

Abstract
Background
Mutated anaplastic lymphoma kinase (ALK) drives the development of advanced non-small cell lung cancer (NSCLC). Most reported small-molecule inhibitors targeting the ALK domain do not display good inhibition of the G1202R solvent front mutation. The solvent front mutation was assumed to hinder drug binding. However, a different fact could be uncovered by the simulations reported in this study through a structural analog of alectinib (JH-VIII-157-02), which demonstrated potent effects against the G1202R mutation.
Methods
Molecular docking, conventional molecular dynamics (MD) simulations, free energy calculations, and umbrella sampling (US) simulations were carried out to make clear the principles of the binding preferences of alectinib and JH-VIII-157-02 toward ALKWT and the ALK G1202R (ALKG1202R) mutation.
Results
JH-VIII-157-02 has similar binding affinities to both ALKWT and ALKG1202R whereas it has has a much lower binding affinity for alectinib to ALKG1202R. Analysis of individual energy terms indicate the major variation involves the van der Waals and entropy terms. Structural analysis reveals that the conformational change of the ATP-binding glycine-rich loop was primarily responsible for the alectinib resistance, not JH-VIII-157-02. In addition, US simulations prove JH-VIII-157-02 has similar dissociative processes from both ALKWT and ALKG1202R, while alectinib is more easily dissociated from ALKG1202R than from ALKWT, thus indicating lesser residence time.
Conclusion
Both the binding affinity and the drug residence time should be emphasized in rational drug design to overcome the G1202R solvent front mutation in ALK resistance.
Keywords: ALK, G1202R, alectinib, JH-VIII-157-02, theoretical study, resistance mechanisms
Introduction
In 2007, the discovery of a fusion of echinoderm microtubule-associated protein-like 4 with anaplastic lymphoma kinase (EML4−ALK) in 3%–7% of patients non-small cell lung cancer (NSCLC) enabled the rapid clinical development and validation of ALK inhibitors as a viable, new cancer therapeutic agent.1 Therefore, after only 4 years, crizotinib – an oral small-molecule inhibitor – was approved by the US Food and Drug Administration (FDA) for patients with ALK-positive NSCLC in 2011.2,3 Since then, several other ALK small-molecule inhibitors, such as ceritinib, alectinib, AP26113, and entrectinib, have also been identified and have entered clinical trials.4–7
Unfortunately, resistance mutations severely hamper the long-term success of ALK small-molecule inhibitors. To date, resistance mutations located in the ALK kinase domain have been identified, including L1152R, C1156Y, I1171T, F1174C, F1174L, L1196M, L1198F, G1202R, S1206Y, and G1269A.8–11 Therefore, to understand such resistance mechanisms is an important activity that will guide drug development and lead to future innovations in the treatment of NSCLC.12,13
The kinase domains of ALK consist of a bilobal structure – a smaller N-terminal lobe and a larger C-terminal lobe – which together create the catalytic site to transfer a phosphate from adenosine triphosphate (ATP) to a substrate hydroxyl (Figure 1A).14 The N-terminal lobe adopts five-stranded antiparallel β-sheets, an important regulatory αC-helix, and an ATP-binding glycine-rich loop (P-loop). In addition, the juxtamembrane segment of the N-terminus forms a β-turn motif, which is unique for ALK. The C-terminal lobe contains eight α-helices (αD-αI) and two β-stranded sheets. The crucial activation loop (A-loop, residues 1,270–1,299) in the C-lobe adopts an overall relatively extended solvent-exposed conformation. The ATP binding site is located in the cleft between the two domains. Meanwhile, the two domains are connected by a flexible linker (kinase hinge).14 Although some resistance mechanisms of ALK mutations have been investigated, few theoretical studies have been conducted to make clear the detailed mechanism of the ALK G1202R solvent front mutation (ALKG1202R).15–18
Figure 1.

Overview of the kinase domain of ALK.
Notes: (A) Overview of the ALK kinase domain structure. G1202R solvent front mutation is colored magenta. Alectinib and JH-VIII-157-02 are stick models, colored blue and green, respectively. (B) Two-dimensional structures of alectinib and JH-VIII-157-02, where the blue cycle-marked atoms are the reaction atoms used in US simulations.
