

Abstract
Polysaccharides are essential and immunologically-relevant components of bacterial cell walls. These biomolecules can be found covalently attached to lipids (e.g., O-polysaccharide contains undecaprenyl, lipopolysaccharide contains Lipid A) or noncovalently associated with cell wells (e.g., capsular polysaccharide). While extensive genetic studies have indicated that the Wzy-dependent biosynthetic pathway is primarily responsible for producing such polysaccharides, in vitro biochemical studies are needed to determine, for example, which gene product is responsible for catalyzing each step in the pathway, and molecular details about the Wzx translocase, Wzy polymerase, and O-PS chain length determinant. Many of these biochemical studies require access to a structurally well-defined polysaccharide repeating unit-pyrophosphate-undecaprenyl (RU-PP-Und), the key building block in this pathway. We describe herein the chemoenzymatic synthesis of Escherichia coli (serotype O157) RU-PP-Und. This involves 1) chemical synthesis of precursor N-acetyl-d-galactosamine (GalNAc)-PP-Und (2 weeks), and 2) enzymatic extension of the precursor to afford RU-PP-Und (2 weeks). Undecaprenol phosphate and peracetylated GalNAc-1-phosphate are prepared from commercially available undecaprenol and peracetylated GalNAc. Chemical coupling of these two products followed by structural confirmation (mass spectrometry and NMR) and deprotection affords GalNAc-PP-Und. This compound is then sequentially modified by enzymes in the E. coli serotype O157 O-PS biosynthetic pathway. Three glycosyltransferases are involved (WbdN, WbdO and WbdP) and they transfer glucose (Glc), L-fucose (L-Fuc), and N-acetylperosamine (PerNAc) onto GalNAc-PP-Und to form the intact RU-PP-Und in a step-wise manner. Final compounds and intermediates are confirmed by mass spectrometry. The procedure can be adapted to the synthesis of analogues with different polysaccharide or lipid moieties.
INTRODUCTION
Cell surface polysaccharides, including lipopolysaccharides (LPS), capsular polysaccharides (CPS) and extracellular polysaccharides (EPS), are important structural and functional components of bacteria, and are normally essential in pathogenic processes, such as bacteria-host interactions, resistance to serum-mediated killing, and regulation of the host immune response1–3. These polysaccharides furthermore possess a high level of diversity with regards to their chemical structures. For example, Escherichia coli isolates contain ~170 different O-serotypes and ~80 K-serotypes with each one producing a structurally unique LPS O-polysaccharide (O-PS, also known as O-antigen) or CPS2. Polysaccharide formation follows one of three major biosynthetic pathways: the Wzy-dependent pathway, the ABC-transporter-dependent pathway and the synthase-dependent pathway. Discovered nearly five decades ago4, the Wzy-dependent pathway may account for the syntheses of the majority heteropolysaccharides across bacterial phyla5.
Extensive genetic studies have made it clear that Wzy-dependent biosynthetic pathways of all know bacterial O-PS are initiated by a member of one of two polyprenol-phosphate sugar-1-phosphate transferase families6–8(Fig. 1). The polyprenol-phosphate sugar-1-phosphate transferase adds a monosaccharide-1-phosphate onto the universal lipid carrier undecaprenyl phosphate (Und-P), forming a precursor monosaccharide-pyrophosphate-undecaprenyl (MS-PP-Und). The MS-PP-Und is then extended by a series of glycosyltransferases (GTs) to form a key oligosaccharide repeating unit-pyrophosphate-undecaprenyl (RU-PP-Und) intermediate9. These two steps occur on the cytoplasmic face of inner membrane, where sugar nucleotides are available. Upon translocation onto the periplasmic side of the inner membrane by Wzx10,11, the repeating units are polymerized by the polymerase Wzy12,13, forming O-PS or CPS. Meanwhile, the strain-specific chain length modality of polysaccharides is controlled by Wzz14–16 in a manner that is yet to be clarified. It should further be noted that the genes involved in the Wzy-dependent pathway of bacterial O-PS are typically assembled in a gene cluster named rfb which has been used in sequencing-based molecular serotyping of bacteria17.
Figure 1.
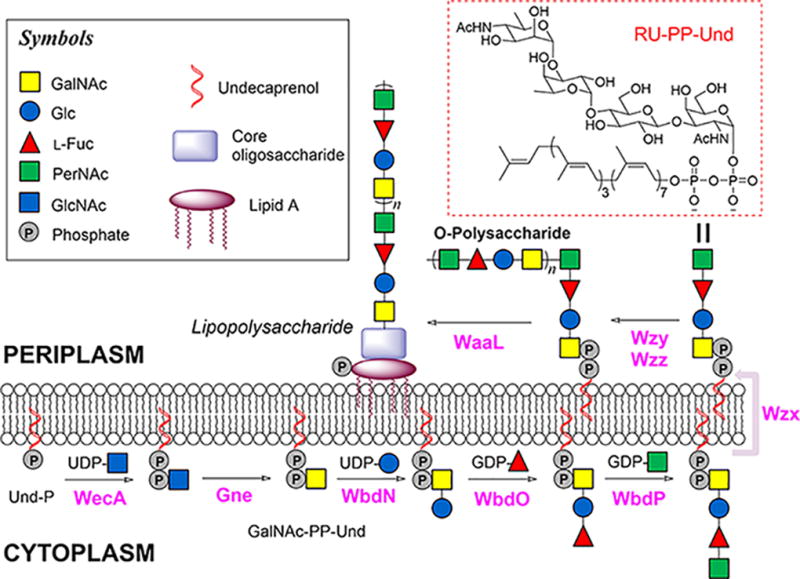
Proposed Wzy-dependent biosynthetic pathway of E. coli O157 LPS. The biosynthesis is initiated with the formation of GlcNAc-PP-Und, which is later epimerized to GalNAc-PP-Und54. Subsequently, the RU-PP-Und is assembled by three GTs (WbdN, WbdO and WbdP) and then translocated to the periplasmic face of the inner membrane by Wzx, where the repeating units are polymerized to form O-PS by Wzy in a block transfer mechanism that is regulated by Wzz. The last step includes WaaL catalyzed transfer of O-PS onto an individually synthesized Lipid A core structure to form the intact LPS.
Despite significant genetic evidence, the functions of Wzy and Wzz were only recently confirmed by in vitro biochemical studies. In these studies, we developed a chemoenzymatic approach to access structurally defined RU-PP-Und and analogues, and then used these substances to biochemically reconstitute the E. coli serotype O86 (E. coli O86) LPS biosynthetic pathway9,18. Undecaprenyl pyrophosphate linked sugars (Sugar-PP-Und), such as the RU-PP-Und used in our Wzy-pathway studies, are common sugar donors of bacterial biosynthetic pathways for cell wall peptidoglycan, teichoic acid, and protein N-/O-glycosylation among others19–21. The use of synthesized Sugar-PP-Und substrates has thus proven invaluable in deciphering the molecular details of these pathways22–28. For example, using chemoenzymatically prepared E. coli O86 RU-PP-Und and analogues, it was revealed that PglL, the oligosaccharyltransferase from the Neisseria meningitidis O-glycosylation pathway, is extremely promiscuous and can transfer a wide range of sugars onto proteins27,28. Given that RU-PP-Und and other Sugar-PP-Und are direct substrates of key enzymes involved in bacterial polysaccharide, cell wall and teichoic acid biosynthesis as well as protein glycosylation (e.g., Wzx, Wzy, WaaL, PglL), these compounds can be readily used to reveal the finer details of these enzymes, including activity, kinetics, substrate specificity, biochemical properties, and catalytic mechanisms among others. Given this level of significance, similar methodologies have been developed to afford Sugar-PP-Und in vitro as summarized in Table 129–33. Nevertheless, to date, our report represents the only example of the in vitro synthesis of O-antigen RU-PP-Und9.
Table 1.
A survey of reports for in vitro Sugar-PP-Und synthesis.
| Reports | Woodward, et al9 | Holkenbrink, et al33 | Lebar, et al31 | Huang, et al32 | Huang, et al29 | Gale, et al30 |
|---|---|---|---|---|---|---|
| Target Compounds | RU-PP-Und & analogues | Trisaccharide-PP-Und | m-DAP Lipid II precursor | Lipid II analogues | Lipid II-PP-Und analogues | GlcNAc-PP-Und |
| Biosynthetic Pathway Involved | O-PS, LPS | S-layer Glycoprotein | Cell wall Peptidoglycan | Cell wall Peptidoglycan | Cell wall Peptidoglycan | Teichoic acid |
| Source of Starting Lipid(s) | Commercial Und; Synthetic lipids | Commercial Und-P | Synthetic Heptaprenol | Synthetic lipids | Extracted and commercial lipids | Commercial Und |
| Chemical Synthesis | Monosaccharide-1-phosphate and Und-P formation; pyrophosphate bond formation; final deprotection | Trisaccharide-1-phosphate formation; pyrophosphate bond formation; final deprotection | Monosaccharide-1-phosphate and lipid phosphate formation; pyrophosphate bond formation; final deprotection | Disaccharide-1-phosphate and lipid phosphate formation; pyrophosphate bond formation; final deprotection | N/A | Monosaccharide-1-phosphate and Und-P formation; pyrophosphate bond formation; final deprotection |
| Enzymatic Synthesis | 4 GTs; Step-wise synthesis | NA | 1 GT | N/A | Multi-enzyme; One-pot synthesis | 2 GTs; Step-wise synthesis |
| Structure Confirmation (final product) | LRMS(ESI) or MALDI-MS | HRMS(ESI) | LRMS(ESI) | HRMS(ESI) 1H NMR | HRMS(ESI) | HRMS(ESI); MS/MS |
| Scale | mg scale | 1 mg | <100 µg | mg scale | mg scale | mg scale; MS detectable |
| Advantage | Synthetic yields; notably pyrophosphate bond formation (59%) | Avoided use of enzymes which need expression and purification | Use of a catalyst in pyrophosphate bond formation greatly improves rate (1/2 day) | --------- | Avoided a lengthy chemical synthesis route | --------- |
| Disadvantage | Pyrophosphate bond formation requires 3 days | Low overall yield; Pyrophosphate bond formation requires 1 week | Pyrophosphate bond formation yields using a truncated lipid were below those reported herein | Low overall yield | Enzymatic pyrophosphate bond formation limits the scope of this approach | Low synthetic yields and pyrophosphate bond formation (reaction time = 1 week) |
In this protocol, we use E. coli serotype O157 (E. coli O157) as a model to describe the experimental details for the chemoenzymatic synthesis of bacterial RU-PP-Und and analogues on a milligram scale. This protocol includes the following features: 1) step-by-step chemical synthesis of MS-PP-Und starting with a commercially available monosaccharide and undecaprenol (or other polyprenols); 2) biochemical identification of responsible GTs, and using these GTs to perform sequential one-pot enzymatic extensions of GalNAc-PP-Und to synthesize RU-PP-Und (Box 2); and 3) an application of the RU-PP-Und in an activity assay for WaaL. Given that most common sugar donors (sugar nucleotides) are commercially available, their synthesis will not be discussed here. The synthesis of uncommon sugar nucleotides found in various bacteria have been summarized elsewhere34,35. As long as the structural information of a bacterial polysaccharide (see http://csdb.glycoscience.ru/bacterial/ for determined structures) and sugar nucleotides are available, and the corresponding GTs genes (usually in a gene cluster such as rfb) have been sequenced, a similar protocol can be applied for the chemoenzymatic synthesis of desired RU-PP-Und and other Sugar-PP-Und. The protocol can also be applied for the synthesis of RU-PP-Und derivatives, as long as the GTs can tolerate modified substrates. In these cases, initial small-scale tests are recommended to check whether the modified substrates are tolerated. Furthermore, GTs responsible for the addition of the second monosaccharide were found to be promiscuous towards the lipid moiety of MS-PP-Und36–42. Given that other GTs involved in the assembly of RU-PP-Und usually do not require the lipid moiety43–47, this procedure can thus be used for the preparation of a vast array of RU-PP-Lipids analogues without compromising reaction yields (see Step 1).
