

Abstract
Leber’s hereditary optic neuropathy (LHON) is a maternally inherited disorder that causes severe loss of sight in young adults, and is typically associated to mitochondrial DNA (mtDNA) mutations. Heteroplasmy of primary LHON mutations, presence of ‘ancillary’ mtDNA mutations, and mtDNA copy number are probably correlated with the penetrance and the severity of the disease. In this study, we performed a mutational screening in an Apulian cohort of LHON patients and we found that 41 out of 54 subjects harbored the m.11778G>A mutation, and 13 harbored the m.3460G>A mutation. Whole mtDNA sequencing was performed in three affected subjects belonging to three unrelated m.11778G>A pedigrees to evaluate the putative synergistic role of additional mtDNA mutations in determining the phenotype. Our study suggests to include haplogroup T as a possible genetic background influencing LHON penetrance and to consider the increase of mtDNA copy number as a protective factor from vision loss regardless the hetero/homoplasmic status of LHON primary mutations.
Key words: LHON, heteroplasmy, homoplasmy, Mitochondrial DNA mutation, mtDNA copy number
Introduction
Leber hereditary optic neuropathy (LHON, MIM#535000) is the most frequent inherited mitochondrial disorder due to mitochondrial DNA (mtDNA) mutations, with a prevalence ranging between 0.2 to 0.4 cases per 100,000 in Europe (1-5). Typically, LHON arises in males (6) during young adulthood with painless loss of central vision in one eye followed by loss of vision in the second eye within a short time (7, 8). LHON is most frequently associated to one of three mtDNA point mutations affecting NADH-ubiquinone oxidoreductase (complex I; EC 1.6.5.3) subunits, i.e. m.3460G>A in MT-ND1; m.11778G>A in MT-ND4; m.14484T>C in MT-ND6(9). Fifteen further mutations have been identified as pathogenic for LHON (http://www.mitomap.org/MITOMAP) with some of them affecting MT-ND subunits of complex I, and associating with phenotypes overlapping MELAS (10), Leigh syndrome (11) and deafness (12-14). Unlike the majority of mtDNA mutations which are heteroplasmic (a mixture of both mutant and normal molecules) in mitochondrial diseases, LHON mutations are frequently homoplasmic (only mutant mtDNA); nonetheless, heteroplasmic mutations, particularly the m.3460G>A, have been detected in about 14% of the families observed (15-19). Although LHON has been the first disease to be identified as caused by mutations in the mtDNA, there are several puzzling features of LHON, i.e. the incomplete penetrance. All the matrilineal members of a LHON pedigree harbor mtDNA mutations, but only some individuals develop blindness implying that the primary mutation is a necessary but not sufficient condition to develop optic neuropathy. Among the genetic factors affecting penetrance, the homo/heteroplasmic condition is one of the pathological features which still needs to be completely elucidated (20). It was suggested that a contributing factor to the complexity of LHON is determined by additional mtDNA mutations, defined as ‘secondary’, that may well act in synergy with the primary ones – so far several nucleotide variants have been reported as such (21-25) – though the significance of such variants still remains controversial. Furthermore, as mtDNA is a multicopy genome, it has been proposed that the fine-tuning of mtDNA ‘copy number’ (20-26) may respond to an alteration of the bioenergetics request (27). Other genetic risk factors as well as environmental triggers – e.g. smoking – have been reported as significantly associated with increased risk of visual loss (28, 29).
Herein, we present molecular and genetic data of a cohort of LHON patients collected from the Apulia Region and we propose some ‘ancillary’ mtDNA mutations and mtDNA copy number as putative factors that may significantly affect LHON penetrance.
Materials and methods
Ophthalmologic examination and sample collection
A total of 54 subjects were enrolled in the study: 46 subjects were from the Ophthalmology Clinic, Policlinico Hospital of Bari, and 8 subjects were from the I.R.C.C.S. Casa Sollievo Della Sofferenza Hospital, San Giovanni Rotondo, Italy. Prior written and informed consent was obtained from each subject according to Institutional Guidelines. Among 54 subjects, 31 had already been partially analyzed in previous studies (20, 26). The control group consisted of 90 unrelated Italian subjects. Slit-lamp biomicroscopy dilated stereoscopic examination of the optic nerve head and fundus, visual field (VF) test (when possible), optical coherence tomography (OCT) (when possible) and fluorescein angiography were performed. The peripapillary RNFL thickness was measured using a spectral-domain Cirrus HD-OCT. The results from the VF tests were considered reliable if the fixation losses were less than 20% and false positive and false negative rates were less than 15%.
Mitochondrial genetics and statistical analysis
Total genomic DNA was extracted by standard methods from peripheral blood of the patients and their relatives with suspicion of LHON and from healthy control subjects. To detect the m.3460G>A, m.11778G>A and m.14484T>C mutations, convenient fragments were amplified by PCR performing a final last cycle of super-extension for 5 min at 72°C, to minimize the possible formation of heteroduplexes between mutant and wild-type strands. The presence of mutations was detected by PCR-RFLP (30) in all the subjects and, when present, confirmed by direct sequencing (ABI prism 310, Applied Biosystems). Entire mtDNA sequencing was performed in three affected individuals as described previously (31). All sequences were analyzed by comparison with mitochondrial reference sequence (Reconstructed Sapiens Reference Sequence – RSRS (32). Mitochondrial haplogroups were defined by the web-based bioinformatic platform Mitochondrial Disease Sequence Data Resource (https://mseqdr.org/). Relative quantification of mtDNA copy number was performed (33). All the data were analyzed by GraphPad Prism and Medcalc (MedCalc Statistical Software version 17.5.5 (MedCalc Software bvba, Ostend, Belgium) applying the chi-square test or the Fisher’s exact test as appropriate to compare percentages of independent groups. To compare quantitative variables ANOVA test in conjunction with Bonferroni test was used and description was done by means and standard deviation if data approach Gaussian distribution. If data were not Gaussian distributed description was done by median and interquartile range, Kruskal-Wallis non parametric analysis of variance were applied to compare groups and non-parametric test according to Conover was used for post-hoc comparisons. Statistical significance was set at P < 0.05.