Abbreviations: A-loop, crucial activation loop; ALK, anaplastic lymphoma kinase; ATP, adenosine triphosphate; P-loop, ATP-binding glycine-rich loop.
Understanding and overcoming drug resistance is an important task for the future development of potent inhibitors targeting the ALKG1202R; as James Black famously stated, “the best way to discover a new drug is to start with an old one.”19 In the present study, alectinib (RO5424802, also formerly known as CH5424802) and its structural analog JH-VIII-157-02 were used to understand the drug-resistance mechanism of ALKG1202R (Figure 1B).20,21 Alectinib is an oral drug, approved by the FDA in December 2015, for the treatment of patients with advanced ALK-positive NSCLC whose disease worsened after, or those who could not tolerate, treatment with crizotinib.20 JH-VIII-157-02 is a structural analog of alectinib that is a potent inhibitor against the G1202R solvent front mutation reported by Hatcher et al.21 Thus, in this study, umbrella sampling (US) simulations in conjunction with conventional molecular dynamics (MD) simulations and Molecular Mechanics/Generalized Born Solvent Area (MM/GBSA) free energy calculations were applied to explore the dissociative processes and the interaction profiles of alectinib and JH-VIII-157-02 to ALKWT and ALKG1202R. This study may provide some clues to elucidate mutation-induced drug resistance at the molecular level and may have important implications for the rational design of novel ALK inhibitors to combat the drug resistance of the ALK G1202R solvent front mutation in NSCLC treatment.
Materials and methods
Protein structure preparation
The three-dimensional structure of human ALKWT/alectinib (PDB ID: 3AOX) was retrieved from the Protein Data Bank (PDB), available at www.pdb.org.5 ALKG1202R was constructed by using EasyModeller software.22,23 Furthermore, both the crystal structure and modeled structure were refined by PyMol software and the Assisted Model Building with Energy Refinement 16 (Amber 16) simulation package, including removal of all non-bonded hetero-atoms, water molecules, addition of missing hydrogen atoms, and energy minimization. Then, the prepared structures were used to undertake molecular docking and MD simulations.
Construction of ALKG1202R/alectinib, ALKWT/JH-VIII-157-02, and ALKG1202R/JH-VIII-157-02 models
Molecular modeling is a vital component in structure-based drug design. In this study, molecular docking – a method which predicts the preferred orientation of one molecule to another – was used to construct the ALKG1202R/alectinib, ALKWT/JH-VIII-157-02, and ALKG1202R/JH-VIII-157-02 models by the latest version of the AutoDock 4.2.6 package.24 AutoDock is a flexible docking program, which is one of the most cited docking software in the research community. It is especially effective for protein–ligand docking. Prior to the docking step, atomic radii of AutoDock4 were assigned to the prepared ALKWT and ALKG1202R protein. Meanwhile, Gasteiger partial charges were assigned to alectinib and JH-VIII-157-02. A grid of 60, 60, and 60 with a grid spacing of 0.375 was created in x, y, and z directions, respectively, surrounding the binding site. The affinity maps of ALKWT and ALKG1202R were calculated using AutoGrid4 software. The docking protocol was as follows: trials of 100 dockings which were clustered according to the root mean square deviation (RMSD) tolerance of 2.0 Å, a population size of 300, with a maximum number of evaluations of 25,000,000, mutation rate set to 0.02, and other parameters set to default settings. AutoDockTools and PyMol were used to analyze the docking results.24,25
Conventional MD simulations
The Amber 16 simulation package was employed for both the conventional MD and US simulations. The X-ray crystal structure of ALKWT/alectinib (PDB ID: 3AOX) and modeled structures of ALKG1202R/alectinib, ALKWT/JH-VIII-157-02, and ALKG1202R/JH-VIII-157-02 were used as the initial structures for conventional MD simulations.5 Before conventional MD simulations, the ligands and proteins were constructed by antechamber and LEaP modules in the Amber 16 simulation package. The proteins were described by the Amber ff14SB force field.26 Both alectinib and JH-VIII-157-02 employed the generalized Amber force field (GAFF), with partial charges assigned by a restrained electrostatic potential (RESP) fitting method based on the electrostatic potentials computed at the Hartree–Fock (HF) SCF/6-31G* level of theory.27 The box dimensions ensured that any protein atom was at least 20 Å away from the wall of the box with periodic boundary condition and also was solvated by TIP3P water molecules. Besides, appropriate numbers of sodium ions were added to neutralize all systems.