Box 2. One-pot multi-step synthesis of E. coli O157 RU-PP-Und -- TIMING 6 days.
Once RU-PP-Und has been successfully synthesized in a step-wise manner, an alternative one-pot approach may be applied for further synthesis. In this case, purification, lyophilization and resuspension steps for intermediates can be omitted to simply the procedure.
Prepare crude disaccharide-PP-Und starting from GalNAc-PP-Und as described in steps 121–130.
Transfer the supernatant into a new 15-mL centrifuge tube.
Transfer 48 µL of distilled H2O, 8 µL of Tris-HCl (1 M, pH 8.0), 20 µL of MnCl2 (200 mM), 120 µL of GDP-L-Fuc (100 mM), and 4.4 µL of rSAP (1000 U/mL) into the tube. Mix well by shaking manually after the addition of each component.
-
Adjust pH of the above mixture to pH 7.5 to 8.5.
CRITICAL STEP: Transfer 3 µL of the mixture onto a small piece of pH indicator strip. If the pH value is lower than 7.5, add 1 M NaOH in 10 µL increments to adjust the pH.
-
Take out WbdO from a −20 °C freezer and allow it to thaw on ice. Take out 100 mM GDP-L-Fuc mother solution from a −20 °C freezer and allow it to thaw at room temperature (25 °C).
CRITICAL STEP: Refer to Step 121.
Transfer 200 µL of WbdO (5 mg/mL) into the tube and gently shake the tube to mix well. The total volume is 4.4 mL.
Incubate the mixture and monitor the reaction as described in steps 141–142.
Quench the reaction as described in Steps 129–130.
Transfer the supernatant into a new 15-mL centrifuge tube.
Transfer 148 µL of distilled H2O, 8 µL of Tris-HCl (1 M, pH 8.0), 20 µL of MnCl2 (200 mM), 120 µL of GDP-PerNAc (100 mM), and 4.8 µL of rSAP (1000 U/mL) into the tube. Mix well by shaking manually after the addition of each component.
-
Adjust pH of the above mixture to pH 7.5 to 8.5.
CRITICAL STEP: Transfer 3 µL of the mixture onto a small piece of pH indicator strip. If the pH value is lower than 7.5, add 1 M NaOH in 10 µL increments to adjust the pH.
-
Take out WbdP from a −20 °C freezer and allow it to thaw on ice. Take out 100 mM GDP-PerNAc mother solution from a −20 °C freezer and allow it to thaw at room temperature (25 °C).
CRITICAL STEP: Refer to Step 121.
Transfer 100 µL of WbdP (5 mg/mL) into the tube and gently shake the tube to mix well. The total volume is 4.8 mL.
Incubate the mixture and monitor the reaction as described in steps 141–142.
-
Quench the reaction as described in Steps 129–130.
PAUSE POINT: After lyophilization, the mixture can be sealed with parafilm and stored at −20 °C for up to two weeks or until purification can be performed.
Purify the RU-PP-Und by reverse phase chromatography as described in Steps 105–112.
Lyophilize the fractions to afford E. coli O157 RU-PP-Und as a white powder.
-
Confirm the identity of the isolated product using mass spectrometry.
PAUSE POINT: After lyophilization, the compound can be sealed and stored at −20 °C indefinitely or until the next use.
Development of the Procedure
E. coli O157, the most notorious isolate of Shiga toxin-producing E. coli, has been responsible for the majority of E. coli outbreaks during the past several decades48. Given the worldwide public health concerns associated with such outbreaks, E. coli O157 represents an especially relevant model strain for the analysis of the LPS biosynthetic pathway. The O-PS repeating unit of E. coli O157 consists of four sugar residues (N-acetyl-d-perosamine (PerNAc), L-Fuc, Glc, and GalNAc), which are assembled into a structure of (PerNAc-α1,3-L-Fuc-α1,4-Glc-β1,3-GalNAc-α1,2-)49 (Fig. 1), a structure shared by Salmonella enterica O30 and Citrobacter freundii F90 O-PSs50. The corresponding rfb gene cluster has been sequenced51, and orf1 (wbdN), orf2 (wbdO), and orf6 (wbdP) (Fig. 2) were predicted to encode the GTs needed for RU-PP-Und assembly. WbdN was recently identified as the β1,3-glucosyltransferase36, thus leaving WbdO and WbdP as the presumed α1,4-fucosyltransferase and α1,3-N-acetylpersaminyltransferase. Given that sequence analysis cannot usually predict GT function, it was essential to biochemically identify the remaining two GTs (see Box 1 as an example for GT identification). This approach also applies to any unknown GT involved in the synthesis of RU-PP-Und for the first time.
Figure 2.

E. coli O157 rfb gene cluster. The gene cluster contains genes responsible for assembly of RU-PP-Und (wbdN, wbdO, wbdP), the polymerase gene wzy, the flippase gene wzx, and genes for sugar nucleotide synthesis.
BOX 1. Activity assignment of WbdO and WbdP – TIMING ~ 4 h.
Activity of each unknown GT should be identified before first time use as described in the following procedures.
Using two 0.2-mL PCR tubes, transfer 3 µL of the solution from Step 137 (contains 6 µg of E. coli O157 Disaccharide-PP-Und) into each one.
Transfer 12.6 µL of distilled H2O, 0.4 µL of Tris-HCl (1 M, pH 8.0), 1 µL of MnCl2 (200 mM) and 1 µL of GDP-L-Fuc (100 mM) into each tube. Mix well by pipetting.
To one tube, add 2 µL of WbdO (5 mg/mL), and mix well by pipetting. To the other tube add 2 µL of WbdP (5 mg/mL), and mix well by pipetting.
The total volume of the reaction mixtures is 20 µL, containing 20 mM Tris-HCl (pH 8.0), 2.5 mM Disaccharide-PP-Und, 5 mM GDP-L-Fuc (2 equiv.), 10 mM MnCl2, 1% Triton X-100 (vol/vol), 5% DMSO (vol/vol), and 0.5 mg/mL WbdO or WbdP.
Incubate the reaction mixtures at 37 °C in a Fisher water bath for 3 h.
Quench the reactions by the addition of equal volumes of ice-cold methanol.
Centrifuge at 15,000 × g at room temperature (20 to 25 °C) for 10 min.
-
Analyze the supernatants (5 µL) by ESI-MS under negative mode, searching m/z peaks corresponding to Trisaccharide-PP-Und.
CRITICAL STEP: The enzyme that can catalyze the formation of Trisaccharide-PP-Und is the α1,4-fucosyltransfearse (in this case, WbdO), while the other GT, WbdP is an α1,3-N-acetylpersaminyltransferase. A similar small scale reaction using E. coli O157 Trisaccharide-PP-Und as an acceptor and GDP-PerNAc as a donor should also be performed to confirm the activity of WbdP. The activity of any GT involved in O-PS synthesis can be identified using a similar system with the corresponding Sugar-PP-Und and sugar nucleotide. Calculate the approximate reaction yield and use this information to determine the amount of enzymes to be used in large scale synthetic efforts.
In order to perform this identification, a GalNAc-PP-Und building block had to first be chemically synthesized. Chemical synthesis of GalNAc-PP-Und (10) is initiated by the preparation of GalNAc-1-phosphate (5)24,52,53 and Undecaprenyl-phosphate (8) (Fig. 3). Specifically, GalNAc (1) is first peracetylated using excess acetic anhydride and triethylamine to give peracetylated GalNAc (2). Selective anomeric deprotection of 2 with hydrazine acetate then affords anomerically-deprotected peracetylated GalNAc (3). Treatment of 3 with dibenzyl N,N-diisopropylphosphoramidite in the presence of the acid/nucleophilic catalyst tetrazole yields the dibenzyl-protected phosphate of peracetylated GalNAc (4). Immediately prior to the coupling step needed to form GalNAc-PP-Und, this derivative undergoes hydrogenolysis to form peracetylated GalNAc-1-phosphate as the diisopropylethylammonium salt (5). Similar phosphoramidite chemistry is applied to Und (6) to furnish protected Und-P (7)52 (Fig. 3). However, the specific reagent has to necessarily be altered to the bis(2-cyanoethyl)-N,N-diisopropylphosphoramidite equivalent so as to preserve the isoprene-based olefins in the deprotection step. Use of this phosphoramidite allows deprotection to then proceed via a base-mediated reaction to afford Und-P (8). Finally, coupling of peracetylated GalNAc-1-phosphate and Und-P makes use of a classical pyrophosphate bond forming strategy53. Specifically, compound 5 is first activated for coupling through treatment with 1,1′-carbonyldiimidazole (CDI) (Fig. 3). Addition of the activated species as a solution in tetrahydrofuran (THF) to compound 8 yields peracetylated GalNAc-pyrophosphate-undecaprenyl (9) after a period of three days. Base-catalyzed deprotection of this substrate then completes the chemical synthesis of GalNAc-PP-Und (10).
Figure 3.
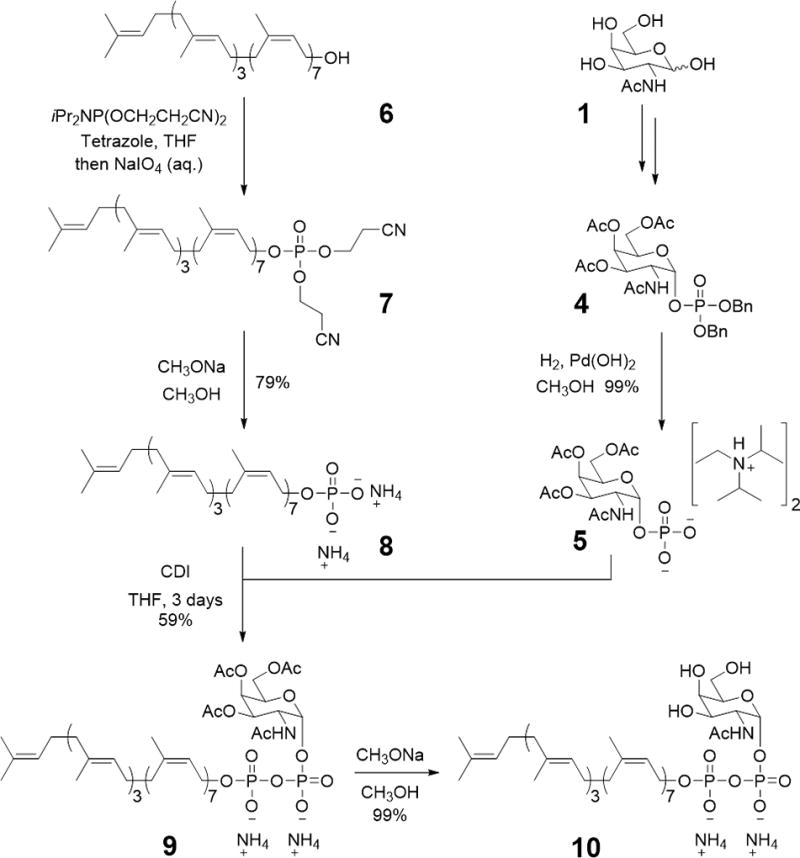
Chemical synthesis of GalNAc-pyrophosphate-undecaprenyl (10) starting with GalNAc (1) and undecaprenol (6).
Upon successful synthesis of GalNAc-PP-Und, we were then able to identify the remaining two GTs and fully assemble the repeating unit donor.
Experimental Design
The protocol described herein details 1) biochemical identification of GTs and 2) how chemically prepared GalNAc-PP-Und can be synthesized and then utilized to generate E. coli O157 RU-PP-Und using the three GTs in a step-wise manner (Fig. 4). A full overview of this experimental approach can be seen as a flowchart in Figure 5.
