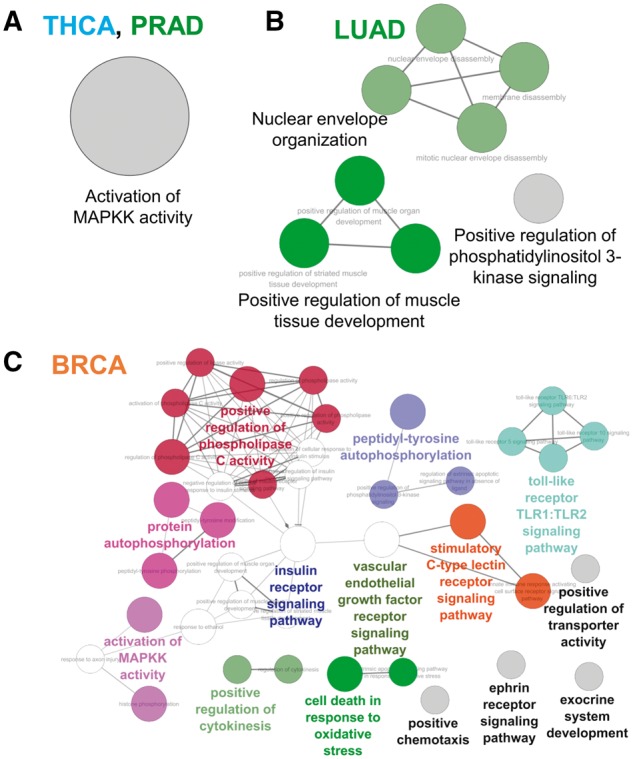
Enriched biological processes of KDR kinases per cancer type. We used the ClueGO app in CytoScape (adjusted P-value <0.05, hypergeometric test followed by multiple test correction using the Bonferroni method).
Discussion
The recurrence of FGs and the KDR provide the most fascinating rationale for prioritizing candidate driver KFGs. Based on KDRs, we performed a pan-cancer analysis of 913 KFGs. The observed striking differences in the number of KDRs between 5′-KFGs and 3′-KFGs, combinatorial effect between 3′-KDR kinases and their 5′-partners and a signal transduction-specific DNA sequence motif in the break point intron sequences showed positive selection of 3′-KDR FGs in cancer. Here, the importance of 3′-KDR FGs was identified again, through a comparison between the KDR kinases and 2719 essential genes from the Online Gene Essentiality database [51]. Essential genes belong to an important gene set whose knockouts induce lethality of the cell. In 5′-KDR FGs, there was no overlapping with essential genes, but 3′-KDR FGs had four essential genes: MAP3K1, MAPK1, PTK2 and STK3.
Among the 10 traditional human kinase groups, the TK group was most frequently observed in KDR kinases. The two key processes to switch on the kinase domain of tyrosine–KFGs are enforced oligomerization and inactivation of inhibitory domains. Then, the activated TK fusions show their signals via transduction cascades [24]. To precede the signal transduction by phosphorylation of tyrosine, the kinase domain is necessary. This mechanism of action may explain why cancers preferred 3′-end location of kinase in FGs. Furthermore, the TK group is known for two type’s kinases with the RTKs in transmembrane signaling and non-receptor tyrosine kinases (non-RTKs) in signal transduction to the nucleus [52]. In the 3′-KDR kinases, there were 11 RTKs (ALK, FGFR1, IGF1R, MERTK, MET, NTRK1, NTRK2, NTRK3, PTK7, RET and ROTS1) and four cytoplasmic TKs (non-RTKs; FYN, FGR, JAK1 and JAK2). For the 5′-KDR kinases, there were only three RTKs: EGFR, FGFR2 and FGFR3.
Notably, analysis of the exon-level gene expression patterns found seven kinases that belonged to ‘Class A’ in 3′-KDR FGs. Most of them (six of seven) were high-DoF scoring kinases (Supplementary Tables S7 and S8). These might be because of different gene expression regulation between normal and cancer cells for the constitutive expression of kinase domains. To draw gene expression plots, we used RPKM value per exon. However, if we can draw RNA-seq coverage plots for every nucleotide using both mapped and unmapped reads from both of the matched tumor and normal samples, we might be able to find much clear and more accurate expression change patterns. Furthermore, the abundance of spanning reads by the alignment of unmapped reads can show the exact abundance for translocated genes including gene fusion, trans-splicing and exon-skipping events.