Results
Population and clinical features
The cohort consisted of 54 subjects including 42 subjects belonging to 12 families, and 12 unrelated subjects. Male: female ratio was overall 28:26, 19: 8 for patients and 9: 18 for unaffected subjects. We counted 28 LHON affected subjects, representing the 52% of the entire cohort, in which the molecular genetic diagnosis was positive for one of the primary mutations (Table 1). All the clinical findings, sex, age and age at onset, molecular genetics test, and therapy as Idebenone administration, risk factors and recovery of vision for all the subjects, when available, are reported in Table 1. Among the 54 subjects tested, 6 (II-1 FAM-A1; II-1 FAM-A8; I-1 FAM-A14; I-1 FAM-A14; III-1 FAM-B2; I-1 FAM-B3) were examined at the acute phase and the diagnosis of LHON was clinically based on the unilateral and severe visual decline followed by a declining vision in the contralateral eye within a few weeks. Alteration of the visual field for the presence of a centrocecal scotoma, an ophthalmoscopic appearance of the fundus with the presence of an edematous, hyperemic optic nerve head, vascular tortuosity and telangiectasia were examined. Fluorescein angiography was performed in all affected subjects, and highlighted vascular telangiectasia in those examined during the acute phase without phenomena of leakage or staining, whereas it showed non-specific alterations in those observed during the atrophic stage. Interestingly, three m.11778G>A patients and one m.3460G>A patient experienced visual recovery: one (II-1 FAM-A2) homoplasmic and one (II-5 FAM-A4) heteroplasmic patient without Idebenone treatment, and two (I-1 FAM-A14; I-1 FAM-B3) homoplasmic patients, including an m.3460G>A, out of the 13 Idebenone treated-affected subjects. In all the cases, the diagnosis was confirmed by mtDNA genetic analysis. Since LHON patients were characterized by a sudden and devastating vision loss, the asymptomatic carriers, who had normal vision, were considered unaffected by the disease. Hereafter, we will refer to the subjects as Carriers, Affected and Controls. The control group consisted of 90 (male: female ratio 47:43) unrelated subjects who did not show any sign of optic neuropathy. The mean age resulted significantly different among LHON subjects, aged 45.3 ys ± 15.9, Carriers aged 47.8 ± 20.5 and Controls aged 37.9 ys ± 11.9 (F = 5.675; p = 0.004).
Table 1.
Clinical and genetic findings in subjects carrying the primary LHON mutations. Ophthalmological findings, sex, age, possible exposure to environmental triggers (i.e. alcohol, smoking and illicit drugs) and mtDNA LHON mutations, percentage of heteroplasmy and copy number are indicated. M, male; F, female; RE, right eye; LE, left eye; LP, light perception; N, no; Y, yes; n.a., not available; HOM, Homplosmic; HET, Heteroplasmic. Number in brackets near Subject ID indicates the reference of papers where the mtDNA copy number was previously reported.
| Subject ID | Family ID | Relation | Sex | Age (ys) | Visual condition at the first examination | Currently visual condition | Level of Visual Loss | Recovery | Idebenone treatment | Age at test (ys) | Age at onset (ys) | Clinical Features | Risk factors | Primary mtDNA mutation | mtDNA Mutation type (%mut) | Copy number (mtDNA/nDNA) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RE | LE | RE | LE | |||||||||||||||
| I-1(26) | FAM-A1 | Relative | F | 62 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 56 | - | Normal vision | - | m.11778G>A | HOM | 431 ± 49 |
| II-1(26) | Proband | M | 38 | 20/200 | 20/40 | LP | LP | Profound | N | Y | 32 | 32 | Bilateral optic atrophy; RNFL: LE 80.17µm; psychotic | Illicit drug and tobacco abuse | m.11778G>A | HOM | 196 ± 16 | |
| II-2(26) | Relative | M | 36 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 30 | - | Normal vision RNFL: RE 87µm; LE 88µm | - | m.11778G>A | HOM | 256 ± 47 | |
| I-1(26) | FAM-A2 | Relative | F | 61 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 58 | - | Normal vision | - | m.11778G>A | HOM | 685 ± 271 |
| II-1(26) | Proband | M | 35 | 20/200 | 20/200 | 20/200 | 20/200 | Moderate | Y | N | 24 | n.a. | Bilateral optic atrophy | - | m.11778G>A | HOM | 348 ± 35 | |
| I-1 | FAM-A3 | Proband | F | 66 | na | na | 20/40 | LP | Moderate | N | Y | 54 | 4 | RE optic subatrophy LE optic atrophy | - | m.11778G>A | HOM | 177 ± 46 |
| II-1(26) | Relative | M | 30 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | n.a. | n.a. | 29 | n.a. | Normal vision | - | m.11778G>A | HOM | 316 ± 62 | |
| II-5(20) | FAM-A4 | Proband | M | 51 | n.a. | n.a. | 20/20 | 20/20 | Normal vision | Y | N | 47 | n.a. | Bilateral optic subatrophy | Alcohol | m.11778G>A | HET (60%) | 149 ± 40 |
| III-2(20) | Proband | M | n.a. | n.a. | n.a. | LP | LP | Profound | n.a. | N | n.a. | n.a. | Bilateral optic subatrophy | - | m.11778G>A | HOM | n.a. | |