Figure 4.

Experimental design for the enzymatic synthesis of E. coli O157 RU-PP-Und. Shrimp alkaline phosphatase (rSAP) is added to drive reactions forward by digesting byproduct nucleoside-diphosphates. See REAGENT SETUP for details of Box 1.
Figure 5.

Flow diagram for chemoenzymatic synthesis of RU-PP-Und.
Chemical Synthesis of GalNAc-PP-Und (Steps 1–120)
The three-step synthesis of the dibenzyl-protected phosphate of peracetylated GalNAc from GalNAc can be readily performed on a gram-scale as described herein. However, should more material be needed for numerous coupling reactions, the scale of these steps can be adjusted without significant adjustments in experimental protocol. We do not recommend, however, adjusting the scale of the subsequent step in which peracetylated GalNAc-1-phosphate is formed via hydrogenolysis. This approach allowed us to avoid large-scale hydrogenations while still providing us with sufficient material for each coupling reaction as the lipid-phosphate was typically available in limiting quantities. Larger scale production of the lipid-phosphate as well as the coupled product, peracetylated GalNAc-PP-Und, will require further optimization of the corresponding reactions. Nevertheless, we have been able to apply this synthetic approach to various GalNAc-PP-Lipid and GlcNAc-PP-Lipid analogs which incorporate the following lipids: Solanesol, Heptaprenol, Pentaprenol, cis-Pentaprenol, Mono-Saturated Pentaprenol, and Farnesol. Use of these lipids and/or a different sugar residue did not noticeably alter reaction yields.
Enzymatic Synthesis of E. coli O157 RU-PP-Und (Boxes 1 & 2 and Steps 121–158)
As shown in Figure 5 with identified glycosyltransferases, enzymatic synthesis of RU-PP-Und and analogues can be carried out either in a step-wise manner (Steps 121–158) or in a one-pot manner (Box 2). However, for first time synthesis, it is recommended that one adopts the step-wise manner to gain familiarity with each enzyme catalyzed reaction (e.g., how long each reaction takes, and how much enzyme is needed). Milligrams of RU-PP-Und were synthesized in this protocol, an amount which is typically sufficient for biological applications. However, should more material be needed, the enzyme catalyzed reaction can simply be scaled up (up to 200 mg product) with appropriate adjustment of enzyme quantities and reaction times. In addition, using this protocol, various RU-PP-Lipids can be readily prepared by simply replacing the starting material (GalNAc-PP-Und) with the desired GalNAc-PP-Lipid. Certain steps may be changed or optimized for this purpose (e.g., DMSO and Trition X-100 can be omitted when lipids are highly soluble). We have been able to prepare various RU-PP-Lipid analogues which include the following lipids: Solanesol, Heptaprenol, Pentaprenol, cis-Pentaprenol, Mono-Saturated Pentaprenol, and Farnesol.
MATERIALS
REAGENTS
Acetic anhydride (Sigma-Aldrich, cat. no. 242845)
Acetone (Fisher Scientific, cat. no. A18-4)
Acetonitrile, anhydrous (Sigma-Aldrich, cat. no. 271004)
Ammonium bicarbonate (Sigma-Aldrich, cat. no. A6141)
Bis(2-cyanoethyl)-N,N-diisopropylphosphoramidite (Sigma-Aldrich, cat. no. 766305)
C-18 Reversed phase silica gel (Sigma-Aldrich, cat. no. 60757)
1,1'-Carbonyldiimidazole (Alfa Aesar, cat. no. A14688)
Celite (Sigma-Aldrich, cat. no. 06858)
Chloroform (Fisher Scientific, cat. no. C298-4)
Chloroform-d (Sigma-Aldrich, cat. no. 151823)
Cis-Farnesol (Sigma-Aldrich, cat. no. 80720)
DEAE-cellulose (Sigma-Aldrich, cat. no. D3764)
Dibenzyl N,N-diisopropylphosphoramidite (Sigma-Aldrich, cat. no. 416436)
Dichloromethane (Fisher Scientific, cat. no. D37-4)
Dichloromethane, anhydrous (Sigma-Aldrich, cat. no. 270997)
E. coli O157 anti-serum (Pro-Lab Diagnostics, cat. no. PL.6250)
Ethyl acetate (Fisher Scientific, cat. no. E124-20)
Farnesol (Sigma-Aldrich, cat. no. F203)
Glacial acetic acid (Fisher Scientific, cat. no. A38-212)
Guanosine 5'-diphospho-β-L-fucose disodium salt (GDP-L-Fuc) (Carbosynth, cat. no. MG01912)
Hexanes (Fisher Scientific, cat. no. H292-4)
Hydrazine acetate (Sigma-Aldrich, cat. no. 259748)
1 N Hydrochloric acid (Sigma-Aldrich, cat. no. H9892)
Manganese (II) chloride (MnCl2) (Sigma-Aldrich, cat. no. 244589)
Methanol (Fisher Scientific, cat. no. A412-4)
Methanol, anhydrous (Sigma-Aldrich, cat. no. 322415)
Methyl sulfoxide (DMSO) (Sigma-Aldrich, cat. no. W387520)
m-Chloroperoxybenzoic acid (Sigma-Aldrich, cat. no. 273031)
N-Acetyl-d-galactosamine (Sigma-Aldrich, cat. no. A2795)
N,N-Diisopropylethylamine (Sigma-Aldrich, cat. no. 387649)
N,N-Dimethylformamide, anhydrous (Sigma-Aldrich, cat. no. 227056)
Palladium hydroxide on carbon (Sigma-Aldrich, cat. no. 212911)
pH-indicator strips pH 5.0–10.0 (EMD-Millipore, cat. no. 1.09533.0001)
Proteinase K (Sigma, cat. no. P2308)
Pyridine (Sigma-Aldrich, cat. no. 270970)
Shrimp Alkaline Phosphatase (rSAP) (New England Biolabs, cat. no. M0371)
Sodium bicarbonate (Sigma-Aldrich, cat. no. S6014)
Sodium chloride (Sigma-Aldrich, cat. no. S3014)
1 N Sodium hydroxide (Sigma-Aldrich, cat. no. 72082)
Sodium periodate (Sigma-Aldrich, cat. no. 311448)
Sodium sulfate (Sigma-Aldrich, cat. no. 798592)
Sodium sulfite (Sigma-Aldrich, cat. no. S0505)
Tetrahydrofuran (Sigma-Aldrich, cat. no. 401757)
Tetrazole (Sigma-Aldrich, cat. no. 17234)
Triethylamine (Sigma-Aldrich, cat. no. T0886)
UltraPure 1M Tris-HCI, pH 7.5 (Life Technologies, cat. no. 15567-027)
UltraPure 1M Tris-HCI, pH 8.0 (Life Technologies, cat. no. 15568-025)
Triton X-100 (Sigma-Aldrich, cat. no. X100)
UltraPure DNase/RNase-Free Distilled Water (Life Technologies, cat. no. 10977)
Undecaprenol (American Radiolabeled Chemicals, cat. no. ARCD 0126)
Uridine 5’-diphosphoglucose disodium salt hydrate (UDP-Glc) (Sigma-Aldrich, cat. no. U4625)
EQUIPMENT
Benchtop centrifuge (Eppendorf, cat. no. 5415 D)
Bransonic Tabletop Cleaner (Branson, cat. no. 100-951-010)
Buchi rotary evaporator (Fisher Scientific, cat. no. 04-987-222)
Chromatography columns (Chemglass Life Sciences, cat. no. CG-1192-04/07/16)
Discovery balance DV215CD (Ohaus, cat. no. 80104139)
Freeze Dryer (Labconco, cat. no. 7753000)
Isotemp economy analog-control water bath Model 115 (Fisher, cat. no. 15-460-15Q)
REAGENT SETUP
Sugar nucleotides
The O-PS of E. coli O157 contains common sugars Glc and L-Fuc (corresponding sugar nucleotides UDP-Glc and GDP-L-Fuc are commercially available) and one rare sugar residue PerNAc, the sugar nucleotide form of which is GDP-PerNAc. GDP-PerNAc is prepared as previously described (see Supplementary Method 1). UDP-Glc, GDP-L-Fuc and GDP-PerNAc should all be dissolved in distilled H2O to a final concentration of 100 mM and stored at −20 °C. For the synthesis of RU-PP-Und from other bacteria, most common sugar nucleotides are commercially available, and synthesis of uncommon sugar nucleotide has been reviewed elsewhere34,35. CRITICAL STEP: GDP-L-Fuc is less stable than other sugar nucleotides. Avoid storing at −20 °C for over 3 months. Unused GDP-L-Fuc should be lyophilized for long-term storage.
Enzymes
The β1,3-glycosyltransferase WbdN, and the other two putative GTs (WbdO and WbdP) are cloned into expression vectors by using a standard cloning approach, and the vectors are transformed into E. coli BL21(DE3) for overexpression and purification (please refer to Supplementary Method 2 for detailed experiments). Given that most GTs involved in the assembly of RU-PP-Und are membrane-bound proteins that tend to precipitate, it is recommended that 0.1–0.5% of Triton X-100 (vol/vol) or/and 5–20% glycerol (vol/vol) be utilized during purification to obtain soluble forms of these enzymes. High concentrations of Trition X-100 should be removed, however, prior to performing reactions in the synthesis of RU-PP-Und. If the soluble form of certain GTs is still not accessible, it is recommended that a fusion protein strategy be applied. For example, WbdO can be fused to maltose binding protein (MBP) for soluble expression (Supplementary Method 2). Before use of the enzymes for synthetic steps in the Procedure, the activity of all putative GTs should be checked via small scale reactions as described in Box 1. E. coli O157 WaaL (the LPS ligase) is identical to that of E. coli O86, which was previously successfully overexpressed and purified in our laboratory18. Shrimp Alkaline phosphatase (rSAP) is commercially available in a concentration of 1000 U/mL. All enzymes are stored at −20 °C with 20% glycerol (vol/vol) and appropriate buffers for long-term use.
Lipid A core
The Lipid A core structure was previously prepared in our laboratory18, and should be dissolved to a final concentration of 40 µM using distilled H2O. CAUTION: Given that Lipid A is an endotoxin, one should avoid contact with any wounds. CRITICAL STEP: If visible particles are observed, sonicate for up to 20 min to promote dissolving of Lipid A core.
EQUIPMENT SETUP
Preparation of a DEAE-cellulose ion-exchange column
Add 100 g of DEAE-cellulose to a Buchner funnel and use vacuum filtration to wash the DEAE-cellulose with 170 mL of 1 M HCl (aq.), 170 mL of deionized H2O, 170 mL of 1 M NaOH (aq.), and 170 mL of deionized H2O. Repeat this washing process two additional times. Wash the DEAE-cellulose with 300 mL of CH3OH. Dry the DEAE-cellulose for at least 24 h in a desiccator placed under vacuum. Add the dried DEAE-cellulose to a large beaker and add excess glacial acetic acid (the acid level should remain above that of the DEAE-cellulose). Allow the DEAE-cellulose to soak for at least 24 h. Thoroughly swirl the beaker and pour a portion of the slurry into a chromatography column (¾ inch ID × 12 inch E.L.). Allow the slurry to settle until a bed of DEAE-cellulose of at least 6 inches is formed. Wash the column with deionized H2O until a neutral pH is reached. Wash the column with 50 mL each of CH3OH, 1:1 CH3OH/CHCl3, and 1:3 CH3OH/CHCl3 (vol:vol). CAUTION: Acetic acid is a known irritant and is corrosive. Personal protective equipment including goggles and gloves should be worn when it is used. CRITICAL STEP: Prepare the DEAE-cellulose column immediately before use.
Preparation of a silica gel column
Add silica gel to a chromatography column (1 ½ inch ID × 18 inch E.L.; the amount of silica gel and column size used is dependent on the scale of the particular reaction) to a height of at least 7 inches. Pack the column under pressure using a solvent appropriate for the given reaction (first concentration of a series). CRITICAL STEP: Prepare immediately before use.