Our study primarily aimed to select unique features of effective kinases in FGs. DoF scores were derived from the number of partner genes, cancer types and break points for each kinase, all of which are important factors to characterize the relative recurrence of each kinase in FG events. For example, BRAF, the highest DoF-scored kinase (DoF score: 576), appeared in four cancer types [LGG, PRAD, skin cutaneous melanoma (SKCM) and THCA) with 12 partners and 12 break points in 3′-KDR FGs. Additionally, BRAF is known to have mutually exclusive patterns with other activating somatic mutations in the MAP kinase signaling pathway. Consistent with a previous study [53], the 14 samples harboring BRAF fusions did not carry the V600E base substitution, the most important driver mutation found in melanoma so far.
Finally, with our annotations focusing on KDR, we organized a comprehensive gene fusion network. This weighted network provides a systematic view and from which we may select kinases with potential clinical implications. We found 31 recurrent kinases in multiple cancer types, and among these kinases, 17 were TK associated. Although PRKCB, PRKCA, ROCK1, SGK2, ULK1, WNK1, PAK1, CDK1 and RPS6KB1 were not TK associated, these kinases had comparable high DoF scores (8 ≤ DoF ≤ 125) in multiple cancer types. Using this method, we suggest several effective but understudied kinases. Here, we denote understudied gene when we found <10 articles of the FG study in PubMed. These genes, including PRKCB, SGK2, WNK1, PRKCH, MELK and CDK12, warrant future investigation.
Conclusion
The KDR is essential in the driver gene fusions inducing amplified cell proliferation. Our study is the first analysis covering the multiple signatures of kinases and FGs that is based on KDR for large sample sets encompassing multiple tumor lineages. This systematic annotation of KFGs can highlight candidate driver FGs and effective kinases in targeted molecular therapy for personalized medicine.
Materials and methods
TCGA pan fusion gene data
Pan-cancer FG data were obtained from the TCGA Fusion Gene Data Portal (http://54.84.12.177/PanCanFusV2, December 2014) [23]. This portal provides detailed description of candidate FGs identified by Pipeline for RNA sequencing Data Analysis [54]. Although not experimentally validated, the candidates were reported with high confidence using stringent criteria in their methods. As a result, 7993 FGs were reported in 13 cancer types from 4366 primary tumor samples: bladder carcinoma (BLCA), breast carcinoma (BRCA), glioblastoma multiforme (GBM), head and neck squamous carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC), acute myeloid leukemia (LAML), low grade glioma (LGG), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), ovarian serous cystadenocarcinoma (OV), prostate adenocarcinoma (PRAD), skin cutaneous melanoma (SKCM), and thyroid cancer (THCA). For these FGs, the following information was collected: TCGA sample ID, FG name of its two partner genes, fusion protein frame information and exon junction break point information at the genomic level. We followed the definition of FG direction for the 5′- and 3′-partner genes to this data set.
Analyzing kinase domain retention
We downloaded 538 human kinase genes from the Human Kinome database [26]. For each of the 7993 FGs from the TCGA Fusion Gene Data Portal, we first screened for kinase partner(s) and referred those who had kinase partner(s) as KFGs. Among the 914 KFGs we identified, there were 525 and 389 FGs annotated as 5′- and 3′-KFGs, respectively. We found 13 KFGs where two kinases fused. However, these KFGs were removed in the next step, as these FGs had frameshifts in their reading frame. For all KFGs, we further applied two filtering criteria based on the locations of break points. First, we required that the break points did not obstruct the reading frames of the partner genes. The TCGA Fusion Gene Data Portal annotated the reading frame for each fusion transcript at each break point. For a systematic analysis, we excluded ‘out-of-frame’ cases and the cases not having coding regions (CDS) in its kinase located segment. For example, for the 5′-KFGs, we selected fusion events categorized as ‘in-frame’, ‘CDS-5UTR’, ‘CDS-3UTR’, ‘3UTR-5UTR’, ‘3UTR-CDS’ or ‘3UTR-3UTR’. Meanwhile, we included FGs with categories of ‘in-frame’, ‘CDS-5UTR’, ‘3UTR-5UTR’, ‘3UTR-CDS’, ‘5UTR-5UTR’ or ‘5UTR-CDS’ for 3′-KFGs. This filtering retained 357 5′-KFGs and 256 3′-KFGs. Next, we required that the kinase domain be kept intact in chimeric FGs rather than being broken. That is, we looked for KFGs with KDR. To this end, we downloaded the kinase domain annotation information including kinase domain loci in protein sequence from UniProt using the UniProtKB search module [55]. Among the 312 kinases involving gene