| I-1(20) | FAM-A5 | Proband | M | 67 | 20/300 | 20/300 | LP | LP | Profound | N | Y | 60 | 56 | Bilateral optic subatrophy | - | m.11778G>A | HET (70%) |
n.a. |
| I-1(26) | FAM-A6 | Proband | F | 51 | 20/25 | 20/32 | 20/25 | 20/32 | Mild | N | Y | 39 | 30 | Bilateral optic subatrophy with hemeralopia; psychiatric signs | - | m.11778G>A | HOM | 311 ± 30 |
| II-1(26) | Proband | F | 25 | 20/40 | 20/32 | 20/40 | 20/40 | Mild | N | Y | 13 | 11 | RE optic atrophy, LE optic subatrophy |
- | m.11778G>A | HOM | 366 ± 43 | |
| I-1(26) | FAM-A7 | Proband | M | 66 | LP | LP | LP | LP | Profound | n.a. | N | 65 | 28 | Bilateral optic atrophy | - | m.11778G>A | HOM | 183 ± 35 |
| I-2(26) | Proband | M | 45 | LP | 20/1000 | LP | 20/800 | Severe | n.a. | N | 44 | 25 | Bilateral optic atrophy | - | m.11778G>A | HOM | 429 ± 75 | |
| I-1 | FAM-A8 | Proband | F | 52 | n.a. | n.a. | 20/40 | 20/200 | Moderate | N | N | n.a. | Bilateral optic subatrophy | - | m.11778G>A | HOM | 486 ± 56 | |
| II-1 | Proband | M | 21 | 20/200 | 20/200 | LP | 20/800 | Severe | N | Y | 20 | 20 | Bilateral optic atrophy; RNFL: RE 95.12µm; LE 98.34µm | - | m.11778G>A | HOM | 190 ± 33 | |
| II-2 | Relative | F | 15 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 221 ± 48 | |
| I-1 | FAM-A9 | Relative | F | 61 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | n.a. | Normal vision | - | m.11778G>A | HOM | 510 ± 77 |
| II-1 | Proband | M | 45 | 20/200 | 20/40 | LP | 20/200 | Severe | n.a. | n.a. | - | n.a. | Bilateral optic atrophy | - | m.11778G>A | HOM | 327 ± 45 | |
| II-2 | Relative | F | n.a. | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | n.a. | - | - | n.a. | Normal vision | - | m.11778G>A | HOM | 430 ± 94 | |
| I-2 | FAM-A10 | Relative | F | 89 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 453 ± 52 |
| II-2 | Relative | F | 59 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 562 ± 68 | |
| III-1 | Proband | M | 34 | n.a | n.a. | LP | LP | Mild | n.a. | n.a. | - | n.a | Bilateral optic subatrophy | - | m.11778G>A | HOM | 334 ± 29 | |
| III-2 | Relative | M | 28 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 484 ± 85 | |
| I-1 | FAM-A11 | Relative | M | 74 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 165 ± 47 |
| I-2 | Relative | F | 68 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 161 ± 39 | |
| I-3 | Relative | F | 66 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 167 ± 25 | |
| I-4 | Relative | F | 58 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 241 ± 73 | |
| I-5 | Relative | F | 55 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 182 ± 59 | |
| II-1 | Relative | M | 34 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 380 ± 18 | |
| II-2 | Proband | M | 38 | n.a. | n.a. | LP | LP | Profound | n.a. | Y | - | n.a. | Bilateral optic atrophy | - | m.11778G>A | HOM | 201 ± 45 | |
| II-3 | Relative | F | 38 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 263 ± 55 | |
| I-1(26) | FAM-A12 | Proband | F | 55 | 20/40 | 20/25 | 20/40 | 20/25 | Mild | N | N | 51 | 26 | Bilateral optic subatrophy with hemeralopia | - | m.11778G>A | HOM | 246 ± 31 |
| I-1(26) | FAM-A13 | Proband | F | 42 | 20/25 | 20/32 | 20/25 | 20/32 | Mild | N | Y | 40 | 30 | Bilateral optic subatrophy with hemeralopia | - | m.11778G>A | HOM | 187 ± 26 |
| I-1(26) | FAM-A14 | Proband | M | 41 | 20/80 | 20/160 | 20/200 | 20/40 | Mild | Y | Y | 28 | 28 | Bilateral optic subatrophy | Tobacco and alcohol abuse | m.11778G>A | HOM | 739 ± 121 |
| I-1 | FAM-A15 | Proband | M | 34 | n.a. | n.a. | n.a. | n.a. | Severe | n.a. | n.a. | 21 | n.a. | Bilateral optic atrophy | - | m.11778G>A | HOM | n.a. |
| I-1(26) | FAM-A16 | Proband | M | 41 | 20/20 | 20/200 | 20/200 | 20/800 | Severe | Y | Y | 33 | 33 | Bilateral optic Subatrophy | - | m.11778G>A | HOM | 136 ± 36 |
| I-1(26) | FAM-A17 | Proband | M | 26 | 20/20 | 20/200 | LP. | LP. | Profound | N | Y | 19 | 19 | Bilateral optic atrophy | - | m.11778G>A | HOM | 313 ± 44 |
| I-1(26) | FAM-A18 | Proband | M | 53 | 20/800 | 20/800 | 20/800 | 20/800 | Profound | N | N | 50 | 20 | Bilateral optic atrophy | - | m.11778G>A | HOM | 268 ± 108 |
| I-1(26) | FAM-A19 | Unrelated | M | n.a. | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 582 ± 76 |
| I-1(26) | FAM-A20 | Unrelated | F | n.a. | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | - | - | Normal vision | - | m.11778G>A | HOM | 303 ± 78 |
| I-1(26) | FAM-B1 | Proband | F | 61 | 20/32 | 20/28 | 20/32 | 20/28 | Mild | N | N | 54 | 40 | Bilateral optic subatrophy | - | m.3460G>A | HOM | 279 ± 36 |
| I-2(26) | Proband | F | 70 | 20/40 | 20/32 | 20/40 | 20/32 | Mild | N | Y | 63 | 35 | Bilateral optic subatrophy | - | m.3460G>A | HOM | 259 ± 27 | |
| II-1(26) | Proband | M | 49 | 20/40 | 20/40 | 20/40 | 20/40 | Mild | N | Y | 42 | 27 | Bilateral optic subatrophy, severe visual impairment with hemeralopia | - | m.3460G>A | HOM | 305 ± 38 | |