Preparation of a C-18 reverse phase column
Add silica gel 60 RP-18 to a chromatography column (½ inch ID × 8 inch E.L.) to a height of at least 5 inches. Pack the column under pressure using CH3OH as the eluent. Wash the column with 10 mL of deionized H2O and repeat two additional times. CRITICAL STEP: Prepare immediately before use.
Preparation of the p-anisaldehyde staining solution
Add a stir bar and 200 mL of absolute ethanol to a 500-mL Erlenmeyer flask. Cool the ethanol to 0 °C and stir at this temperature for 5 min. Carefully add 2.5 mL of glacial acetic acid and 5.5 mL of p-anisaldehyde to the flask. Using a 60-mL separatory funnel, slowly add 7.5 mL of concentrated sulfuric acid dropwise over a period of 5 minutes. Continue to stir the solution for 5 min, after which it can transferred to an amber storage vial and be stored at 4 °C for up to three months. CAUTION: Preparation of the staining solution requires the handling of concentrated acids. Personal protective equipment including goggles and gloves should be worn when it is used.
Mass spectrometry
In this protocol, all electrospray ionization mass spectral data was collected using a Shimadzu LCMS-2010A liquid chromatograph mass spectrometer under negative mode with direct infusion. Other mass spectrometers could also be utilized.
PROCEDURE
Chemical synthesis of undecaprenyl phosphate – TIMING 4 days
-
1)
Charge a 10-mL round bottom flask with 30 mg of undecaprenol and seal it with a septum.
CRITICAL STEP: For the synthesis of other GalNAc-PP-Lipids, simply replace Und with same number of moles (39 µmol) of different Lipids.
-
2)
Briefly place the flask under vacuum to evacuate the flask, fill the flask with argon using an argon-filled balloon, and then repeat this evacuation/filling process two additional times.
-
3)
Add 13 mg of bis(2-cyanoethyl)-N,N-diisopropylphosphoramidite and cool the reaction to 0 °C.
-
4)
Add 11 mg of tetrazole and stir the reaction at 0 °C for 5 min.
-
5)
Warm the reaction to room temperature (25 °C) and stir for an additional 1 h.
-
6)
Cool the reaction to 0 °C and add 10 µL of pyridine and 17 mg of NaIO4 sequentially.
CRITICAL STEP: It is imperative to cool the flask prior to addition of these reagents to avoid a sudden increase in temperature and concomitant decrease in overall yield.
-
7)
Stir the reaction for 15 min at 0 °C.
-
8)
Warm the reaction to room temperature and stir for an additional 1 h.
-
9)
Dilute the reaction with 10 mL of ethyl acetate and transfer the solution into a 60-mL separatory funnel.
-
10)
Wash the organic fraction successively with 10 mL each of deionized H2O, 5% Na2SO3 (wt/vol) and saturated NaCl (aq.).
-
11)
Transfer the organic fraction into a 25-mL Erlenmeyer flask and dry the organic layer over Na2SO4 (a quantity sufficient to yield freely moving salt granules) for 10 min.
-
12)
Filter the organic fraction through a funnel containing a cotton plug into a 25-mL round bottom flask.
-
13)
Remove the solvent in vacuo using a water bath temperature of 25 °C.
-
14)
Dissolve the residue in 10 mL of 1% NaOCH3 (wt/vol) in CH3OH solution.
-
15)
Stir the reaction at room temperature for 3 days.
-
16)
Remove the solvent in vacuo using a water bath temperature of 25 °C.
PAUSE POINT: The 25-mL round bottom flask can be sealed with a septum and stored at −20 °C for one week or until the purification can be performed.
Purification of undecaprenyl phosphate – TIMING ~2 h
-
17)
Dissolve the residue from Step 16 in 1 mL of CH3OH.
-
18)
Open the stopcock of a DEAE-cellulose ion-exchange column (0.75 inch ID × 12 inch E.L.) to allow the solvent used during packing to reach the top of the ion-exchange bed.
-
19)
Slowly add the solution from Step 17 to the top of the column, allowing the solution to flow into the column until it reaches the top of the ion-exchange bed.
-
20)
Rinse the round bottom flask with an additional 0.5 mL of CH3OH and slowly add this solution to the top of the column, allowing the solution to flow into the column until it reaches the top of the ion-exchange bed.
-
21)
Add 100 mL of 1:3 CH3OH:CHCl3 (vol:vol) to the column to begin the elution. Collect the eluent over 20 10-mL test tubes.
-
22)
Add 100 mL of a 30 mM NH4HCO3 in CH3OH solution and continue the elution. Collect the eluent over 20 10-mL test tubes.
-
23)
Dilute 5 µL of each fraction in 0.5 mL of CH3OH and analyze each sample using mass spectrometry under negative mode to discern the location of the desired product (m/z = 846).
-
24)
Combine the product-containing fractions into a 50-mL round bottom flask.
-
25)
Remove the solvent in vacuo using a water bath temperature of 25 °C to afford Und-P as a white powder. A representative yield is 26 mg (79%). TROUBLESHOOTING
PAUSE POINT: The 25-mL round bottom flask can be sealed with a septum and stored at 4 °C indefinitely or until the subsequent coupling step can be performed.
Preparation of peracetylated GalNAc – TIMING ~ 5 ½ h
CRITICAL: Currently, quite a few peracetylated monosaccharides are commercially available at reasonable prices including peracetylated GalNAc (CAS No: 76375-60-5, $390/10 g from Carbosynth Limited) and peracetylated GlcNAc (CAS No: 7772-79-4, $80/10 g from Carbosynth Limited). If commercial peracetylated monosaccharides are used, Steps 26–48 can be skipped.
-
26)
Charge a 100-mL round bottom flask with 0.900 g of N-Acetyl-d-galactosamine and a stir bar.
-
27)
Seal the flask with a septum and briefly place it under vacuum to evacuate the flask.
-
28)
Fill the flask with N2 using an N2-filled balloon and then repeat this evacuation/filling process two additional times.
-
29)
Add 25 mL of anhydrous CH3CN and 6.65 mL of triethylamine, and then cool the reaction to 0 °C.
-
30)
Add 6.65 mL of acetic anhydride dropwise and stir the reaction at 0 °C until the solution becomes clear (~ 2 h).
-
31)
Warm the reaction to room temperature and stir for an additional 2 h.
-
32)
Cool the reaction to 0 °C and add 9.1 mL of CH3OH.
-
33)
Stir the reaction for 30 min at 0 °C.
-
34)
Remove the solvent in vacuo using a water bath temperature of 25 °C and then dissolve the residue in 50 mL of CH2Cl2.
-
35)
Transfer the contents of the flask to a 250-mL separatory funnel.
-
36)
Wash the organic fraction successively with 25 mL of 1 M HCl (aq.), 2×25 mL of 5% NaHCO3 (aq.), and 25 mL of saturated NaCl (aq.).
-
37)
Transfer the organic fraction into a 250-mL Erlenmeyer flask and dry the organic layer over Na2SO4 (a quantity sufficient to yield freely moving salt granules) for 10 min.
-
38)
Filter the organic fraction through a funnel containing a cotton plug into a 100-mL round bottom flask.
-
39)
Remove the solvent in vacuo using a water bath temperature of 25 °C.
PAUSE POINT: The 250-mL round bottom flask can be sealed with a septum and stored at 4 °C indefinitely or until the subsequent reaction can be performed.
Purification of peracetylated GalNAc – TIMING ~ 2 h
-
40)
Dissolve the residue from Step 39 in 5 mL of 2:1 hexane:acetone (vol:vol).
-
41)
Open the stopcock of a silica gel column (1 ½ inch ID × 18 inch E.L.) to allow the solvent used during packing (2:1 hexane:acetone (vol:vol)) to reach the top of the silica gel bed.
-
42)
Slowly add the solution from Step 40 to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed
-
43)
Rinse the round bottom flask with an additional 2 mL of 2:1 hexane:acetone (vol:vol) and slowly add this solution to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed.
-
44)
Add 100 mL of 2:1 hexane:acetone (vol:vol) to the column to begin the elution. Collect the eluent over 15 10-mL test tubes.
-
45)
Add 100 mL of 1:1 hexane:acetone (vol:vol) and continue the elution. Collect the eluent over 15 10-mL test tubes.
-
46)
Analyze the fractions using Thin Layer Chromatography (25 mm × 75 mm plate) with a 1:1 hexane:acetone (vol:vol) mobile phase. Stain the TLC plates with a p-anisaldehyde solution and apply heat via a heat gun to discern the location of the desired product (brown spot at R = 0.25).
CAUTION: The staining solution contains concentrated acids and is thus corrosive. Personal protective equipment including goggles and gloves should be worn when it is used.
-
47)
Combine the product-containing fractions into a 100-mL round bottom flask.
-
48)
Remove the solvent in vacuo using a water bath temperature of 25 °C to afford peracetylated GalNAc as a white foam. A representative yield is 1.450 g (92%).
PAUSE POINT: The 100-mL round bottom flask can be sealed with a septum and stored at 4 °C indefinitely or until the subsequent reaction can be performed.
Preparation of anomerically-deprotected peracetylated GalNAc – TIMING ~ 1 h
-
49)
Charge a 100-mL round bottom flask with 1.450 g of peracetylated GalNAc and a stir bar.
-
50)
Add 20 mL of anhydrous DMF and 0.412 g of hydrazine acetate.
-
51)
Stir the reaction for 40 min at room temperature.
-
52)
Remove the solvent in vacuo using a water bath temperature of 25 °C.
PAUSE POINT: The 100-mL round bottom flask can be sealed with a septum and stored at 4 °C indefinitely or until the subsequent reaction can be performed.
Purification of anomerically-deprotected peracetylated GalNAc – TIMING ~ 2 h
-
53)
Dissolve the residue from Step 52 in 5 mL of 1:1 hexane:acetone (vol:vol).
-
54)
Open the stopcock of a silica gel column (1 ½ inch ID × 18 inch E.L.) to allow the solvent used during packing (1:1 hexane:acetone (vol:vol)) to reach the top of the silica gel bed.
-
55)
Slowly add the solution from Step 53 to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed
-
56)
Rinse the round bottom flask with an additional 2 mL of 1:1 hexane:acetone (vol:vol) and slowly add this solution to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed.
-
57)
Add 100 mL of 1:1 hexane:acetone (vol:vol) to the column to begin the elution. Collect the eluent over 15 10-mL test tubes.
-
58)
Add 100 mL of 1:2 hexane:acetone (vol:vol) and continue the elution. Collect the eluent over 15 10-mL test tubes.
-
59)
Analyze the fractions using Thin Layer Chromatography (25 mm × 75 mm plate) with a 1:1 hexane:acetone (vol:vol) mobile phase. Stain the TLC plates with a p-anisaldehyde solution and apply heat via a heat gun to discern the location of the desired product (brown spot at R = 0.2).
CAUTION: The staining solution contains concentrated acids and is thus corrosive. Personal protective equipment including goggles and gloves should be worn when it is used.
-
60)
Combine the product-containing fractions into a 100-mL round bottom flask.
-
61)
Remove the solvent in vacuo using a water bath temperature of 25 °C to afford anomerically-deprotected peracetylated GalNAc as a white foam. A representative yield is 1.031 g (80%).
PAUSE POINT: The 100-mL round bottom flask can be sealed with a septum and stored at 4 °C indefinitely or until the subsequent reaction can be performed.
Preparation of the dibenzyl-protected phosphate of peracetylated GalNAc – TIMING ~ 18 h
-
62)
Charge a 100-mL round bottom flask with 1.031 g of anomerically-deprotected peracetylated GalNAc, 2.080 g of tetrazole, and a stir bar.