fusions, 301 kinases had annotation of their kinase domain loci. As such domain loci information was based on amino acid sequence, the genomic break point information was converted to the amino acid level for each kinase while considering all UniProt protein accessions, transcript isoforms and multiple break points for one kinase. To map the kinase domain to the human genome sequence, we used the RefSeq gene model of human reference genome (hg19) available from the UCSC Genome Browser [56, 57]. For 5′-KFGs, we considered the kinase domain to be successfully retained in the FG if the break points occurred on the 3′-end of the kinase domain, and such 5′-KFGs were referred to as 5′-KDR FGs (Figure 1A). On the contrary, if the kinase domain was not included completely in the resultant 5′-KFGs, we referred such FGs as 5′-non-KDR FGs. Similarly, for 3′-KFGs, we considered the FG to have retained the kinase domain if the break points occurred on the 5′-end of the kinase domain region and referred such 3′-KFGs as 3′-KDR FGs while the remaining 3′-KFGs as 3′-non-KDR FGs. As a result, 35 5′-KDR FGs and 136 3′-KDR FGs remained. In summary, as shown in Figure 1B, there are four groups of KFGs according to their break point locations relative to kinase domain regions: 5′-KDR FGs and 5′-non-KDR FGs for 5′-KFGs, and 3′-KDR FGs and 3′-non-KDR FGs for 3′-KFGs. Detailed annotations including kinase domain loci and break point loci on the amino acid sequence for each FG are described in Supplementary Table S1 according to KDR FG groups.
Finding short DNA sequence motifs in the break point introns
We used the tool DREAM to search for potential DNA motifs around break points. As a part of the MEME suite [34], DREAM discovers short ungapped motifs enriched in the input nucleotide sequences compared with shuffled sequences. Here, we collected the introns in which break points supposed to occur in 5′- and 3′-KDR FGs separately and searched for potential DNA motifs using DREAM. For the motifs discovered in 5′- and 3′-KDR FGs, we performed GO enrichment analysis using GOMO, also a tool of the MEME toolkit. GOMO scans promoter sequences of all human genes for the presence of the nucleotide motifs provided by the users. Thus, we used GOMO to determine if any of the motifs we found in KDR FGs were also significantly enriched in genes linked to a particular GO term [35, 36].
TCGA RNA-seq data acquisition and drawing expression depth plot
Gene expression data were obtained from TCGA (5 January 2015) [41]. The normalized gene expression data, measured in RPKM mapped reads, from RNASeqV2 was extracted using the R package TCGA-Assembler [58]. To draw RNA-seq expression plot, we used pyplot function of the matplotlib module in Python 2.7.2 [59]. We collected expression levels of all exons in wild-type kinase gene structure. We then compared exon expression that occurred before the break point (on the 5′-end) and that after the break point (on the 3′-end). We distinguished the KDR FGs into three groups according to the expression patterns of the deleted exons by gene fusion in the corresponding kinases: low expression (exon expression levels were all zero or close to zero, Class A), moderate expression (exon expression levels were <30% of the maximum RPKM, Class B) or high expression (Class C; see Results section). To scale these expressional level changes per kinase, we assigned a weight for each class. Classes A, B and C were given the weight of 3, 2 and 1, respectively.
Constructing a KDR gene fusion network
We built a network using KDR FGs only. In this network, each node represents a partner gene or kinase gene, and each edge represents a gene fusion event. A gene fused with different partners would have multiple edges. A FG could also occur in different cancer types; thus, we allowed multiple edges to represent the same FG in different cancer types through distinguishable edge colors. We used Cytoscape (version 3.2.1) [60] for visualization and analysis of the network. To identify the enriched GO biological process terms in each cancer type, we used a Cytoscape plug-in to decipher functionally grouped GO and pathway annotation networks (ClueGO) [61].
Key Points
We presented a comprehensive landscape of the multiple signatures of kinases and FGs that is based on KDR for large sample sets encompassing 13 tumor lineages, including 914 KFGs covering 312 kinases.
We found multiple lines of evidence supporting a positive selection on 3′-kinase fusion genes (3′-KFGs) in cancer, such as more KDR than 5′-kinase fusion genes (5′-KFGs), combinatorial effect between 3′-KDR kinases and their 5′-partners and a signal transduction-specific DNA sequence motif in the break point intron sequences.
The kinases with high DoF scores tended to undergo strong gene expression alteration at the break points.