| I-2 | FAM-B2 | Relative | F | 71 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 70 | - | Normal vision | - | m.3460G>A | HET (35%) |
206 ± 15 |
| II-1(26) | Relative | F | 47 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 39 | - | Mild mental retardation | - | m.3460G>A | HOM | 636 ± 74 | |
| II-2 | Relative | F | 46 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 38 | - | Borderline mental functioning | - | m.3460G>A | HET (40%) | 713 ± 144 | |
| II-3(26) | Relative | M | 44 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 43 | - | Normal vision | Tobacco abuse | m.3460G>A | HOM | 213 ± 9 | |
| III-1(26) | Proband | M | 25 | 20/200 | 20/200 | 20/800 | L.P. | Profound | N | Y | 17 | n.a. | Bilateral optic atrophy epilepsies, mild mental retardation | - | m.3460G>A | HOM | 240 ± 86 | |
| III-2(26) | Relative | M | 21 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 17 | - | Hyperemic optic disk, borderline mental functioning | - | m.3460G>A | HOM | 604 ± 149 | |
| III-3(26) | Relative | F | 18 | 20/25 | 20/25 | 20/25 | 20/25 | Normal vision | - | - | 12 | - | Borderline mental functioning | - | m.3460G>A | HOM | 667 ± 72 | |
| III-4 | Relative | M | 18 | 20/20 | 20/20 | 20/20 | 20/20 | Normal vision | - | - | 10 | - | Borderline mental functioning | Tobacco abuse | m.3460G>A | HET (40%) |
739 ± 184 | |
| I-1(26) | FAM-B3 | Proband | M | 16 | 20/80 | 20/200 | 20/40 | 20/80 | Moderate | Y | Y | 13 | 13 | Bilateral optic subatrophy; several maternal relatives with optic atrophy | - | m.3460G>A | HOM | 140 ± 38 |
| I-1(20) | FAM-B4 | Proband | M | 77 | 20/32 | 20/25 | 20/32 | 20/25 | Mild | N | Y | 69 | 30 | Bilateral optic subatrophy with hemeralopia | Illicit drug Alcohol and tobacco abuse | m.3460G>A | HET (15%) | 285 ± 24 |
Genetic analysis of mtDNA
If we consider a total of 54 LHON mutation-positive subjects, they represent an overall observed prevalence of 1: 75,503 in the Apulia population with sex proportion of 29 males (1: 68,250 male) versus 25 females (1: 83,916 female) (December 31, 2016). On the basis of clinical features and genetic mitochondrial analysis, we diagnosed 28 subjects as Affected and 26 as Carriers. Among the 54 subjects, 76% harbored the m.11778A mutation and 24% carried the m.3460G>A mutation. Moreover, the 95% of subjects who harbored m.11778G>A were homoplasmic (51% Affected; 49% Carriers) and 5% were heteroplasmic (all Affected); whereas among the subjects who harbored m.3460G>A, 70% were homoplasmic (56% Affected; 44% Carriers) and 30% were heteroplasmic (25% Affected; 75% Carriers). The difference of frequency of homoplasmic subjects between m.11778G>A and m.3460G>A resulted statistically significant at Fisher exact test (p=0.0248). Among the heteroplasmic Affected, I-1 FAM-A5 and II-5 FAM-A4 (Family11) were both heteroplasmic for m.11778G>A with mutant load estimated to be ~75% (34) and ~60%, respectively; I-1 FAM-B4 was heteroplasmic for m.3460G>A with an estimated load of ~15%. Among the heteroplasmic Carriers, I-2, II-2, III-4 all belonging to FAM-B2 harbored the m.3460G>A with a mutant load estimated as ~30%, ~40%, and ~40%, respectively. None of them carried the m.14484T>C primary mutation. Among all 54 LHON subjects, 33 (Table 1) were reported in our previous study (20, 26) and the relatives of FAM-B2 (Table 1) will be described in the near future (Manuscript in preparation).
The penetrance rate of both LHON mutations in our cohort was: 52% (28/54), i.e. 71% (20/28) in males and 29% (8/28) in females. Among the 41 subjects (22 males; 19 females) who harbored the m.11778G>A mutation, 24 (16 males; 8 females) developed typical optic neuropathy thus showing 59% phenotype penetrance. In the remaining 13 subjects (7 males; 6 females) who harbored m.3460G>A mutation, 6 (4 males; 2 female) were Affected thus showing 46% phenotype penetrance. The penetrance rate did not result significantly different (chi-square = 1.375; p = 0.2409), but there was a difference of percentage of affected between males and females respectively 32% (8/25) vs 68.97% (20/29) that resulted statistically significant (chi-square = 7.348, p = 0.0067).
We performed Sanger sequencing of the entire mtDNA genome of three Affected carrying homoplasmic m.11778G>A mutation: II-1 FAM-A1; II-1 FAM-A2; I-1 FAM-A3. The criterion of selection of these three patients was based on the fact that they showed a quite peculiar manifestation. II-1 FAM-A1 reported a history of alcohol, tobacco, and drug abuse and he had been diagnosed as psychotic following psychiatric examination. II-1 FAM-A2 had reported visual recovery without having been treated with Idebenone; I-1 FAM-A3 had experienced an early onset of LHON with an acute and painless loss of central vision already at 4 ys of age.