-
63)
Seal the flask with a septum and briefly place it under vacuum to evacuate the flask.
-
64)
Fill the flask with N2 using an N2-filled balloon and then repeat this evacuation/filling process two additional times.
-
65)
Add 35 mL of CH2Cl2 and cool the reaction to −40 °C.
-
66)
Add 4.99 mL of dibenzyl N,N-diisopropylphosphoramidite dropwise over a period of 5 min.
-
67)
Stir the reaction for 3 h as it warms to room temperature.
-
68)
Cool the reaction to −78 °C and add 5.125 g of mCPBA in two portions.
-
69)
Allow the reaction to warm to room temperature overnight.
-
70)
Transfer the contents of the flask to a 125-mL separatory funnel.
-
71)
Wash the organic fraction successively with 15 mL each of saturated Na2SO3 (aq.), deionized H2O, and saturated NaCl (aq.).
-
72)
Transfer the organic fraction into a 125-mL Erlenmeyer flask and dry the organic layer over Na2SO4 (a quantity sufficient to yield freely moving salt granules) for 10 min.
-
73)
Filter the organic fraction through a funnel containing a cotton plug into a 100-mL round bottom flask.
-
74)
Remove the solvent in vacuo using a water bath temperature of 25 °C.
PAUSE POINT: The 100-mL round bottom flask can be sealed with a septum and stored at 4 °C overnight.
Purification of the dibenzyl-protected phosphate of peracetylated GalNAc – TIMING ~ 2 ½ h
-
75)
Dissolve the residue from Step 74 in 5 mL of CH2Cl2.
-
76)
Open the stopcock of a silica gel column (1 ½ inch ID × 18 inch E.L.) to allow the solvent used during packing (CH2Cl2) to reach the top of the silica gel bed.
-
77)
Slowly add the solution from Step 75 to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed
-
78)
Rinse the round bottom flask with an additional 2 mL of CH2Cl2 and slowly add this solution to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed.
-
79)
Add 100 mL of CH2Cl2 to the column to begin the elution. Collect the eluent over 15 10-mL test tubes.
-
80)
Add 100 mL of a 1% (vol/vol) CH3OH in CH2Cl2 solution and continue the elution. Collect the eluent over 15 10-mL test tubes.
-
81)
Add 100 mL of a 2% (vol/vol) CH3OH in CH2Cl2 solution and continue the elution. Collect the eluent over 15 10-mL test tubes.
-
82)
Analyze the fractions using Thin Layer Chromatography (25 mm × 75 mm plate) with a 2% CH3OH in CH2Cl2 mobile phase. Stain the TLC plates with a p-anisaldehyde solution and apply heat via a heat gun to discern the location of the desired product (brown spot at R = 0.2).
CAUTION: The staining solution contains concentrated acids and is thus corrosive. Personal protective equipment including goggles and gloves should be worn when it is used.
-
83)
Combine the product-containing fractions into a 100-mL round bottom flask.
-
84)
Remove the solvent in vacuo using a water bath temperature of 25 °C to afford the dibenzyl-protected phosphate of peracetylated GalNAc as a white foam. A representative yield is 1.262 g (70%).
PAUSE POINT: The 100-mL round bottom flask can be sealed with a septum and stored at 4 °C indefinitely or until the hydrogenolysis reaction can be performed. The scale of these first three reactions provides sufficient material to perform several iterations of the subsequent reactions which may make use of Und-P (8) or other lipid phosphates.
Preparation of peracetylated GalNAc-1-phosphate – TIMING ~ 2 h
-
85)
Charge a 25-mL round bottom flask with 15 mg of 20% Pd(OH)2 (wt/vol) on carbon and a stir bar.
-
86)
Seal the flask with a septum and briefly place it under vacuum to evacuate the flask.
-
87)
Fill the flask with N2 and then repeat this evacuation/filling process two additional times.
-
88)
After moistening the catalyst with a minimal amount of ethyl acetate, slowly add 4.5 mL of CH3OH to the sealed flask.
-
89)
Dissolve 90 mg of the dibenzyl-protected phosphate of peracetylated GalNAc with 0.5 mL of CH3OH and add this solution to the flask.
-
90)
Evacuate the flask and then fill it with H2 using an H2-filled balloon.
CAUTION: Hydrogen gas is highly flammable.
-
91)
Repeat Step 90 two additional times.
-
92)
Stir the reaction for 30 min at room temperature.
-
93)
Carefully remove the balloon and bubble N2 through the solution.
CAUTION: Hydrogen gas is highly flammable.
-
94)
Add 0.1 mL of N,N-diisopropylethylamine and 5 mL of CH3OH, and allow the reaction to stir for 30 min.
-
95)
Filter the solution through a syringe containing a pad of Celite into a 25-mL round bottom flask.
CAUTION: Be sure to not filter to dryness as this represents a flammability hazard. After filtration is completed, several mL of H2O should be added to the filter before disposing of it in a specified waste container.
-
96)
Remove the solvent in vacuo using a water bath temperature of 25 °C to afford the crude peracetylated GalNAc-1-phosphate which will be utilized directly in the coupling reaction.
PAUSE POINT: The 25-mL round bottom flask can be sealed with a septum and stored at 4 °C indefinitely or until the coupling step can be performed.
Preparation of GalNAc-PP-Und – TIMING 7 days
CRITICAL: These steps can also be used to prepare Solanesol, Heptaprenol, Pentaprenol, cis-Pentaprenol, Mono-Saturated Pentaprenol, and Farnesol-based products.
-
97)
Charge a 25-mL round bottom flask with 23 mg of 1,1'-carbonyldiimidazole (CDI) and 23 mg of peracetylated GalNAc-1-phosphate, sealing it with a septum.
-
98)
Briefly place the flask under vacuum to evacuate the flask, fill the flask with argon using an argon-filled balloon, and then repeat this evacuation/filling process two additional times.
-
99)
Add 3 mL of anhydrous THF and stir the reaction at room temperature for 2 h.
CRITICAL STEP: Formation of CDI-activated peracetylated GalNAc-1-phosphate can be confirmed by removing 5 µL of the reaction solution, diluting it with 0.5 mL CH3OH, and analyzing the sample using mass spectrometry under negative mode (m/z = 476).
-
100)
Add 42 µL of anhydrous CH3OH and stir the reaction at room temperature for 1h.
CRITICAL STEP: This step will remove the excess CDI to prevent a reaction between CDI and undecaprenyl phosphate upon its introduction.
-
101)
Remove the solvent in vacuo using a water bath temperature of 25 °C to afford CDI-activated peracetylated GalNAc-1-phosphate
CRITICAL STEP: CDI-activated peracetylated GalNAc-1-phosphate should be used immediately in the next step.
-
102)
Add 3 mL of anhydrous THF to the residue and transfer this solution to a separate 25-mL round bottom flask (sealed with a septum and flushed with argon) containing 23 mg of undecaprenyl phosphate. TROUBLESHOOTING
-
103)
Stir the reaction at room temperature for 3 days.
-
104)
Remove the solvent in vacuo using a water bath temperature of 25 °C.
-
105)
Add 1 mL of H2O to the residue from Step 104. The solution will be cloudy in appearance.
-
106)
Open the stopcock of a C-18 reverse phase column (0.5 inch ID × 8 inch E.L.) to allow the water used during packing to reach the top of the silica gel bed.
-
107)
Slowly add the solution from Step 105 to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed.
-
108)
Rinse the round bottom flask with an additional 0.5 mL of H2O and slowly add this solution to the top of the column, allowing the solution to flow into the column until it reaches the top of the silica gel bed.
-
109)
Add 10 mL of 1.7% NH4HCO3 (aq.) (wt/vol) to the column to begin the elution. Collect the eluent over two 10-mL test tubes.
-
110)
Add 10 mL of a 5% solution of isopropanol (vol:vol) in 1.7% NH4HCO3 (aq.) (wt/vol) and continue the elution. The amount of isopropanol should be increased by 5% for each 10 mL of eluent used until a 50% solution is used. Collect the eluent over two 10-mL test tubes for each 10 mL of eluent used.
CRITICAL STEP: Isopropanol is a suitable organic solvent for the purification of Undecaprenol and Solanesol-based products, while acetonitrile is a suitable organic solvent for Heptaprenol, Pentaprenol, cis-Pentaprenol, Mono-Saturated Pentaprenol, and Farnesol-based products.
-
111)
Dilute 5 µL of each fraction in 0.5 mL of CH3OH and analyze each sample using mass spectrometry under negative mode to discern the location of the desired product (m/z = 1254).
-
112)
Combine the product-containing fractions into a 50-mL round bottom flask.
-
113)
Lyophilize the fractions to afford peracetylated GalNAc-PP-Und as a white powder. A representative isolated yield is 20 mg (59%).
-
114)
Due to the amphiphilic nature of GalNAc-PP-Und, 1H, 13C, and 31P-NMR characterization should be performed on the peracetylated precursor isolated in Step 113. Mass spectrometry analysis can be performed on both molecules.
-
115)
Add 3 mL of a 0.1% NaOCH3 in CH3OH solution (wt/vol) to a 10-mL round bottom flask containing the white powder obtained in Step 113.
-
116)
Seal the flask with a septum and stir the reaction at room temperature for 40 min.
-
117)
Remove the solvent in vacuo using a water bath temperature of 25 °C.
-
118)
Repeat Steps 105–112 to ensure purity of the material obtained in step 117.
-
119)
Lyophilize the fractions to afford GalNAc-PP-Und as a white powder. A representative isolated yield is 18 mg (99%).
-
120)
Confirm the identity of the isolated product using mass spectrometry.
PAUSE POINT: The lyophilized GalNAc-PP-Und can be sealed and stored at −20 °C indefinitely or until the enzymatic reactions can be performed.
Preparation of Disaccharide-PP-Und – TIMING 3 days
CRITICAL: Prior to performing large scale syntheses, it is highly recommended that small scale reactions (e.g., 20 µL total volume reaction) be performed to confirm the activity of each enzyme. For the synthesis of RU-PP-Lipids, simply replace GalNAc-PP-Und with the same number of moles of different GalNAc-PP-Lipids.
-
121)
Take out WbdN from a −20 °C freezer and allow it to thaw on ice. Take out the 100 mM UDP-Gal from a −20 °C freezer and allow it to thaw at room temperature.
CRITICAL STEP: WbdN should be stored in 0.1 mL aliquots at −20 °C to avoid repeated freezing and thawing.
-
122)
Add 11.7 mg (10 µmol) of chemically synthesized GalNAc-PP-Und to a 15-mL centrifuge tube.
-
123)
Transfer 200 µL of DMSO and 400 µL of 10% (vol/vol) Triton X-100 into the tube. Vortex the tube for 1 min, and then place the tube into the water bath (room temperature) of the Bransonic Tabletop Cleaner. Sonicate to completely dissolve GalNAc-PP-Und. The sonication may take up to 30 min.
CRITICAL STEP: Be sure to make 10% Trition X-100 first and add this solution. Do not add pure Trition X-100 and distilled H2O. Monitor the dissolution of GalNAc-PP-Und to ensure that there are no visible particles. For GalNAc-PP-Lipids with high water solubility, for example GalNAc-PP-Farnesyl, use H2O to dissolve and skip this step.
-
124)
Consecutively transfer 2.9 mL of distilled H2O, 80 µL of Tris-HCl (1 M, pH 8.0), 200 µL of MnCl2 (200 mM), 120 µL of UDP-Glc (100 mM), and 4 µL of rSAP (1000 U/mL) into the tube. Mix well by shaking manually after the addition of each component.
CRITICAL STEP: The reaction can be scaled down or up by volume (200 µL to 20 mL) as long as the final concentration of each component is kept consistent. For GalNAc-PP-Lipids with high water solubility, DMSO and Trition X-100 can be omitted.
-
125)
Transfer 100 µL of WbdN (5 mg/mL) into the tube and gently shake the tube to mix well.
