

Abstract
VCP-proteinopathy is a multisystem neurodegenerative disorder caused by mutations in valosin containing protein. Here, we report the first Greek case of VCP-proteinopathy in a 62 year old patient with a slowly progressing muscular weakness since his mid-40s and a severe deterioration during the last year. He also manifested dementia with prominent neuropsychiatric symptoms, including aggression, apathy, palilalia and obsessions. Brain MRI revealed frontal atrophy, while muscle MRI showed diffuse muscle atrophy. Family history was positive and several members of the family had been diagnosed with motor neuron disease, dementia or behavioral symptoms. Sequencing of the VCP gene revealed a pathogenic heterozygous missense mutation p.R159H. Conclusively, the present report highlights the intrafamilial variability and broadens the phenotypic spectrum of VCP-proteinopathy.
Key words: VCP, ALS, dementia
Introduction
Valosin Containing Protein (VCP) mutations have been well recognized as a cause of dominantly inherited Inclusion Body Myopathy, Paget’s Disease and Frontotemporal Dementia (IBMPFD), although the term multisystemic proteinopathy has been proposed as a prevailing nomenclature in order to also include non-VCP IBMPFD and other rarer phenotypes, such as parkinsonism and peripheral neuropathy (1). IBMPFD (OMIM 167320) was firstly described in 2000 by Kimonis et al. (2) and the disease locus (VCP gene; 601023) was mapped to chromosome 9p13.3-p12(3). VCP belongs to the cytosolic chaperone AAA class of ATPases of the endoplasmic reticulum-associated degradation (ERAD) system and is involved in many different cellular functions and signaling pathways, with its most important role facilitating proteasome-mediated degradation of misfolded polypeptides (4).
The clinical picture of the disease, as implied by its acronym, includes myopathy, Paget’s disease and frontotemporal dementia with incomplete penetrance. Myopathy is the most common manifestation (~90%) and muscle weakness of variable pattern, either proximal or distal, may be usually the presenting symptom with onset in 3rd to 4th decade. Bone involvement is observed in almost half of the patients, while dementia of the frontotemporal type is a later onset symptom affecting approximately a third of the patients. However, the phenotypic spectrum of the disease has now broadened to include less frequent manifestations such as motor neuron disease, parkinsonism and Charcot-Marie-Tooth - like disease (1).
Herein, we describe the first Greek case of myopathy combined with an early onset progressive cognitive decline in a patient with a VCP mutation and a positive family history of neurodegenerative disorders, such as dementia and ALS.
Case report
The index patient is a 62 year old male with a personal history of arterial hypertension and an operation for herniated disc at the L4-L5 level at the age of 48 years. The symptoms began approximately in his mid-forties with steppage gait. His walking difficulties progressively worsened, especially during the last year and at the time of admission, he was no longer able to walk without aid. He also developed neuropsychiatric symptoms and in particular episodes of aggression and obsessions treated with atypical neuroleptics, such as quetiapine, with a satisfactory response, while he thereafter developed palilalia and apathy which still persist.
His mother had been diagnosed with “presenile dementia” and died at the age of 63 y (Fig. 1). His older brother had been diagnosed in his forties with classical ALS with a combination of upper and lower motor neuron involvement and subsequently died at 53 y. Another brother has been reported with behavioral symptoms since the age of 55 y.
Figure 1.
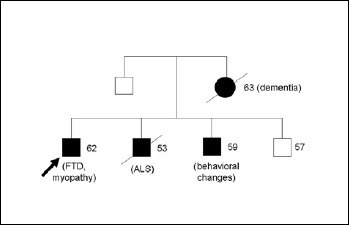
Pedigree of the patient’s family (arrow indicates the index patient). Main symptoms and present age or age at death are indicated.
On examination, the patient was unable to stand and walk without bilateral assistance. Although strength assessment of individual muscles was quite difficult due to poor cooperation, there was a severe symmetrical muscular weakness and a diffuse wasting in all muscles of upper and lower extremities, particularly prominent distally in lower limbs. Deep tendon reflexes were traced in upper and lower extremities, while no pathologic reflexes were elicited. Neuropsychological evaluation was abnormal with impaired conceptualization, mental flexibility and motor programming.