All the mtDNA nucleotide variants identified in the three patients were analyzed by MtoolBox which performs prioritization taking into account the pathogenicity of each mutated allele with different algorithms, and the nucleotide variability of each variant site (34). For all three patients the m.11778G>A mutation was prioritized with a high score of pathogenicity (Supplemental Table 1). II-1 FAM-A1 showed 56 additional variants of which 45 mutational events help to define the sample haplogroup (K1a). The prioritization process recognized 7 variants annotated in Mitomap and they were predicted as not having a deleterious effect (Supplemental Table 1A). II-1 FAM-A2 showed 61 variants, of which 38 contributed to defining the sample haplogroup (T2e2a) and 8 further variants were prioritized (Supplemental Table 1B). We focused on two out of the eight variants: m.4136A>G/MT-ND1 (p.Y277C) and m.9139G>A/MT-ATPase6 were predicted to have a deleterious effect (Supplemental Table 1B) and already reported as associated to LHON disease (http://www.mitomap.org/MITOMAP). We noticed that the patient carried two additional mutations, m.4216T>C/MT-ND1 (p.Y304H) and m.4917A>G/MT-ND1 (p.N150D), previously defined as ‘secondary’ LHON mutations (35), that were not prioritized because they are indeed polymorphic variants and also T2e2a haplogroup markers (http://www.phylotree.org/). I-1 FAM-A3 showed 78 variants, of which 64 contributed to defining the sample haplogroup (T2e2a) and 9 variants were further prioritized (Supplemental Table 1C). Among the nine mutations, the m.4136A>G/MT-ND1 and m.9139G>A/MT-ATPase6 mutations are the same deleterious variants also identified in the above mentioned II-1 FAM-A2. In the I-1 FAM-A3 patient we noticed the presence of m.4216T>C and m.4917A>G mutations which define the same haplogroup T2e2a. Interestingly, the same MT-ND1 variants defining haplogroup T2e2a had been identified also in I-1 FAM-A5(36). None of the Affected fitted to haplogroup J previously suggested as increasing the penetrance of the m.11778 LHON mutation (37).
Supplemental Table 1.
List of mtDNA mutations prioritized by MToolBox in three LHON patients. All variants identified in each LHON subject were prioritized as potentially deleterious (score closer to 1 is more likely to be damaging) the mutations which do not contribute to the macro-haplogroup definition and, if non-synonymous, can be predicted as disease-associated by at least one of the pathogenicity prediction methods. A. II-1 FAM-A1; B. II-1 FAM-A2; C. I-1 FAM-A3.
| A. II-1 FAM-A1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variant Allele | Locus | Nt Variability | Codon Position | AA Change | AA Variability | Disease Score | Mitomap Associated Disease(s) | Somatic Mutations | dbSNP ID | Haplogroup |
| 11179G | MT-ND4 | 0.00021 | 3 | syn | 1.0 | K1a | ||||
| 15653T | MT-CYB | 0.00149 | 1 | M303L | 0.0028 | 0.124 | ||||
| 309.CCT | MT-DLOOP | 0.00196 | ||||||||
| 11778A | MT-ND4 | 0.00368 | 2 | R340H | 0.0605 | 0.853 | LHON/Progressive Dystonia | rs199476112 | ||
| 513.CA | MT-DLOOP | 0.0681 | ||||||||
| 5046A | MT-ND2 | 0.079 | 1 | V193I | 0.21 | 0.127 | ||||
| 310C | MT-DLOOP | 0.283 | Normal buccal swab | rs66492218 | ||||||
| 150T | MT-DLOOP | 0.419 | Longevity / Cervical Carcinoma / HPV infection risk | Elderly fibroblasts/leukocytes, lung, thyroid, prostate tumors | rs62581312 | |||||
| B. II-1 FAM-A2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variant Allele | Locus | Nt Variability | Codon Position | AA Change | AA Variability | Disease Score | Mitomap Associated Disease(s) | Somatic Mutations | dbSNP ID | Haplogroup |
| 309.CT | MT-DLOOP | 0.00196 | T2e2a | |||||||
| 11778A | MT-ND4 | 0.00368 | 2 | R340H | 0.0605 | 0.853 | LHON/Progressive Dystonia | rs199476112 | ||
| 9139A | MT-ATP6 | 0.00431 | 1 | A205T | 0.0051 | 0.893 | LHON | |||
| 4136G | MT-ND1 | 0.00641 | 2 | Y277C | 0.0226 | 0.76 | LHON | rs199476121 | ||
| 6026A | MT-CO1 | 0.0479 | 3 | syn | 0.0 | rs41474553 | ||||
| 16293G | MT-DLOOP | 0.106 | Glioblastoma | |||||||
| 310C | MT-DLOOP | 0.283 | Normal buccal swab | rs66492218 | ||||||
| 150T | MT-DLOOP | 0.419 | Longevity / Cervical Carcinoma / HPV infection risk | Elderly fibroblasts/leukocytes, lung, thyroid, prostate tumors | rs62581312 | |||||
| C. I-1 FAM-A3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variant Allele | Locus | Nt Variability | Codon Position | AA Change | AA Variability | Disease Score | Mitomap Associated Disease(s) | Somatic Mutations | dbSNP ID | Haplogroup |
| 11778A | MT-ND4 | 0.00277 | 2 | R340H | 0.0402 | 0.853 | LHON / Progressive Dystonia | rs199476112 | T2e2a | |
| 9139A | MT-ATP6 | 0.00376 | 1 | A205T | 0.0043 | 0.893 | LHON | |||
| 309.CT | MT-DLOOP | 0.0043 | ||||||||
| 4136G | MT-ND1 | 0.00639 | 2 | Y277C | 0.0232 | 0.76 | LHON | rs199476121 | ||
| 8222C | MT-CO2 | 0.0147 | 1 | syn | 0.0021 | |||||
| 16153A | MT-DLOOP | 0.038 | rs2853512 | |||||||