-
126)
The total volume of the reaction mixture is 4 mL, containing 20 mM Tris-HCl (pH 8.0), 2.5 mM GalNAc-PP-Und, 3 mM UDP-Glc (1.2 equiv.), 10 mM MnCl2, 1% Triton X-100 (vol/vol), 5% DMSO (vol/vol), 1 U/mL rSAP and 0.125 mg/mL WbdN.
-
127)
Incubate the reaction mixture at 37 °C in a Fisher water bath for 6 h with occasional gentle shaking.
-
128)
Monitor the reaction by mass spectrometry. Take out 1 µL of the reaction mixture and mix with 9 µL of 1:1 H2O:CH3OH (vol:vol) in an 1.5-mL Eppendorf tube. Centrifuge the mixture at 15,000 × g at room temperature for 5 min with an Eppendorf centrifuge, and inject 5 µL of the supernatant into an ESI-MS for analysis under negative mode. Starting material (GalNAc-PP-Und) is typically consumed within 6 h depending upon the activity of the corresponding GT. If not, monitor the reaction every 6 h until totally consumed. TROUBLESHOOTING.
CRITICAL STEP: Check the mass spectrum for m/z values corresponding to substrate and product with both single and double charges (Fig. 6).
-
129)
When the starting material disappears or corresponding m/z values become minute compared with that of the product, quench the reaction by boiling for 5 min in a water bath.
-
130)
Transfer the reaction mixture to four 1.7-mL Eppendorf tubes, and centrifuge at 15,000 × g at room temperature for 10 min.
-
131)
Transfer the supernatant into a 15-mL centrifuge tube, and lyophilize the mixture using a Freeze Dryer.
Figure 6.

Step-wise enzymatic synthesis of E. coli O157 RU-PP-Und. Scheme and yields (a) and mass spectra (b–d) are included.
PAUSE POINT: After lyophilization, the tube can be sealed with parafilm and stored at −20 °C for up to two weeks or until purification is performed.
-
132)
Purify the Disaccharide-PP-Und by reverse-phase chromatography as described in Steps 105–112.
-
133)
Lyophilize the fractions to afford E. coli O157 Disaccharide-PP-Und as a white powder.
-
134)
Confirm the identity of the isolated product using mass spectrometry.
PAUSE POINT: After lyophilization, the compound can be sealed and stored at −20 °C indefinitely or until the next step can be performed.
Preparation of Trisaccharide-PP-Und – TIMING 3 days
As identified in Box 1, WbdO is responsible for the addition of the L-Fuc to trisaccharide-PP-Und.
-
135)
Take out WbdO from a −20 °C freezer and allow it to thaw on ice. Take out 100 mM GDP-L-Fuc mother solution from a −20 °C freezer and allow it to thaw at room temperature (25 °C).
CRITICAL STEP: Refer to Step 121.
-
136)
Add 10.6 mg (8 µmol) of Disaccharide-PP-Und from Step 133 to a 15-mL centrifuge tube.
CRITICAL STEP: It is recommended that a small portion of the Disaccharide-PP-Und be kept for later or further use. In this case, 1.5 µmol was kept, and 8 µmol was used as the substrate for Trisaccharide-PP-Und synthesis.
-
137)
Transfer 160 µL of DMSO and 320 µL of 10% (vol/vol) Triton X-100 into the tube. Vortex the tube for 1 min, and place the tube into the water bath (room temperature) of the Bransonic Tabletop Cleaner. Sonicate to completely dissolve Disaccharide-PP-Und.
CRITICAL STEP: Refer to Step 123. With an increased number of monosaccharides, the Sugar-PP-Und will become easier to dissolve.
-
138)
Consecutively transfer 2.24 mL of distilled H2O, 64 µL of Tris-HCl (1 M, pH 8.0), 160 µL of MnCl2 (200 mM), 96 µL of GDP-L-Fuc (100 mM), and 3.2 µL of rSAP (1000 U/mL) into the tube. Mix well by shaking manually after the addition of each component.
-
139)
Transfer 160 µL of WbdO (5 mg/mL) into the tube and gently shake the tube to mix well.
-
140)
The total volume of the reaction mixture is 3.2 mL, containing 20 mM Tris-HCl (pH 8.0), 2.5 mM Disaccharide-PP-Und, 3 mM GDP-L-Fuc (1.2 equiv.), 10 mM MnCl2, 1% Triton X-100 (vol/vol), 5% DMSO (vol/vol), 1 U/mL rSAP and 0.25 mg/mL WbdO.
CRITICAL STEP: Refer to Step 126.
-
141)
Incubate the reaction mixture at 37 °C in a Fisher water bath for 6 h with occasional gentle shaking.
-
142)
Monitor the reaction by mass spectrometry. Take out 1 µL of the reaction mixture and mix with 9 µL of 1:1 H2O:CH3OH (vol:vol) in an Eppendorf tube. Centrifuge the mixture at 15,000 × g at room temperature for 5 min with an Eppendorf centrifuge, and inject 5 µL of the supernatant into an ESI-MS for analysis under negative mode. Starting material (Trisaccharide-PP-Und) is typically consumed within 6 h depending upon the activity of the corresponding GT.
TROUBLESHOOTING. CRITICAL STEP: Refer to Step 128.
-
143)
Quench the reaction and lyophilize as described in Steps 129–131.
PAUSE POINT: After lyophilization, the tube can be sealed with parafilm and stored at −20 °C for up to two weeks or until purification can be performed.
-
144)
Purify the Trisaccharide-PP-Und by reverse phase chromatography as described in Steps 105–112.
-
145)
Lyophilize the fractions to afford E. coli O157 Trisaccharide-PP-Und as a white powder.
-
146)
Confirm the identity of the isolated product using mass spectrometry.
PAUSE POINT: After lyophilization, the compound can be sealed and stored at −20 °C indefinitely or until the next step can be performed.
Preparation of E. coli O157 RU-PP-Und – TIMING 3 days
-
147)
Take out WbdP from a −20 °C freezer and allow it to thaw on ice. Take out 100 mM GDP-PerNAc mother solution from a −20 °C freezer and allow it to thaw at room temperature (25 °C).
CRITICAL STEP: Refer to Step 121.
-
148)
Add 8.85 mg (6 µmol) of Trisaccharide-PP-Und from Step 145 to a 15-mL centrifuge tube.
CRITICAL STEP: It is recommended that a small portion of the Trisaccharide-PP-Und be kept for later or further use. In this case, 1.2 µmol was kept, and 6 µmol was used as the substrate for RU-PP-Und synthesis.
-
149)
Transfer 120 µL of DMSO and 240 µL of 10% (vol/vol) Triton X-100 into the tube. Vortex the tube for 1 min, and place the tube into the water bath (room temperature) of the Bransonic Tabletop Cleaner. Sonicate to completely dissolve the Trisaccharide-PP-Und.
CRITICAL STEP: Refer to Step 137.
-
150)
Consecutively transfer 2.1 mL of distilled H2O, 48 µL of Tris-HCl (1 M, pH 8.0), 120 µL of MnCl2 (200 mM), 72 µL of GDP-PerNAc (100 mM), and 2.4 µL of rSAP (1000 U/mL) into the tube. Mix well by shaking manually after the addition of each component.
-
151)
Transfer 60 µL of WbdP (5 mg/mL) into the tube and gently shake the tube to mix well.
-
152)
The total volume of the reaction mixture is 2.4 mL, containing 20 mM Tris-HCl (pH 8.0), 2.5 mM Trisaccharide-PP-Und, 3 mM GDP-PerNAc (1.2 equiv.), 10 mM MnCl2, 1% Triton X-100 (vol/vol), 5% DMSO (vol/vol), 1 U/mL rSAP and 0.125 mg/mL WbdP.
CRITICAL STEP: Refer to Step 126.
-
153)
Incubate the reaction mixture at 37 °C in a Fisher water bath for 6 h, with occasional gentle shaking.
-
154)
Monitor the reaction by mass spectrometry. Take out 1 µL of the reaction mixture and mix with 9 µL of 1:1 H2O:CH3OH (vol:vol) in an Eppendorf tube. Centrifuge the mixture at 15,000 × g at room temperature for 5 min with an Eppendorf centrifuge, and inject 5 µL of the supernatant into an ESI-MS for analysis under negative mode. Starting material usually can be consumed within 6 h depending on the activity of the corresponding GT.
TROUBLESHOOTING. CRITICAL STEP: Refer to Step 128.
-
155)
Quench the reaction and lyophilize the product as described in Steps 129–131.
PAUSE POINT: After lyophilization, the tube can be sealed with parafilm and stored at −20 °C for up to two weeks or until purification can be performed.
-
156)
Purify the RU-PP-Und by reverse phase chromatography as described in Steps 105–112.
-
157)
Lyophilize the fractions to afford E. coli O157 RU-PP-Und as a white powder.
-
158)
Confirm the identity of the isolated product using mass spectrometry.
PAUSE POINT: After lyophilization, the final product can be sealed with parafilm and stored at −20 °C indefinitely or until the next use.
Activity Assay of WaaL using synthesized RU-PP-Und – TIMING 1 day
-
159)
Mass 0.42 mg (0.25 µmol) of E. coli O157 RU-PP-Und with an analytical balance, and transfer it into a 1.7-mL Eppendorf tube.
-
160)
Transfer 50 µL of DMSO and 50 µL of 10% (vol/vol) Triton X-100 into the tube. Vortex the tube for 1 min, and place the tube into the water bath (room temperature) of the Bransonic Tabletop Cleaner. Sonicate to completely dissolve the E. coli O157 RU-PP-Und.
CRITICAL STEP: Refer to Step 123.
-
161)
Transfer 150 µL of distilled H2O into the tube.
CRITICAL STEP: The final concentration of RU-PP-Und is 1 mM. The solution can be stored at −20 °C for up to 3 months or until the next use.
-
162)
Transfer 0.5 µL of RU-PP-Und (1 mM) into a 1.7-mL Eppendorf tube. Transfer 0.5 µL of distilled H2O to another tube as a negative control.
-
163)
To each tube, transfer 71.5 µL of distilled H2O, 5 µL of 1 M Tris-HCl (pH 7.5), and 20 µL of Lipid A core (40 µM), mixing well by pipetting.
-
164)
Take out 48 µL of the mixture from each tube (Reaction time = 0 min) and store at −20 °C for later use.
-
165)
To each tube transfer 2 µL of WaaL (1 mg/mL) and mix well by pipetting.
-
166)
Incubate the two tubes at 30 °C for 12 h.
CRITICAL STEP: Such a reaction time and WaaL amounts will yield complete transfer of RU from RU-PP-Und onto Lipid A core. For experiments such as biochemical characterization, kinetic studies, or activity assays of other enzymes (Wzy, Wzz, etc.), reaction times, WaaL amounts and RU-PP-Und amounts may be optimized accordingly.
-
167)
Transfer 1 µL of Proteinase K (20 mg/mL) to each tube, and incubate at 56 °C for 1 h.
-
168)
Analyze the reactions and controls (reactions at 0 min) using western blot (E. coli O157 anti-serum as first antibody) and SDS-PAGE/Silver staining (Fig. 7) as described previously18.
Figure 7.

Activity assay of WaaL. Result using silver-staining (a) and western blot (b) with E. coli O157 anti-serum as the first antibody. After the reaction, a band was observed above the one corresponding to Lipid A core, thus dicating the presence of Lipid A core with one repeating unit.