Prior to MD productive simulations, an equilibration protocol was carried out, including an initial minimization comprising 5,000 steps of steepest descent and 5,000 steps of conjugate gradient to the solvent molecules. Afterward, the side chains of proteins were relaxed with harmonic restraints of 10 kcal mol−1 Å−2 comprising 5,000 steps of steepest descent and 5,000 steps of conjugate gradient. Then, all molecules were relaxed in the water box, including 5,000 steps of steepest descent and 5,000 steps of conjugate gradient. Thereafter, all systems were heated from 0 K to 300 K using a time constant at a constant volume over a period of 500 ps. Subsequently, all systems were equilibrated at a constant pressure of 1 bar for 1 ns. Finally, each system was submitted to 100 ns conventional MD simulation in the isothermal-isobaric ensemble without any restraint. During the productive simulations, the Particle Mesh Ewald (PME) algorithm was utilized to consider the long-range electrostatic interactions of a periodic box with a cutoff of 10 Å, and the bonds involved in hydrogen atoms were constrained by the SHAKE algorithm.28,29 Pressure and temperature were maintained using the Langevin temperature scalings.30 A time step of 2 fs was applied, and coordinates were saved every 20 ps for further analysis.
Free energy calculations
The MM/GBSA methodology has been widely used in elucidating the mechanisms of mutation-induced drug resistance.15,17,31–33 The MM/GBSA methodology computes the binding free energy (ΔGbind) by utilizing a thermodynamic cycle that combines the molecular mechanical energies with the continuum solvent approaches.34 The ΔGbind in this study was computed by using the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
where ΔGbind in Equation (1) is the total binding free energy between the ligand and the receptor, which is also equal to Equation (2). ΔGcom, ΔGrec, and ΔGlig are the free energies of the complex, receptor, and ligand, respectively. ΔEMM and ΔGsol represent the molecular mechanics interaction and solvation energies. TΔS represents the change of the conformational entropy upon ligand binding at temperature T. ΔEMM can be split into three terms (Equation 3): intermolecular interaction energy (ΔEint), van der Waals energy (ΔEvdW), and electrostatic energy (ΔEelec). In Equation (4), the solvation free energy (ΔGsol) includes the polar (ΔGGB) and non-polar (ΔGSA) components. In this study, by using the single-trajectory strategy, ΔEint is completely canceled between ligand, receptor, and complex. The polar component (ΔGGB) was calculated using a Generalized Boltzmann (GB) model equal to 5.35,36 The interior and exterior dielectric constants were set to 1 and 80, respectively. The nonpolar component (ΔGSA) accounts for the nonpolar contribution to solvation and was approximated by relating it to the solvent accessible surface area with a coefficient of 0.0072. The contribution of entropy (−TΔS) to ΔGbind was estimated by normal mode analysis using the NMODE module in the AmberTools 16 program.37 The normal mode conformational entropy (SNM) is a combination of the following three components:
| (5) |
In Equation (5), Strans, Srot, and Svib represent changes in translational, rotational, and vibrational freedoms, respectively.
In the present study, trajectories from conventional MD simulations between 80 and 100 ns with 500 snapshots were applied to binding free energy calculations and free energy decompositions. Only a 100 snapshots for each system were used to estimate the contribution of the entropy due to the high computational demand.
US simulations
The equilibrated snapshots extracted from the trajectories were used as initial structures for the US simulations due to the G1202R solvent front mutation that may affect the binding geometries to alectinib and JH-VIII-157-02. In this study, the distance between one atom in the receptor (Cα in Ile-1171 of ALKWT and ALKG1202R) and another carbon atom in the ligand (blue cycle, Figure 1B) were selected as the reaction coordinate (RC).