Laboratory studies, including serum creatine phosphokinase (CPK), transaminases (AST, ALT) and alkaline phosphates (ALP) were normal. Brain MRI revealed frontal lobe atrophy (Fig. 2). Bone radiographs did not reveal any abnormality suggestive of Paget’s disease. Nerve conduction studies showed slightly low amplitudes of peroneal and tibial nerves obviously due to significant axonal loss, while EMG showed diffuse myopathic changes and mild spontaneous activity in the form of fibrillations and positive sharp waves in distal leg muscles. Muscle biopsy of the left vastus lateralis was not informative as it showed severe and non specific end stage changes with fibroadipose tissue replacement. Muscle MRI of lower limbs showed extended atrophy and fatty degeneration in almost all lower leg muscles with a relative sparing of the left biceps femoris at thigh level (Fig. 3), while muscle MRI of upper limbs revealed diffuse atrophy and fibro-adipose tissue replacement especially of the posterior and anterior compartment of the arm (more pronounced in biceps and triceps) and to a lesser extent of the forearm muscles. Diagnosis of IBMPFD was confirmed by direct Sanger sequencing of coding regions and flanking intronic regions in the VCP gene, which revealed a heterozygous missense mutation p.R159H (c.476G>A).
Figure 2.
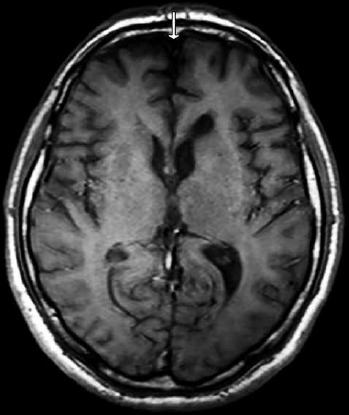
T1 weighted brain magnetic resonance image (MRI) transverse image showing frontal lobe atrophy (arrow).
Figure 3.
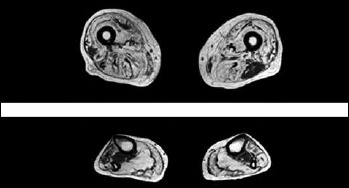
Muscle MRI of thigh and lower legs showing extensive T1w hyperintensity in most muscle groups suggesting severe fatty and/or fibrous degeneration. At thigh level there is an asymmetric relative sparing of the left biceps femoris and to a lesser extent of the left gracilis with a severe involvement of the other muscles and a characteristic patchy appearance particularly in vastus lateralis. At lower leg level there is also a severe involvement of both anterior and posterior compartment with a patchy appearance of anterior tibialis and peroneal muscles and a relative sparing of tibialis posterior muscle.
Discussion
IBMPFD is a rare, clinically heterogeneous disorder transmitted in an autosomal dominant manner (1). Myopathy is by far considered as the prevailing presenting symptom in most cohorts, though with variable pattern of muscle involvement. Although proximal shoulder and pelvic muscle weakness is frequently observed, a scapuloperoneal or a predominant distal phenotype has also been reported, even at the onset of the disease (5-7). A selective pattern of muscle involvement mainly affecting glutei, hamstrings and calf muscles, may be observed on muscle MRI, usually with an inhomogeneous distribution of fatty replacement (8). Early-onset dementia of frontotemporal type may occur in approximately one third of patients, diagnosed at a mean age of 55 years (1). Quite interestingly, cognitive decline and behavioral changes are observed in most affected members of the present family and indeed the mother of the index patient has suffered from a pure dementing syndrome, although some degree of a concomitant myopathy cannot be definitely excluded.
In the present case, the family history with the constellation of symptoms, such as myopathy, dementia and an ALS-like syndrome in members of consecutive generations, which are indicative of an apparently autosomal dominant inheritance pattern, raised the suspicion for a possible VCP-associated syndrome. Sequencing analysis of the VCP gene in the index patient revealed the already known p.R159H pathogenic mutation. This mutation has been previously described in few families with a heterogeneous presentation and severity of disease. More specifically some affected members have been diagnosed with familial ALS (fALS) and/or FTD without signs of myopathy, whereas another family had affected members with a milder phenotype consisting of myopathy and PD without signs of dementia despite their age being far above the mean age at onset of FTD in VCP carriers (9, 10). Since an increasing body of evidence suggests that ALS may be part of a wide clinical spectrum, targeted genetic panel testing may be considered in familial cases of co-occurrence of ALS with dementia syndrome and/or myopathy including VCP, C9orf72, TARDBP, SQSTM1, MATR3, HNRNPA2B1, and CHCHD10 (11).