TIMING
Chemical Synthesis of GalNAc-PP-Und
| Steps 1–16 | 4 days |
| Steps 17–25 | 2 h |
| Steps 26–39 | 5 ½ h |
| Step 40–48 | 2 h |
| Step 49–52 | 1 h |
| Step 53–61 | 2 h |
| Step 62–74 | 18 h |
| Step 75–84 | 2 ½ h |
| Step 85–96 | 2 h |
| Step 97–120 | 7 days |
Enzymatic Synthesis of E. coli O157 RU-PP-Und
Activity assay of WaaL using synthesized RU-PP-Und
| Step 159–161 | 1 h |
| Step 162–166 | 13 h |
| Step 167–168 | 10 h |
TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table.
| Steps | Problems | Solution |
|---|---|---|
| Steps 25 and 102 | Upon removing the solvent from purified undecaprenyl-phosphate, a waxy residue is formed which is difficult to transfer. | This residue will take on a white powder form following the addition of 1–2 mL of hexanes, rotary evaporation, and drying under vacuum. This form greatly facilitates transfer of the product to another container should this process be necessary. |
| Step 128 | Starting material not completely consumed. | Add another 100 µL of WbdN and incubate for 12 h at 37 °C. |
| Step 142 | Starting material not completely consumed. | Add another 160 µL of WbdO, 40 µL of GDP-l-Fuc and incubate for 12 h at 37 °C |
| Step 154 | Starting material not completely consumed. | Add another 60 µL of WbdP and incubate for 12 h at 37 °C. |
ANTICIPATED RESULTS
The Procedure describes the chemical synthesis of GalNAc-PP-Und beginning from GalNAc and Und. Preparation of the dibenzyl-protected phosphate of peracetylated GalNAc entailed a sequence of three chemical steps with an overall yield of 52%, while Und-P was prepared directly from Und in a yield of 79%. Benzyl group removal and subsequent activation of peracetylated GalNAc-1-phosphate enabled pyrophosphate bond formation to be realized. This step initially yielded minimal product due to the physical state of Und-P upon purification (waxy residue). Upon the addition of a small amount of hexane, rotary evaporation, and adequate drying under vacuum, however, a white powder was obtained which allowed yields to be consistently obtained at the 59% level. Subsequent deprotection afforded the desired GalNAc-PP-Und product in 99% yield (18 mg). While these yields represent optimal results, variation among trials did not exceed 10%. It should furthermore be noted that this synthesis can be extended to lipids other than Und, including but not limited to Solanesol, Heptaprenol, Pentaprenol, cis-Pentaprenol, Mono-Saturated Pentaprenol, and Farnesol.
Enzymatic synthesis of RU-PP-Und from the chemically prepared GalNAc-PP-Und was then accomplished in a total yield of over 70% (expecting over 90% yield in each GT catalyzed step) on semi-preparative scales (9 mg) (Fig. 6). RU-PP-Lipids can also be synthesized with over 70% yield on semi-preparative scales (see Supplementary Data 1 for mass spectra of RU-PP-Farnesyl and RU-PP-Mono-saturated-Pentaprenyl). During the enzymatic synthesis of RU-PP-Und, the specific activity of putative GTs involved in the assembly of the repeating units can be identified. For example, in this case, WbdO and WbdP are identified as the α1,4-fucosyltransferase and α1,3-N-acetylperosaminyltransferase, respectively.
The protocol for the activity assay of WaaL using synthesized RU-PP-Und furthermore provides visible results regarding the activity of WaaL. As shown in Figure 7, RU-PP-Und is visible in the western blot (Fig. 7b, lane 5) using E. coli O157 anti-serum as the first antibody. This evidence confirms the structure of synthesized RU-PP-Und given that the anti-serum is specific for E. coli O157 structures. While RU-PP-Und cannot be visualized in SDS-PAGE/silver staining (Fig. 7a, lane 5), Lipid A core can be visualized by both western blot analysis and silver staining (Fig. 7a,b, lane 6). After incubating with WaaL for 12 h, the band corresponding to RU-PP-Und which was initially present (Fig. 7b, lane 3) disappeared and a new band above Lipid A core was observed (Fig. 7b, lane 4) which correspond to the RU-Lipid A core product. SDS-PAGE confirmed the formation of the product (Fig. 7a, lane 4).
Analytical data
Analytical data is provided for peracetylated GalNAc (2), anomerically-deprotected peracetylated GalNAc (3), the dibenzyl-protected phosphate of peracetylated GalNAc (4), and peracetylated GalNAc-pyrophosphate-undecaprenyl (9). NMR spectra are available in Supplementary Data 2.
Peracetylated GalNAc (2)
White foam. 1H NMR (500 MHz, CD3Cl): δ 6.18 (d, J = 3.6 Hz, 1H), 5.48 (d, J = 9.1 Hz, 1H), 5.39 (d, J = 2.7 Hz, 1H), 5.18 (dd, J = 11.5 Hz, J = 3.2 Hz, 1H), 4.72-4.66 (m, 1H), 4.21 (t, J = 6.7 Hz, 1H), 4.10-4.00 (m, 2H), 2.14 (s, 6H), 2.00 (s, 3H), 1.99 (s, 3H), 1.91 (s, 3H); 13C NMR (125 MHz, CD3Cl): δ 171.3, 170.5, 170.4, 170.2, 169.0, 91.5, 68.7, 68.0, 65.9, 61.5, 47.2, 23.3, 21.1, 20.9, 20.8, 20.8.
Anomerically-deprotected peracetylated GalNAc (3)
White foam. 1H NMR (500 MHz, CD3Cl): δ 5.94 (d, J = 9.5 Hz, 1H), 5.35 (d, J = 2.4 Hz, 1H), 5.28 (t, J = 2.9 Hz, 1H), 5.22 (dd, J = 11.4 Hz, J = 3.3 Hz, 1H), 4.53-4.47 (m, 1H), 4.40 (t, J = 6.6 Hz, 1H), 4.29 (bs, 1H), 4.10-4.01 (m, 2H), 2.13 (s, 3H), 2.02 (s, 3H), 1.97 (s, 3H), 1.95 (s, 3H); 13C NMR (125 MHz, CD3Cl): δ 171.3, 171.0, 170.8, 170.6, 92.3, 68.3, 67.8, 66.6, 62.3, 48.3, 23.4, 21.0, 20.9(2).
Dibenzyl-protected phosphate of peracetylated GalNAc (4)
White foam. 1H NMR (500 MHz, CD3Cl): δ 8.59 (bs, 1H), 7.39-7.31 (m, 10H), 5.70 (dd, J = 5.8 Hz, J = 3.3 Hz, 1H), 5.48 (d, J = 9.4 Hz, 1H), 5.36 (d, J = 2.0 Hz, 1H), 5.12-5.00 (m, 4H), 4.63-4.56 (m, 1H), 4.24 (t, J = 6.6 Hz, 1H), 4.05 (dd, J = 11.3 Hz, J = 6.6 Hz, 1H), 3.92 (dd, J = 11.3 Hz, J = 6.5 Hz, 1H), 2.12 (s, 3H), 1.97 (s, 3H), 1.91 (s, 3H), 1.70 (s, 3H); 13C NMR (125 MHz, CD3Cl): δ 170.9, 170.5, 170.4, 170.3, 129.1, 129.0, 129.0, 128.3, 128.3, 97.2, 70.2, 70.1, 70.0, 68.9, 67.5, 67.0, 61.5, 47.7, 23.1, 20.9, 20.9, 20.8; 31P NMR (162 MHz, CD3Cl): δ −3.5.
Peracetylated GalNAc-pyrophosphate-undecaprenyl (9)
White solid. 1H NMR (500 MHz, CD3OD): δ 5.60 (dd, J = 3.2 Hz, J = 7.3 Hz, 1H), 5.42 (t, J = 6.1 Hz, 2H), 5.21 (dd, J = 3.2 Hz, J = 11.3 Hz, 1H), 5.14-5.05 (m, 10H), 4.58 (t, J = 6.9 Hz, 1H), 4.54-4.45 (m, 3H), 4.21 (dd, J = 8.1 Hz, J = 10.9 Hz, 1H), 4.04 (dd, J = 5.8 Hz, J = 10.8 Hz, 1H), 2.10 (s, 3H), 2.08-2.00 (m, 34H), 1.98 (s, 3H), 1.97 (s, 3H), 1.96-1.92 (m, 6H), 1.89 (s, 3H), 1.71 (s, 3H), 1.66-1.62 (m, 21H), 1.58 (s, 3H), 1.57 (s, 9H); 13C NMR (125 MHz, CD3OD): δ 174.4, 172.3, 172.2, 171.9, 140.6, 136.5, 136.4, 136.3, 136.3, 136.1, 135.9, 132.1, 126.3, 126.3, 126.3, 126.0, 125.6, 125.6, 125.6, 123.6, 123.6, 96.5, 70.4, 68.8, 68.7, 63.8, 63.8, 62.3, 41.0, 41.0, 40.9, 34.4, 33.4, 33.4, 33.4, 33.2, 33.0, 30.8, 30.8, 30.7, 30.6, 30.5, 30.5, 30.4, 28.0, 27.8, 27.8, 27.8, 27.7, 27.7, 27.7, 26.1, 24.0, 23.9, 23.8, 23.0, 20.8, 20.8, 20.7, 17.9, 16.3, 14.5, 14.0; 31P NMR (162 MHz, CD3OD): δ −9.7, −12.6; LRMS (m/z): [M-H]- calcd. for C69H110NO15P2, 1254.7; found, 1254.7. Final deprotection with 0.1% NaOCH3 in CH3OH and purification via C-18 reverse phase silica gel afforded GalNAc-PP-Und (10) (18 mg, 99%). LRMS (m/z): [M-H]- calcd. for C63H104NO12P2,1128.7; found, 1128.7.
Supplementary Material
Supplementary Data 1: Mass spectra of RU-PP-Lipids
Supplementary Data 2: NMR spectra of chemically synthesized compounds
Supplementary Method 1: Synthesis of GDP-PerNAc
Supplementary Method 2: Cloning and expression of putative GTs WbdN, WbdO and WbdP
Acknowledgments
This work is supported by the National Institute of Health (R01GM085267 and R01AI083754 to P.G.W., U01GM116263 to P.G.W. and L.L.).