| 6026A | MT-CO1 | 0.0718 | 3 | syn | 0.0 | rs41474553 | ||||
| 16293G | MT-DLOOP | 0.098 | Glioblastoma | |||||||
| 310C | MT-DLOOP | 0.215 | Normal buccal swab | |||||||
| 150T | MT-DLOOP | 0.414 | Longevity / Cervical Carcinoma / HPV infection risk | Elderly fibroblasts/leukocytes, lung, thyroid, prostate tumors | rs62581312 | |||||
Analysis of mtDNA copy number in LHON subjects
The evaluation of mtDNA copy numbers was performed independently from the type of primary mutations in 25 Affected (23 homoplasmic; 2 heteroplasmic) and 26 Carriers (24 homoplasmic; 3 heteroplasmic) (Table 1). mtDNA copy number of thirty-one LHON subjects was evaluated in previous works (20, 26). Mitochondrial copy number values were measured either using MT-ND1 or MT-ND4 as target genes. The analysis of the MT-ND4 region was indeed implemented after assessing the presence of an ancient polymorphism, m.3480A>G/ND1, just within the annealing region of ND1 primers in I-1, II-1, II-2 (FAM-A1), I-2, II-2, III-1, III-2 (FAM-A10) I-1 FAM-B4 LHON subjects, and unexpectedly led to very low values of copy number. Three Affected (I-1 FAM-A5, III-2 FAM-A4 and I-1 FAM-A15) were diagnosed, but we could not perform the quantitation of the mtDNA content due to the scarcity of DNA samples. The values of the mtDNA copy number showed a statistically significant difference (KW = 24.828, p = 0.000004) and the peak of mtDNA content shifted progressively towards higher values from Controls (median 207; interquartile range 155.5-251.25) to Affected (median 268; interquartile range 189.25-328.75) to Carriers (median 380; interquartile range 219-567). The post-hoc analysis showed a statistically significant difference between Controls versus Affected and Controls versus Carriers (Fig. 1). Among the Carriers, we noticed that the relatives belonging to Family11 showed a quite low mtDNA content below the mean of Carriers. Indeed, the age of the Family11 members is relatively higher than the age of the other Carriers and we reasoned that this may explain the lower amount of mtDNA according to the decrease of mtDNA copy number with aging (38); additionally, the age of the oldest women of the family (FAM-A11) was over 60ys which is compatible with a low estrogens condition thus confirming in vivo the loss of the protective role by estrogens in activating mitochondrial biogenesis and mtDNA content in LHON (39). On the other hand, the comparison between the mtDNA copy number of Affected subjects, whose blood samples were obtained in the acute phase of the disease (II-1 FAM-A8, I-1 FAM-A14, I-1 FAM-A17, III-1 FAM B2), and mtDNA copy number of those subjects already affected by optic atrophy in the chronic stage (II-1 FAM-A2, I-1 FAM-A7, I-2 FAM-A7, I-1 FAM-A18), did not reveal any difference.
Figure 1.
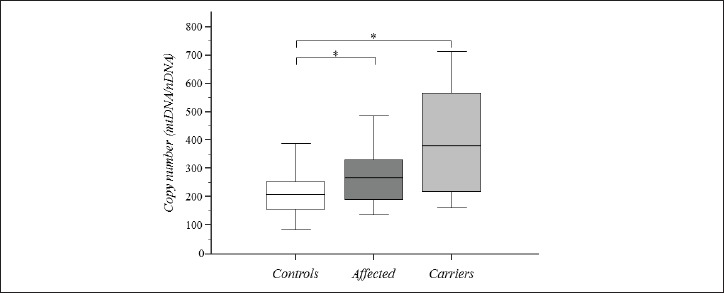
Analysis of mtDNA content in LHON subjects. Box-plot of mtDNA copy number (mtDNA/nDNA) by Affected, Carriers and Controls. Experiments were performed in triplicates for all samples; for thirty-one LHON mutation carriers mtDNA content was evaluated in previous works (20, 26) and herein included. Asterisks indicate statistical significance (p-value#x003C;0.05) at post-hoc comparisons.
Discussion
In the present study, we report on the genetic and molecular characterization of LHON subjects born and living in Apulia, Southern Italy. From our data, the prevalence of subjects carrying the LHON primary mutations can be estimated to be approximately 1 case in 75,503 thus, if compared to the prevalence in the North of England (1/31,000), the Netherlands (1/39,000) (2) and Finland (1/50,000) (19,40), it may indicate that in our Region the disease is very low or remains underestimated. Probably, the reason is that some patients may not be adequately diagnosed or are misdiagnosed or are diagnosed outside the Region. Our mutational screening disclosed two out of the three most common primary LHON mutations, i.e. m.11778G>A and m.3460G>A, either homoplasmic or heteroplasmic. Interestingly, we found that heteroplasmy is present for both primary mutations but it is more frequent for the m.3460G>A than for m.11778G>A mutation, according to a previous estimation in different countries (2, 41). Among the mitochondrial diseases, LHON is notable for the incomplete penetrance since not all the Carriers will develop loss of sight and it is expected that additional genetic and environmental factors may play a role in LHON penetrance (42). As in the majority of mitochondrial disorders, it has been suggested that a certain amount of wild-type mtDNA can compensate for the mutant mtDNA in a cell of a LHON individual (18). In our cohort we found no difference in the LHON manifestations among the Affected, either homoplasmic or heteroplasmic, since there was no clear-cut segregation with either more severe or benign clinical course of the disease, respectively. This finding is supported by a study performed specifically in a LHON heteroplasmic population (20).