We proposed common features of ‘effective’ (highly recurrent) kinases involved in gene fusion such as stronger recurrence, expression alteration at break point, redundant usage in multiple cancer types and 3′-location tendency.
Through our annotations, we pinpointed several kinase candidates for future studies.
Supplementary Material
Acknowledgements
The authors thank the TCGA Fusion Gene Data Portal site for making the fusion gene data available for this work. The authors also thank Drs Christine M. Lovly and Zhiyong Ding for their valuable discussion.
Funding
This work was partially supported by National Institutes of Health grants (grant numbers R01LM011177 and R21CA196508 to Z.Z.). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Pora Kim is a bioinformatics post-doctoral fellow in the Bioinformatics and Systems Medicine Laboratory (BSML), Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston.
Peilin Jia is an assistant professor in Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston. She codirects the Bioinformatics and Systems Medicine Laboratory.
Zhongming Zhao is a professor in the Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston. He directs the Bioinformatics and Systems Medicine Laboratory.
References
- 1. Latysheva NS, Babu MM.. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res 2016;44:4487–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mertens F, Johansson B, Fioretos T, et al. The emerging complexity of gene fusions in cancer. Nat Rev Cancer 2015;15:371–81. [DOI] [PubMed] [Google Scholar]
- 3. Van Allen EM, Wagle N, Levy MA.. Clinical analysis and interpretation of cancer genome data. J Clin Oncol 2013;31:1825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hunter T. Treatment for chronic myelogenous leukemia: the long road to imatinib. J Clin Invest 2007;117:2036–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong S, Witte ON.. The BCR-ABL story: bench to bedside and back. Annu Rev Immunol 2004;22:247–306. [DOI] [PubMed] [Google Scholar]
- 6. Druker BJ. Inhibition of the Bcr-Abl tyrosine kinase as a therapeutic strategy for CML. Oncogene 2002;21:8541–6. [DOI] [PubMed] [Google Scholar]
- 7. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6. [DOI] [PubMed] [Google Scholar]
- 8. Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644–8. [DOI] [PubMed] [Google Scholar]
- 9. Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012;337:1231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitelman F, Johansson B, Mertens F.. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet 2004;36:331–4. [DOI] [PubMed] [Google Scholar]
- 11. Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 2011;39:D945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim P, Yoon S, Kim N, et al. ChimerDB 2.0–a knowledgebase for fusion genes updated. Nucleic Acids Res 2010;38:D81–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar-Sinha C, Kalyana-Sundaram S, Chinnaiyan AM.. Landscape of gene fusions in epithelial cancers: seq and ye shall find. Genome Med 2015;7:129.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoglund M, Frigyesi A, Mitelman F.. A gene fusion network in human neoplasia. Oncogene 2006;25:2674–8. [DOI] [PubMed] [Google Scholar]
- 15. Bohlander SK. Fusion genes in leukemia: an emerging network. Cytogenet Cell Genet 2000;91:52–6. [DOI] [PubMed] [Google Scholar]
- 16. Wu CC, Kannan K, Lin S, et al. Identification of cancer fusion drivers using network fusion centrality. Bioinformatics 2013;29:1174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chmielecki J, Peifer M, Jia P, et al. Targeted next-generation sequencing of DNA regions proximal to a conserved GXGXXG signaling motif enables systematic discovery of tyrosine kinase fusions in cancer. Nucleic Acids Res 2010;38:6985–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Zhao L, Jiang H, et al. Short homologous sequences are strongly associated with the generation of chimeric RNAs in eukaryotes. J Mol Evol 2009;68:56–65. [DOI] [PubMed] [Google Scholar]
- 19. Frenkel-Morgenstern M, Valencia A.. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics 2012;28:i67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abate F, Zairis S, Ficarra E, et al. Pegasus: a comprehensive annotation and prediction tool for detection of driver gene fusions in cancer. BMC Syst Biol 2014;8:97.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shugay M, Ortiz de Mendibil I, Vizmanos JL, et al. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 2013;29:2539–46. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Xia J, Jia P, et al. Application of next generation sequencing to human gene fusion detection: computational tools, features and perspectives. Brief Bioinform 2013;14:506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yoshihara K, Wang Q, Torres-Garcia W, et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015;34:4845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Medves S, Demoulin JB.. Tyrosine kinase gene fusions in cancer: translating mechanisms into targeted therapies. J Cell Mol Med 2012;16:237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Drake JM, Lee JK, Witte ON.. Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol Cell Biol 2014;34:1722–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science 2002;298:1912–34. [DOI] [PubMed] [Google Scholar]
