

Abstract
Congenital pulmonary airway malformation (CPAM) is a developmental malformation of the lower respiratory tract. We report the case of a male newborn diagnosed with cystic lung disease during prenatal ultrasound. A cesarean section was performed at the 32nd gestational week because of premature rupture of the membranes, and soon after the delivery the newborn developed respiratory failure and died. The aim of this study is to report an autopsy case because of its rarity, and to briefly discuss the CPAM subtypes and differential diagnosis of cystic lung diseases of childhood.
Keywords: Cystic Adenomatoid Malformation of Lung, Congenital; Respiratory Distress Syndrome; Newborn; Autopsy
CASE REPORT
A 27-year-old Gravida 4, Para 3, Abortus 0 patient was admitted to the obstetrics ward of a referral hospital at the 32nd gestational week, with the diagnosis of premature rupture of the membranes and fetal malformations. The prenatal care was initiated during the second month, and serological tests for toxoplasmosis, rubella, syphilis, and human immunodeficiency virus were negative. The fetal ultrasonography at 29 weeks’ gestation showed reduced left thoracic size and a large cystic mass occupying the right hemithorax. A cesarean section was performed for fetal distress, and a male newborn was delivered weighing 2030 g. The baby presented cardiorespiratory arrest immediately after birth and death was certified at 37 minutes of life. Consent for autopsy was granted by the parents.
AUTOPSY FINDINGS
On opening the chest, a mediastinal shift to the left hemithorax was noted. The heart and lungs together weighed 38 grams. The right lung measured 9 cm × 7 cm × 3 cm and showed a hyperinflated aspect exhibiting multiple cystic formations with up to 7 cm in diameter ( Figure 1 ). The left lung measured 4 cm × 2cm × 1 cm and appeared immature. The heart had normal anatomical connection with the large arteries, but on opening, a 2 mm atrial septal defect was evident. No other systemic malformation was present.
Figure 1. Gross view of the hyperinflated right lung with multiple cysts.
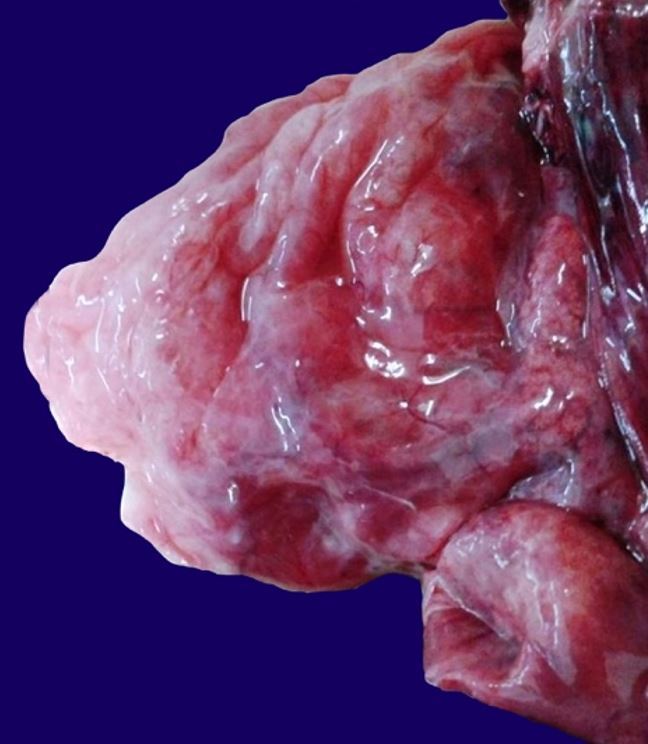
The thoracic organs were fixed in formalin for subsequent dissection ( Figure 2A ). The right lung cut surface showed to be markedly altered by the presence of numerous contiguous cystic formations outlined by thin walls, imparting a honeycomb appearance. It additionally showed two lobes, both affected by the malformation. The left lung had two lobes and appeared compact ( Figure 2B ).
Figure 2. A – Front view of the thoracic organs after formalin fixation; B – Posterior view of the thoracic organs. Note the right lung with numerous contiguous cystic formations outlined by thin walls with honeycomb appearance (arrow) and the immature left lung with the two lobes (arrowhead).