With the aim of better investigating LHON penetrance we performed the whole mtDNA sequencing of three Affected who had a different course of disease: the first one was a man (II-1 FAM-A1) who was diagnosed as psychotic and, although after the disease onset he had started Idebenone treatment, he did not experience any visual recovery; the second one (II-1 FAM-A2) was a man who experienced visual recovery without Idebenone treatment; the third one (I-1 FAM-A3) was a woman who had an early acute and painless loss of central vision at four years of age. Unexpectedly, the latter two cases who manifested a less severe and a more severe LHON phenotype respectively, shared the co-occurrence of two mutations (m.4136A>G, m.9139G>A), thus suggesting that both mutations do not have an unequivocal effect in worsening the LHON manifestation, although both mutations had been previously reported to play a synergistic role when occurring with the LHON primary mutation (43). Furthermore, we identified the co-occurrence of the m.4136A>G with two haplogroup markers, m.4216T>C and m.4971A>G, in two out of three sequenced patients; the same genotype with the three variants had been previously reported in other three unrelated patients harboring the m.11778G>A mutation, coming from the Apulia Region, described by La Morgia et al. and Torroni et al. (43, 44). Interestingly, the three Apulian patients previously described (43, 44), similarly to our patients, belonged to haplogroup T2. The shared Apulian origin of these families supports the hypothesis that the m.11778G>A together with m.4136A>G, 4216T>C and 4917A>G are indeed associated and acquired by descent from a common maternal ancestor. Several studies have identified the haplogroup J as a risk genetic background for patients with m.11778G>A and m.14484T>C (44-46) and haplogroup K for patients with m.3460G>A; conversely, haplogroup H seemed to have a protective role in patients with m.11778G>A (47). On the basis of the data herein discussed, we suggest that also haplogroup T may play a role in LHON penetrance; this might not be unexpected since haplogroup T, like haplogroup J, belongs to the same macro-haplogroup JT (http://www.phylotree.org/tree/index.htm). Regarding the II-1 FAM-A1 case, the patient did not show any significant or prioritized mutation implying that his extraocular signs (i.e. psychotic trait) might be probably due more to his drug and alcohol addiction.
According to recent evidences (20, 26, 27) supporting the concept that the increase in mitochondrial mass differentiates the LHON unaffected from the affected, we measured mtDNA content in either heteroplasmic or homoplasmic LHON subjects. With the aim of evaluating the mtDNA copy number of the overall cohort of LHON subjects, we also included thirty-one subjects already reported (20, 26), and we found an increase in the number of mtDNA molecules in peripheral blood of Carriers versus Affected despite the homo- or heteroplasmy of LHON mutations. We may claim that such increase can be considered as a compensatory response to the decline in the respiratory function and a way to protect from vision loss. On the other hand, in agreement with previous studies (39, 48) we found that environmental triggers such as tobacco, alcohol, as well as low estrogen conditions and age itself are all contributing factors that affect mitochondrial biogenesis. We cannot rule out that among the Carriers those subjects showing low content of mtDNA might be considered at a high risk of developing the disease. But, of course, this should be monitored over time.
In conclusion, our study on Apulian LHON subjects, despite the small number, suggests the haplogroup T as a possible genetic background influencing LHON penetrance. Furthermore, mtDNA content may be considered as a protective factor from vision loss regardless the hetero/homoplasmic status of LHON primary mutations.
References
- 1.Chinnery PF, Thorburn DR, Samuels DC, et al. The inheritance of mitochondrial DNA heteroplasmy: random drift, selection or both? Trends Genet 2000;16:500-5. [DOI] [PubMed] [Google Scholar]
- 2.Man PY, Griffiths PG, Brown DT, et al. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet 2003;72:333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puomila A, Viitanen T, Savontaus ML, et al. Segregation of the ND4/11778 and the ND1/3460 mutations in four heteroplasmic LHON families. J Neurol Sci 2002;205:41-5. [DOI] [PubMed] [Google Scholar]
- 4.Mascialino B, Leinonen M, Meier T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. Eur J Ophthalmol 2012;22:461-5. [DOI] [PubMed] [Google Scholar]
- 5.Spruijt L, Kolbach DN, de Coo RF, et al. Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol 2006;141:676-82. [DOI] [PubMed] [Google Scholar]
- 6.Dimitriadis K, Leonhardt M, Yu-Wai-Man P, et al. Leber’s hereditary optic neuropathy with late disease onset: clinical and molecular characteristics of 20 patients. Orphanet J Rare Dis 2014;9:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman NJ, Biousse V, David R, et al. Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite. Am J Ophthalmol 2005;140:407-15. [DOI] [PubMed] [Google Scholar]
- 8.Barboni P, Carbonelli M, Savini G, et al. Natural history of Leber’s hereditary optic neuropathy: longitudinal analysis of the retinal nerve fiber layer by optical coherence tomography. Ophthalmology 2010;117:623-7. [DOI] [PubMed] [Google Scholar]
- 9.Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol 2005;140:517-23. [DOI] [PubMed] [Google Scholar]
- 10.Pulkes T, Eunson L, Patterson V, et al. The mitochondrial DNA G13513A transition in ND5 is associated with a LHON/MELAS overlap syndrome and may be a frequent cause of MELAS. Ann Neurol 1999;46:916-19. [DOI] [PubMed] [Google Scholar]
- 11.Fruhman G, Landsverk ML, Lotze TE, et al. Atypical presentation of Leigh syndrome associated with a Leber hereditary optic neuropathy primary mitochondrial DNA mutation. Mol Genet Metab 2011;103:153-60. [DOI] [PubMed] [Google Scholar]
- 12.Wei QP, Zhou X, Yang L, et al. The coexistence of mitochondrial ND6 T14484C and 12S rRNA A1555G mutations in a Chinese family with Leber’s hereditary optic neuropathy and hearing loss. Biochem Biophys Res Commun 2007;357:910-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhang AM, Jia X, Yao YG, et al. Co-occurrence of A1555G and G11778A in a Chinese family with high penetrance of Leber’s hereditary optic neuropathy. Biochem Biophys Res Commun 2008;376:221-4. [DOI] [PubMed] [Google Scholar]