Currently, more than 20 genes were identified to cause ALS and FTD. On the genetic basis, the complex network that underlies the pathogenesis of ALS and FTD was elucidated. Interestingly, disease-related mutant proteins form aberrant aggregates in two essential cellular machineries: The RNA quality and protein quality control machineries. Aggregations are hallmarks of many neurodegenerative disorders, and inclusions of the ubiquitous, highly conserved RNA binding protein TDP-43 represent the most important unifying marker throughout ALS molecular pathology. VCP/p97 is an important TDP-43 interaction partner and VCP proteinopathy contributes to TDP-43 dysregulation. Furthermore, TDP-43 proteinopathy is also predominantly associated with a certain frontotemporal lobar dementia (FTLD) subtype Ub+(FTLD-U)/TDP+ (FTLD-TDP). Therefore, ALS and the FTLD-TDP subtype are thought to represent different clinical manifestations of a common pathological pathway (12). Notably, aggregated, cytoplasmic TDP-43 inclusions have been detected in both hereditary and sporadic ALS with or without TARDBP mutations. Toxic gain of function and loss of function mechanisms for TDP-43 are discussed. Furthermore, at least three ALS-related genes, including VCP, MATR3 and SQSTM1/p62, have been implicated in distal myopathies (11). The evidence of combined ALS/distal myopathy phenotypes in some individuals and the presence of TDP-43 inclusions on muscle biopsy further support the hypothesis of an ALS-FTLD/myopathy continuum.
Conclusively, the present case adds to the phenotypic heterogeneity in VCP proteinopathy and highlights an even striking intrafamilial variation. Myopathy followed by rapidly cognitive decline was the clinical presentation of the index case, while the other siblings presented with different phenotypes, such as dementia and ALS. Overall, the poor genotype-phenotype correlation possibly implies that other modifying factors may contribute to the clinical heterogeneity of VCP mutations. The elucidation of the underlying pathomechanisms may explain the clinical diversity and will be essential in providing relatively accurate prognostic information and genetic counseling.
References
- 1.Evangelista T, Weihl CC, Kimonis V, et al. VCP related diseases Consortium. 215th ENMC International Workshop VCP-related multisystem proteinopathy (IBMPFD) 13-15 November 2015, Heemskerk, The Netherlands. Neuromuscul Disord 2016;26:535-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kimonis VE, Kovach MJ, Waggoner B, et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med 2000;2:232-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovach MJ, Waggoner B, Leal SM, et al. Clinical delineation and localization to chromosome 9p133-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab 2001;74:458-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krause S. Insights into muscle degeneration from heritable inclusion body myopathies. Front Aging Neurosci 2015;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Figueroa-Bonaparte S, Hudson J, Barresi R, et al. Mutational spectrum and phenotypic variability of VCP-related neurological disease in the UK. J Neurol Neurosurg Psychiatry 2015;87:680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stojkovic T, Hammouda el H, Richard P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord 2009;19:316-23. [DOI] [PubMed] [Google Scholar]
- 7.Shi Z, Hayashi YK, Mitsuhashi S, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol 2012;19:501-9. [DOI] [PubMed] [Google Scholar]
- 8.Díaz-Manera J, Llauger J, Gallardo E, et al. Muscle MRI in muscular dystrophies. Acta Myol 2015;34:95-108. [PMC free article] [PubMed] [Google Scholar]
- 9.Koppers M, van Blitterswijk MM, Vlam L, et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:837,e7-13. [DOI] [PubMed] [Google Scholar]
- 10.van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 2009;73:626-32. [DOI] [PubMed] [Google Scholar]
- 11.Sabatelli M, Marangi G, Conte A, et al. New ALS-related genes expand the spectrum paradigm of amyotrophic lateral sclerosis. Brain Pathol 2016;26:266-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Chalabi A, Jones A, Troakes C, et al. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 2012;124:339-52. [DOI] [PubMed] [Google Scholar]