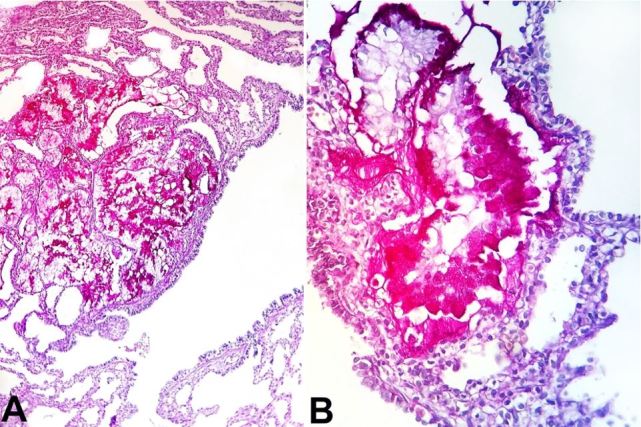
Histological examination of the right lung stained with hematoxylin-eosin (H&E) and Periodic Acid Schiff (PAS) revealed a proliferation of the terminal respiratory structures forming cysts that were lined with ciliated pseudostratified epithelium with polypoid projections of the mucosa, small cartilage foci in the wall of the bronchial structures ( Figure 3A , 3B ), clusters of goblet cells, and mucinous glands ( Figure 4A and 4B ). The left lung showed an immature parenchyma with the predominance of collapsed alveoli. However, cartilage around the bronchi was present.
Figure 3. Photomicrograph of the right lung revealing in A – Cystic dilated bronchiolar structures (H&E, 200X); and B – Polypoid projections of the mucosa and rare cartilage foci in the bronchial wall (H&E, 200X).
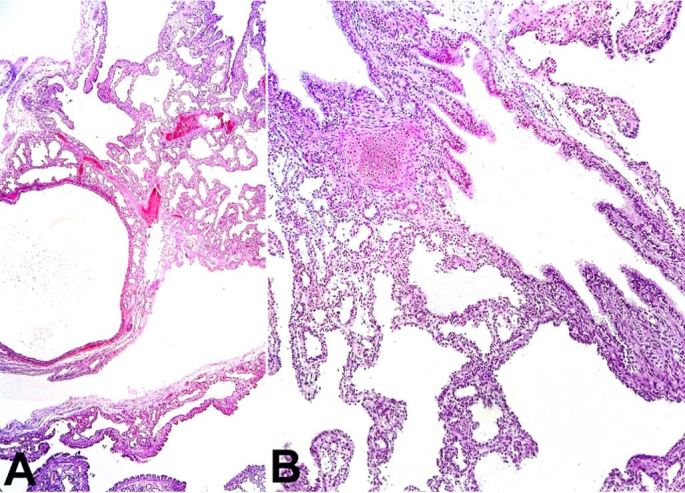
Figure 4. A – Photomicrograph of the right lung revealing clusters of mucinous glands (PAS, 200X); and B – Presence of the goblet cells (PAS, 400X).
There were no significant changes in the vascularization of both lungs. The placenta did not present abnormalities on histological examination.
DISCUSSION
The incidence of congenital lung malformations ranges from 30 to 42 cases per 100,000 inhabitants per year, and cystic diseases account for 25%-30% of this total. 1
Congenital pulmonary airway malformations (CPAMs) are a heterogeneous group of cystic and non-cystic lung lesions that result from early airway maldevelopment with the incidence ranging between 1:10,000 and 1:35,000 newborns. 2 - 6 This entity was first classified by Ch’in and Tang 7 in 1949 as congenital cystic adenomatoid malformation (CCAM). Although originally described as “congenital cystic adenomatoid malformation,” this entity now has the designation of “congenital pulmonary airway malformation” because the lesions described as “cystic” were present in only three of the five subtypes and “adenomatoid” in only one subtype. 6
The accepted pathogenesis for CPAM is that an abnormal airway patterning and branching during lung morphogenesis results in the appearance of lung cysts. 5 It is considered a hamartomatous abnormality of the bronchial tree by some authors, whereas others favor the etiology of an arrest in the development of the fetal bronchial tree with airway obstruction. 8 Although the exact cellular mechanisms involved in the pathogenesis are unknown, many potential genes have been associated with the formation of lung cysts. 5
In 1977, CPAM was classified into three types (I, II, and III) by Stocker et al. 9 This classification is based on clinical, macroscopic, and microscopic criteria. The microscopic features that distinguish CPAM from normal lung include: (i) proliferation of the terminal respiratory structures forming cysts; (ii) polypoid projections of the mucosa; (iii) increased smooth muscle and elastic tissue within cyst walls; (iv) the absence of cartilage; (v) the presence of mucous-secreting cells; and (vi) the absence of inflammation. 5 In 2009, two subtypes were added to Stocker et al.’s classification, namely: type 0, which corresponds to the proximal bronchial anomaly (acinar dysplasia); and type IV, which corresponds to the peripheral lung anomaly. 2 , 10 Each of the subtypes has characteristics that favor the classification:
Type 0: 1%-3% of frequency, birth presentation, small cysts of 0.5 cm with ciliated pseudostratified epithelial lining and goblet cells, bronchiolar cartilage present, and all lobes involved. 4
Type I: 60%-65% of frequency, newborn respiratory distress, multiple large cysts with 10 cm or a single dominant cyst, ciliated pseudostratified epithelial lining with bronchiolar differentiation, mucinous cells in 33% and cartilage present in 10% of cases, and only one lobe involved. 4 , 9