- 14.Khan MR, Bashir R, Naz S. SLC26A4 mutations in patients with moderate to severe hearing loss. Biochem Genet 2013;51:514-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol 1991;111:750-62. [DOI] [PubMed] [Google Scholar]
- 16.Smith KH, Johns DR, Heher KL, et al. Heteroplasmy in Leber’s hereditary optic neuropathy. Arch Ophthalmol 1993;111:1486-90. [DOI] [PubMed] [Google Scholar]
- 17.Nikoskelainen EK, Marttila RJ, Huoponen K, et al. Leber’s “plus”: neurological abnormalities in patients with Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 1995;59:160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chinnery PF, Andrews RM, Turnbull DM, et al. Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation? Am J Med Genet 2001;98:235-43. [DOI] [PubMed] [Google Scholar]
- 19.Puomila A, Viitanen T, Savontaus ML, et al. Segregation of the ND4/11778 and the ND1/3460 mutations in four heteroplasmic LHON families. J Neurol Sci 2002;205:41-45. [DOI] [PubMed] [Google Scholar]
- 20.Bianco A, Bisceglia L, Russo L, et al. High mitochondrial DNA copy number is a protective factor from vision loss in heteroplasmic Leber’s Hereditary Optic Neuropathy (LHON). Invest Ophthalmol Vis Sci 2017;58:2193-7. [DOI] [PubMed] [Google Scholar]
- 21.Sartore M, Grasso M, Piccolo G, et al. Leber’s hereditary optic neuropathy (LHON)-related mitochondrial DNA sequence changes in italian patients presenting with sporadic bilateral optic neuritis. Biochem Mol Med 1995;56:45-51. [DOI] [PubMed] [Google Scholar]
- 22.Chinnery PF, Howell N, Andrews RM, et al. Mitochondrial DNA analysis: polymorphisms and pathogenicity. J Med Genet 1999;36:505-10. [PMC free article] [PubMed] [Google Scholar]
- 23.Brown MD, Starikovskaya E, Derbeneva O, et al. The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup. J Human Genetics 2002;110:130-8. [DOI] [PubMed] [Google Scholar]
- 24.Bi R, Logan I, Yao YG. Leber Hereditary Optic Neuropathy: a mitochondrial disease unique in many ways. Handb Exp Pharmacol 2016. [DOI] [PubMed] [Google Scholar]
- 25.Lodi R, Montagna P, Cortelli P, et al. ‘Secondary’ 4216/ND1 and 13708/ND5 Leber’s hereditary optic neuropathy mitochondrial DNA mutations do not further impair in vivo mitochondrial oxidative metabolism when associated with the 11778/ND4 mitochondrial DNA mutation. Brain 2000;123(Pt 9):1896-902. [DOI] [PubMed] [Google Scholar]
- 26.Bianco A, Martinez-Romero I, Bisceglia L, et al. Mitochondrial DNA copy number differentiates the Leber’s hereditary optic neuropathy affected individuals from the unaffected mutation carriers. Brain 2016;139. [DOI] [PubMed] [Google Scholar]
- 27.Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 2014;137(Pt 2):335-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudson G, Keers S, Yu-Wai-Man P, et al. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am J Hum Genet 2005;77:1086-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirkman MA, Korsten A, Leonhardt M, et al. Quality of life in patients with leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2009;50:3112-5. [DOI] [PubMed] [Google Scholar]
- 30.Guerriero S, Vetrugno M, Ciraci L, et al. Bilateral progressive visual loss in an epileptic, mentally retarded boy. Middle East Afr J Ophthalmol 2011;18:67-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tommasi S, Favia P, Weigl S, et al. Mitochondrial DNA variants and risk of familial breast cancer: an exploratory study. Int J Oncol 2014;44:1691-8. [DOI] [PubMed] [Google Scholar]
- 32.Behar DM, van Oven M, Rosset S, et al. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet 2012;90:675-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoccolella S, Artuso L, Capozzo R, et al. Mitochondrial genome large rearrangements in the skeletal muscle of a patient with PMA. Eur J Neurol 2012;19:e63-4 [DOI] [PubMed] [Google Scholar]
- 34.Calabrese C, Simone D, Diroma MA, et al. MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 2014;30:3115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johns DR, Berman J. Alternative, simultaneous complex I mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Biochem Biophys Res Commun 1991;174:1324-30 [DOI] [PubMed] [Google Scholar]
- 36.Zoccolella S, Petruzzella V, Prascina F, et al. Late-onset Leber hereditary optic neuropathy mimicking Susac’s syndrome. J Neurol 2010;257:1999-2003 [DOI] [PubMed] [Google Scholar]
- 37.Carelli V, Achilli A, Valentino ML, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet 2006;78:564-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mengel-From J, Thinggaard M, Dalgard C, et al. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Human Genetics 2014;133:1149-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giordano L, Deceglie S, d’Adamo P, et al. Cigarette toxicity triggers Leber’s hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huoponen K, Vilkki J, Aula P, et al. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet 1991;48:1147-53. [PMC free article] [PubMed] [Google Scholar]
- 41.Brown MD, Trounce IA, Jun AS, et al. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber’s hereditary optic neuropathy mitochondrial DNA mutation. J Biol Chem 2000;275:39831-6. [DOI] [PubMed] [Google Scholar]
- 42.Meyerson C, Van Stavern G, McClelland C. Leber hereditary optic neuropathy: current perspectives. Clin Ophthalmol 2015;9:1165-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.La Morgia C, Achilli A, Iommarini L, et al. Rare mtDNA variants in Leber hereditary optic neuropathy families with recurrence of myoclonus. Neurology 2008;70:762-70. [DOI] [PubMed] [Google Scholar]
- 44.Torroni A, Petrozzi M, DUrbano L, et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet 1997;60:1107-21. [PMC free article] [PubMed] [Google Scholar]
- 45.Hudson G, Carelli V, Spruijt L, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet 2007;81:228-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carelli V, Achilli A, Valentino ML, et al. Haplogroup effects and recombination of mitochondrial DNA: Novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet 2006;78:564-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Howell N, Herrnstadt C, Shults C, et al. Low penetrance of the 14484 LHON mutation when it arises in a non-haplogroup J mtDNA background. American Journal of Medical Genetics Part A 2003;119A):147-51. [DOI] [PubMed] [Google Scholar]
- 48.Giordano C, Montopoli M, Perli E, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain 2011;134(Pt 1):220-34. [DOI] [PMC free article] [PubMed] [Google Scholar]