- 27. Wang J, Duncan D, Shi Z, et al. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 2013;41:W77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bononi A, Agnoletto C, De Marchi E, et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res 2011;2011:329098.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim P, Zhao J, Lu P, Zhao Z.. mutLBSgeneDB: mutated ligand binding site gene DataBase. Nucleic Acids Res 2016. doi: 10.1093/nar/gkw905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Payne C, Wiffen PJ, Martin S.. Interventions for fatigue and weight loss in adults with advanced progressive illness. Cochrane Database Syst Rev 2012;1:CD008427.. [DOI] [PubMed] [Google Scholar]
- 32. Pecqueur C, Oliver L, Oizel K, et al. Targeting metabolism to induce cell death in cancer cells and cancer stem cells. Int J Cell Biol 2013;2013:805975.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Collins K, Jacks T, Pavletich NP.. The cell cycle and cancer. Proc Natl Acad Sci USA 1997;94:2776–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 2009;37:W202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boden M, Bailey TL.. Associating transcription factor-binding site motifs with target GO terms and target genes. Nucleic Acids Res 2008;36:4108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buske FA, Boden M, Bauer DC, et al. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics 2010;26:860–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li SC, Shiau CK, Lin WC.. Vir-Mir db: prediction of viral microRNA candidate hairpins. Nucleic Acids Res 2008;36:D184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi J, Duan Z, Sun J, et al. Identification and validation of a novel microRNA-like molecule derived from a cytoplasmic RNA virus antigenome by bioinformatics and experimental approaches. Virol J 2014;11:121.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Radha V, Nambirajan S, Swarup G.. Association of Lyn tyrosine kinase with the nuclear matrix and cell-cycle-dependent changes in matrix-associated tyrosine kinase activity. Eur J Biochem 1996;236:352–9. [DOI] [PubMed] [Google Scholar]
- 40. Schaller MD, Borgman CA, Cobb BS, et al. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA 1992;89:5192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cancer Genome Atlas Research Network; Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Whitmarsh AJ. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta 2007;1773:1285–98. [DOI] [PubMed] [Google Scholar]
- 43. Grande E, Bolos MV, Arriola E.. Targeting oncogenic ALK: a promising strategy for cancer treatment. Mol Cancer Ther 2011;10:569–79. [DOI] [PubMed] [Google Scholar]
- 44. Salgia R. Role of c-Met in cancer: emphasis on lung cancer. Semin Oncol 2009;36:S52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsukahara F, Maru Y.. Bag1 directly routes immature BCR-ABL for proteasomal degradation. Blood 2010;116:3582–92. [DOI] [PubMed] [Google Scholar]
- 46. Bonvini P, Gastaldi T, Falini B, et al. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res 2002;62:1559–66. [PubMed] [Google Scholar]
- 47. Bonvini P, Dalla Rosa H, Vignes N, et al. Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res 2004;64:3256–64. [DOI] [PubMed] [Google Scholar]
- 48. Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun 2014;5:4846.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nikiforov YE. Thyroid carcinoma: molecular pathways and therapeutic targets. Mod Pathol 2008;21(Suppl 2):S37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rodriguez-Berriguete G, Fraile B, Martinez-Onsurbe P, et al. MAP kinases and prostate cancer. J Signal Transduct 2012;2012:169170.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen WH, Minguez P, Lercher MJ, et al. OGEE: an online gene essentiality database. Nucleic Acids Res 2012;40:D901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ruetten H, Thiemermann C.. Effects of tyrphostins and genistein on the circulatory failure and organ dysfunction caused by endotoxin in the rat: a possible role for protein tyrosine kinase. Br J Pharmacol 1997;122:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ross JS, Wang K, Chmielecki J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016;138:881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Torres-Garcia W, Zheng S, Sivachenko A, et al. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics 2014;30:2224–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Magrane M, Consortium U.. UniProt knowledgebase: a hub of integrated protein data. Database 2011;2011:bar009.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rosenbloom KR, Armstrong J, Barber GP, et al. The UCSC genome browser database: 2015 update. Nucleic Acids Res 2015;43:D670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pruitt KD, Tatusova T, Maglott DR.. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 2007;35:D61–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhu Y, Qiu P, Ji Y.. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nat Methods 2014;11:599–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng 2007;9:90–5. [Google Scholar]
- 60. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009;25:1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.