Type II: 10%-15% of frequency, first month of life presentation, multiple spaced cysts with 2.5 cm (sponge-like appearance), ciliated cuboidal or columnar epithelial lining, the absence of mucinous cells and cartilage, and the involvement of usually only one lobe. 4 , 9
Type III: 8% of frequency, in uterus or birth respiratory distress, bulky firm mass with adenomatoid appearance and cysts of 1.5 cm size, ciliated cuboidal epithelial lining, the absence of mucous cells and cartilage, and the involvement of usually only one lobe or one lung. 4 , 9
Type IV: 10%-15% of frequency, newborn respiratory distress, pneumothorax, pneumonia or incidental finding, peripheral cysts with acinar-alveolar epithelial differentiation, a 7-cm-sized cyst and flattened alveolar lining cells, the rare absence of mucous cells and cartilage, and usually only one lobe involved. 4
The diagnosis of type I CPAM, in this case, was based on clinical presentation and macroscopic finding characterized by multiple large cysts, which was confirmed by histological examination, and which demonstrated cysts with ciliated pseudostratified epithelial lining with bronchiolar differentiation, a cluster of goblet cells, mucinous glands, and small cartilage foci. The risk of respiratory distress at birth is up to 30% in cases of type I CPAM. 1
The symptoms and prognosis depend on the degree of pulmonary involvement. 3 - 5 , 8 The clinical presentation spectrum varies from asymptomatic cases, recurrent pneumonia, or respiratory insufficiency. 2 In severe cases, patients develop respiratory failure even in the neonatal period, but others have a benign clinical course, with the manifestation of chronic obstructive pulmonary disease or recurrent pneumonia as an adult. 3 - 5 , 8 In our case, the lung malformation was responsible for the early acute respiratory failure and death.
In a literature review on multi-cystic congenital lung diseases, the main differential diagnoses of CPAM should include congenital lobar over-inflation (CLO), 1 , 11 , 12 primary ciliary dyskinesia (PCD), 13 , 14 cystic fibrosis (CF), 15 pulmonary sequestration (PS), 1 and Williams–Campbell syndrome (WCS). 16 , 17
CLO is a rare lung malformation, which is also referred to as congenital lobar emphysema with a frequency between 1:20,000 and 1:30,000 births. It is characterized by the hyperinflation of a lung lobe by air trapping. 1 , 11 , 12 This condition can also evolve with acute respiratory failure at birth, similar to our case. The pulmonary histopathology of CLO is usually normal, with mild alveolar dilatation without destruction of the alveolar septa, 11 which was not observed in our case.
Another hypothesis, based on the newborn’s outcome, is PCD, which is also rare with an estimated incidence between 1:10,000 and 1:20,000 births. 13 PCD is a genetic disorder, causing abnormalities in the structure or function of the cilia, accumulating mucus in the lungs. 13 , 14 Therefore, in these cases, bronchial dilation is secondary to mucus accumulation. This criterion is useful for the diagnosis and was not observed in our case.
In the differential diagnosis, we must also include CF, which is a recessive autosomal disease 15 resulting in thick viscous mucus that obstructs exocrine glands and bronchi lumens. In CF, obstruction and secondary infections are the causes of bronchiectasis, 15 which was not found in our case.
The PS accounts for 0.15%-6.45% of all lung malformations and is characterized by non-functioning lung tissue that has no connection with the bronchial tree and receives its blood supply from the systemic circulation, via the descending thoracic aorta or the abdominal aorta. 1 The absence of anomalous vascularization in this case excludes this hypothesis.