Footnotes
AUTHOR CONTRIBUTIONS
L.L., R.L.W. and P.G.W. designed the research and developed the methods; L.L., R.L.W. and W.H. performed experiments; L.L. and R.L.W. wrote the manuscript; J.Q., J.S. and C.M. revised the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
References
- 1.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitfield C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem. 2006;75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 3.Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 4.Osborn MJ, Weiner IM. Biochemical aspects of structure, differentiation and morphogenesis in microorganisms. Mechanism of biosynthesis of the lipopolysaccharide of Salmonella. Fed Proc. 1967;26:70–76. [PubMed] [Google Scholar]
- 5.Whitfield C. Glycan chain-length control. Nat Chem Biol. 2010;6:403–404. doi: 10.1038/nchembio.376. [DOI] [PubMed] [Google Scholar]
- 6.Amer AO, Valvano MA. The N-terminal region of the Escherichia coli WecA (Rfe) protein, containing three predicted transmembrane helices, is required for function but not for membrane insertion. J Bacteriol. 2000;182:498–503. doi: 10.1128/jb.182.2.498-503.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Liu D, Reeves PR. C-terminal half of Salmonella enterica WbaP (RfbP) is the galactosyl-1-phosphate transferase domain catalyzing the first step of O-antigen synthesis. J Bacteriol. 1996;178:2598–2604. doi: 10.1128/jb.178.9.2598-2604.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitfield C, Trent MS. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem. 2014;83:99–128. doi: 10.1146/annurev-biochem-060713-035600. [DOI] [PubMed] [Google Scholar]
- 9.Woodward R, et al. In vitro bacterial polysaccharide biosynthesis: defining the functions of Wzy and Wzz. Nat Chem Biol. 2010;6:418–423. doi: 10.1038/nchembio.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu D, Cole RA, Reeves PR. An O-antigen processing function for Wzx (RfbX): a promising candidate for O-unit flippase. J Bacteriol. 1996;178:2102–2107. doi: 10.1128/jb.178.7.2102-2107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marolda CL, Vicarioli J, Valvano MA. Wzx proteins involved in biosynthesis of O antigen function in association with the first sugar of the O-specific lipopolysaccharide subunit. Microbiology. 2004;150:4095–4105. doi: 10.1099/mic.0.27456-0. [DOI] [PubMed] [Google Scholar]
- 12.Daniels C, Vindurampulle C, Morona R. Overexpression and topology of the Shigella flexneri O-antigen polymerase (Rfc/Wzy) Mol Microbiol. 1998;28:1211–1222. doi: 10.1046/j.1365-2958.1998.00884.x. [DOI] [PubMed] [Google Scholar]
- 13.Kim TH, et al. Characterization of the O-antigen polymerase (Wzy) of Francisella tularensis. J Biol Chem. 2010;285:27839–27849. doi: 10.1074/jbc.M110.143859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, et al. Overexpression and characterization of Wzz of Escherichia coli O86:H2. Protein Expr Purif. 2006;48:49–55. doi: 10.1016/j.pep.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Franco AV, Liu D, Reeves PR. A Wzz (Cld) protein determines the chain length of K lipopolysaccharide in Escherichia coli O8 and O9 strains. J Bacteriol. 1996;178:1903–1907. doi: 10.1128/jb.178.7.1903-1907.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burrows LL, Chow D, Lam JS. Pseudomonas aeruginosa B-band O-antigen chain length is modulated by Wzz (Ro1) J Bacteriol. 1997;179:1482–1489. doi: 10.1128/jb.179.5.1482-1489.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y, et al. Genetic analysis of the Cronobacter sakazakii O4 to O7 O-antigen gene clusters and development of a PCR assay for identification of all C. sakazakii O serotypes. Appl Environ Microbiol. 2012;78:3966–3974. doi: 10.1128/AEM.07825-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han W, et al. Defining function of lipopolysaccharide O-antigen ligase WaaL using chemoenzymatically synthesized substrates. J Biol Chem. 2012;287:5357–5365. doi: 10.1074/jbc.M111.308486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weerapana E, Glover KJ, Chen MM, Imperiali B. Investigating bacterial N-linked glycosylation: synthesis and glycosyl acceptor activity of the undecaprenyl pyrophosphate-linked bacillosamine. J Am Chem Soc. 2005;127:13766–13767. doi: 10.1021/ja054265v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aas FE, Vik A, Vedde J, Koomey M, Egge-Jacobsen W. Neisseria gonorrhoeae O-linked pilin glycosylation: functional analyses define both the biosynthetic pathway and glycan structure. Mol Microbiol. 2007;65:607–624. doi: 10.1111/j.1365-2958.2007.05806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valvano MA. Undecaprenyl phosphate recycling comes out of age. Mol Microbiol. 2008;67:232–235. doi: 10.1111/j.1365-2958.2007.06052.x. [DOI] [PubMed] [Google Scholar]
- 22.Perlstein DL, Zhang Y, Wang TS, Kahne DE, Walker S. The direction of glycan chain elongation by peptidoglycan glycosyltransferases. J Am Chem Soc. 2007;129:12674–12675. doi: 10.1021/ja075965y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, et al. Synthesis of heptaprenyl-lipid IV to analyze peptidoglycan glycosyltransferases. J Am Chem Soc. 2007;129:3080–3081. doi: 10.1021/ja069060g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginsberg C, Zhang YH, Yuan Y, Walker S. In vitro reconstitution of two essential steps in wall teichoic acid biosynthesis. ACS Chem Biol. 2006;1:25–28. doi: 10.1021/cb0500041. [DOI] [PubMed] [Google Scholar]
- 25.Glover KJ, Weerapana E, Numao S, Imperiali B. Chemoenzymatic synthesis of glycopeptides with PglB, a bacterial oligosaccharyl transferase from Campylobacter jejuni. Chem Biol. 2005;12:1311–1315. doi: 10.1016/j.chembiol.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, et al. Overexpression and topology of bacterial oligosaccharyltransferase PglB. Biochem Biophys Res Commun. 2010;394:1069–1074. doi: 10.1016/j.bbrc.2010.03.126. [DOI] [PubMed] [Google Scholar]
- 27.Musumeci MA, et al. In vitro activity of Neisseria meningitidis PglL O-oligosaccharyltransferase with diverse synthetic lipid donors and a UDP-activated sugar. J Biol Chem. 2013;288:10578–10587. doi: 10.1074/jbc.M112.432815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faridmoayer A, et al. Extreme substrate promiscuity of the Neisseria oligosaccharyl transferase involved in protein O-glycosylation. J Biol Chem. 2008;283:34596–34604. doi: 10.1074/jbc.M807113200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang LY, et al. Enzymatic synthesis of lipid II and analogues. Angew Chem Int Ed Engl. 2014;53:8060–8065. doi: 10.1002/anie.201402313. [DOI] [PubMed] [Google Scholar]
- 30.Gale RT, Sewell EW, Garrett TA, Brown ED. Reconstituting poly (glycerol phosphate) wall teichoic acid biosynthesis in vitro using authentic substrates. Chem Sci. 2014;5:3823–3830. [Google Scholar]
- 31.Lebar MD, et al. Forming cross-linked peptidoglycan from synthetic gram-negative Lipid II. J Am Chem Soc. 2013;135:4632–4635. doi: 10.1021/ja312510m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang SH, et al. New continuous fluorometric assay for bacterial transglycosylase using Forster resonance energy transfer. J Am Chem Soc. 2013;135:17078–17089. doi: 10.1021/ja407985m. [DOI] [PubMed] [Google Scholar]
- 33.Holkenbrink A, Koester DC, Kaschel J, Werz DB. Total Synthesis of α-Linked Rha–Rha–Gal Undecaprenyl Diphosphate Found in Geobacillus stearothermophilus. Eur J Org Chem. 2011;2011:6233–6239. [Google Scholar]
- 34.Wagner GK, Pesnot T, Field RA. A survey of chemical methods for sugar-nucleotide synthesis. Nat Prod Rep. 2009;26:1172–1194. doi: 10.1039/b909621n. [DOI] [PubMed] [Google Scholar]
- 35.Thibodeaux CJ, Melancon CE, 3rd, Liu HW. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew Chem Int Ed Engl. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Y, et al. Biochemical characterization of WbdN, a β1,3-glucosyltransferase involved in O-antigen synthesis in enterohemorrhagic Escherichia coli O157. Glycobiology. 2012;22:1092–1102. doi: 10.1093/glycob/cws081. [DOI] [PubMed] [Google Scholar]
- 37.Yi W, et al. The wbnH gene of Escherichia coli O86:H2 encodes an α1,3-N-acetylgalactosaminyl transferase involved in the O-repeating unit biosynthesis. Biochem Biophys Res Commun. 2006;344:631–639. doi: 10.1016/j.bbrc.2006.03.181. [DOI] [PubMed] [Google Scholar]
- 38.Brockhausen I, et al. Characterization of two β1,3-glucosyltransferases from Escherichia coli serotypes O56 and O152. J Bacteriol. 2008;190:4922–4932. doi: 10.1128/JB.00160-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riley JG, Xu C, Brockhausen I. Synthesis of acceptor substrate analogs for the study of glycosyltransferases involved in the second step of the biosynthesis of O-antigen repeating units. Carbohydr Res. 2010;345:586–597. doi: 10.1016/j.carres.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 40.Brockhausen I, Riley JG, Joynt M, Yang X, Szarek WA. Acceptor substrate specificity of UDP-Gal: GlcNAc-R β1,3-galactosyltransferase (WbbD) from Escherichia coli O7:K1. Glycoconj J. 2008;25:663–673. doi: 10.1007/s10719-008-9127-7. [DOI] [PubMed] [Google Scholar]
- 41.Xu C, et al. Biochemical characterization of UDP-Gal:GlcNAc-pyrophosphate-lipid β1,4-Galactosyltransferase WfeD, a new enzyme from Shigella boydii type 14 that catalyzes the second step in O-antigen repeating-unit synthesis. J Bacteriol. 2011;193:449–459. doi: 10.1128/JB.00737-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, et al. Characterization of two UDP-Gal:GalNAc-diphosphate-lipid β1,3-galactosyltransferases WbwC from Escherichia coli serotypes O104 and O5. J Bacteriol. 2014;196:3122–3133. doi: 10.1128/JB.01698-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi W, et al. Escherichia coli O86 O-antigen biosynthetic gene cluster and stepwise enzymatic synthesis of human blood group B antigen tetrasaccharide. J Am Chem Soc. 2005;127:2040–2041. doi: 10.1021/ja045021y. [DOI] [PubMed] [Google Scholar]
- 44.Engels L, Elling L. WbgL: a novel bacterial α1,2-fucosyltransferase for the synthesis of 2'-fucosyllactose. Glycobiology. 2014;24:170–178. doi: 10.1093/glycob/cwt096. [DOI] [PubMed] [Google Scholar]
- 45.Yi W, et al. Characterization of a bacterial β1,3-galactosyltransferase with application in the synthesis of tumor-associated T-antigen mimics. Biochemistry. 2008;47:1241–1248. doi: 10.1021/bi7020712. [DOI] [PubMed] [Google Scholar]
- 46.Li M, et al. Identification of a new α1,2-fucosyltransferase involved in O-antigen biosynthesis of Escherichia coli O86:B7 and formation of H-type 3 blood group antigen. Biochemistry. 2008;47:11590–11597. doi: 10.1021/bi801067s. [DOI] [PubMed] [Google Scholar]
- 47.Li M, et al. Characterization of a novel α1,2-fucosyltransferase of Escherichia coli O128:b12 and functional investigation of its common motif. Biochemistry. 2008;47:378–387. doi: 10.1021/bi701345v. [DOI] [PubMed] [Google Scholar]
- 48.Bavaro MF. E. coli O157:H7 and other toxigenic strains: the curse of global food distribution. Curr Gastroenterol Rep. 2012;14:317–323. doi: 10.1007/s11894-012-0264-6. [DOI] [PubMed] [Google Scholar]
- 49.Perry MB, MacLean L, Griffith DW. Structure of the O-chain polysaccharide of the phenol-phase soluble lipopolysaccharide of Escherichia coli O157:H7. Biochem Cell Biol. 1986;64:21–28. doi: 10.1139/o86-004. [DOI] [PubMed] [Google Scholar]
- 50.Samuel G, Hogbin JP, Wang L, Reeves PR. Relationships of the Escherichia coli O157, O111, and O55 O-antigen gene clusters with those of Salmonella enterica and Citrobacter freundii, which express identical O antigens. J Bacteriol. 2004;186:6536–6543. doi: 10.1128/JB.186.19.6536-6543.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Reeves PR. Organization of Escherichia coli O157 O antigen gene cluster and identification of its specific genes. Infect Immun. 1998;66:3545–3551. doi: 10.1128/iai.66.8.3545-3551.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Branch CL, Burton G, Moss SF. An expedient synthesis of allylic polyprenyl phosphates. Synth Commun. 1999;29:2639–2644. [Google Scholar]
- 53.Danilov LL, Maltsev SD, Shibaev VN, Kochetkov NK. Synthesis of polyprenyl pyrophosphate sugars from unprotected mono-and oligo-saccharide phosphates. Carbohydr Res. 1981;88:203–211. [Google Scholar]
- 54.Rush JS, Alaimo C, Robbiani R, Wacker M, Waechter CJ. A novel epimerase that converts GlcNAc-P-P-undecaprenol to GalNAc-P-P-undecaprenol in Escherichia coli O157. J Biol Chem. 2010;285:1671–1680. doi: 10.1074/jbc.M109.061630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data 1: Mass spectra of RU-PP-Lipids
Supplementary Data 2: NMR spectra of chemically synthesized compounds
Supplementary Method 1: Synthesis of GDP-PerNAc
Supplementary Method 2: Cloning and expression of putative GTs WbdN, WbdO and WbdP