The congenital form of WCS is a cystic lung disease associated with a deficiency of cartilage in the bronchial tree. The diagnosis is made by excluding other common causes of bronchiectasis. 16 The bilateral involvement of the lung and the association with other anomalies—particularly with congenital cardiac diseases—is often observed. 16 , 17 The absence of both criteria does not favor this diagnosis for the described case.
CONCLUSION
This case is particularly interesting because it emphasizes the importance of the differential diagnosis of congenital cystic diseases of the lung, which are identified on gestational ultrasound. An accurate diagnosis is fundamental for prenatal counseling and better medical management to reduce mortality and to improve prognosis.
Procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from the participant of the study.
Footnotes
How to cite: Ursini WP, Ponce CC. Congenital pulmonary airway malformation. Autops Case Rep [Internet]. 2018; 8(2): e2018022. http://dx.doi.org/10.4322/acr.2018.022
Financial support: None
References
- 1. Andrade CF, Ferreira HPC, Fischer GB. Congenital lung malformations. J Bras Pneumol. 2011;37(2):259-71. 10.1590/S1806-37132011000200017. [DOI] [PubMed] [Google Scholar]
- 2. Reis AR, Ribeiro FB, Schultz R. Congenital cystic adenomatoid malformation type I. Autops Case Rep. 2015;5(3):21-6. 10.4322/acr.2015.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chong Y, Rhee YJ, Han SJ, Cho HJ, Kang SK, Kang MW. Life-threatening congenital cystic adenomatoid malformation in the premature neonate. Korean J Thorac Cardiovasc Surg. 2016;49(3):210-3. 10.5090/kjtcs.2016.49.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sfakianaki AK, Copel JA. Congenital cystic lesions of the lung: congenital cystic adenomatoid malformation and bronchopulmonary sequestration. Rev Obstet Gynecol. 2012;5(2):85-93. [PMC free article] [PubMed] [Google Scholar]
- 5. David M, Lamas-Pinheiro R, Henriques-Coelho T. Prenatal and postnatal management of congenital pulmonary airway malformation. Neonatology. 2016;110(2):101-15. 10.1159/000440894. [DOI] [PubMed] [Google Scholar]
- 6. Bolde S, Pudale S, Pandit G, Ruikar K, Ingle SB. Congenital pulmonary airway malformation: A report of two cases. World J Clin Cases. 2015;3(5):470-3. http://dx.doi.org/10.12998/wjcc.v3.i5.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ch’in KY, Tang MY. Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol (Chic). 1949;48(3):221-9. [PubMed] [Google Scholar]
- 8. Giubergia V, Barrenechea M, Siminovich M, Pena HG, Murtagh P. Congenital cystic adenomatoid malformation: clinical features, pathological concepts and management in 172 cases. J Pediatr (Rio J). 2012;88(2):143-8. 10.2223/JPED.2177. [DOI] [PubMed] [Google Scholar]
- 9. Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8(2):155-71. 10.1016/S0046-8177(77)80078-6. [DOI] [PubMed] [Google Scholar]
- 10. Durell J, Lakhoo K. Congenital cystic lesions of the lung. Early Hum Dev. 2014;90(12):935-9. 10.1016/j.earlhumdev.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 11. Cataneo DC, Rodrigues OR, Hasimoto EN, Schmidt AF Jr, Cataneo AJ. Congenital lobar emphysema: 30-year case series in two university hospitals. J Bras Pneumol. 2013;39(4):418-26. 10.1590/S1806-37132013000400004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biyyam DR, Chapman T, Ferguson MR, Deutsch G, Dighe MK. Congenital lung abnormalities: embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics. 2010;30(6):1721-38. 10.1148/rg.306105508. [DOI] [PubMed] [Google Scholar]
- 13. Mirra V, Werner C, Santamaria F. Primary ciliary dyskinesia: an update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front Pediatr. 2017;5:135. 10.3389/fped.2017.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013;188(8):913-22. 10.1164/rccm.201301-0059CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jain M, Goss CH. Update in cystic fibrosis 2013. Am J Respir Crit Care Med. 2014;189(10):1181-6. 10.1164/rccm.201402-0203UP. [DOI] [PubMed] [Google Scholar]
- 16. Williams H, Campbell P. Generalized bronchiectasis associated with deficiency of cartilage in the bronchial tree. Arch Dis Child. 1960;35(180):182-91. 10.1136/adc.35.180.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wayne KS, Taussig LM. Probable familial congenital bronchiectasis due to cartilage deficiency (Williams–Campbell syndrome). Am Rev Respir Dis. 1976;114(1):15-22. [DOI] [PubMed] [Google Scholar]