The RCs of these systems were extended 20 Å from the initial distance and separated into 41 windows by a step of 0.5 Å. For each window, 8-ns MD simulations were applied to each window to guarantee the convergence for each system. Additionally, the harmonic potential was used as the original potential for each window to create a thermodynamic state from one state to another. In this study, an elastic constant of 5 kcal mol−1 Å−2 was applied to all the windows to pull each ligand away from the binding cavity at a constant speed and force. The weighted histogram analysis method (WHAM) was carried out to calculate the potential of mean force (PMF) along the RCs.38 Each RC was split into 2,000 bins, and the temperature was set to 300 K for the WHAM calculation.
Results and discussion
RMSD analysis of the receptor–ligand backbones
In this study, homologous modeling and molecular docking were used to generate the initial structures of ALKG1202R/alectinib, ALKWT/JH-VIII-157-02, and ALKG1202R/JH-VIII-157-02. It is essential to investigate the structural stability of the three modeled complexes. Therefore, 100-ns conventional MD simulations were carried out to probe the structural stability of the modeled complexes and the crystal structure of ALKWT/alectinib, selected as the control. To monitor the stability and flexibility of these systems, the RMSDs of the Cα atoms from their initial position were analyzed. As shown in Figure 2, the RMSD of protein backbones of all systems quickly reached a steady state after 20–50 ns of simulation. All ligands in each system reached equilibrium and stabilization during the last 50 ns. Moreover, superposition of the conformations between the initial structure and last snapshot show a similar pattern, with minor adjustments. However, it can also be observed that the conformational change of the modeled systems was larger than that of the crystal structure (Figure 3). Overall, these results can be explained as an induced-fit phenomenon that the modeled complexes undergo conformational change to reach the optimal binding mode, and the selection of the initial conformations were reasonable and satisfactory for further analysis.
Figure 2.

RMSD analysis of alectinib and JH-VIII-157-02 to ALKWT and ALKG1202R.
Notes: (A) ALKWT and alectinib; (B) ALKG1202R and alectinib; (C) ALKWT and JH-VIII-157-02; and (D) ALKG1202R and JH-VIII-157-02.
Abbreviations: ALK, anaplastic lymphoma kinase; ATP, adenosine triphosphate; RMSD, root mean square deviation.
Figure 3.

Alignment of the initial structure (cyan) and the last snapshot (yellow).
Notes: (A) ALKWT and alectinib; (B) ALKG1202R and alectinib; (C) ALKWT and JH-VIII-157-02; and (D) ALKG1202R and JH-VIII-157-02.
Abbreviations: ALK, anaplastic lymphoma kinase; ATP, adenosine triphosphate.
Interestingly, RMSD for the heavy atoms of alectinib in ALKG1202R was more fluctuant than for the other three systems during the whole conventional MD simulation. One might, therefore, conclude that alectinib in ALKG1202R is more flexible to a degree when compared to the wild type (WT). Afterward, the root mean square fluctuations of the protein backbone of all subunits were calculated, and we noticed that the P-loop region of ALKG1202R/alectinib exhibits amplified fluctuations when compared to other systems. Therefore, the distribution of the degree of opening of the P-loop, which represents the degree of opening of the binding pocket, was undertaken. The degree of opening was defined as the center of mass of the P-loop to the center of mass of the ligand. As shown in Figure 4A and C, the P-loop region of ALKG1202R/alectinib shows a higher degree of opening than the ALKWT/alectinib because the G1202R mutation would require rearrangement of the protein and rigid structure of alectinib. For comparison, the more flexible structure of JH-VIII-157-02 in ALKWT and ALKG1202R shows a similar distribution of the degree of opening (Figure 4B and D), indicating that a more flexible structure may overcome the G1202R solvent front mutation.
Figure 4.

The distribution of the opening degree of the P-loop. The opening degree was defined as the center of mass of the P-loop to the center of mass of the ligand.
Notes: (A) Alignment of the most populated conformations between ALKWT/alectinib (gray) and ALKG1202R/alectinib (magenta); (B) alignment of the most populated conformations between ALKWT/JH-VIII-157-02 (gray) and ALKG1202R/JH-VIII-157-02 (magenta); (C) the distribution of the opening degree of the P-loop between ALKWT/alectinib and ALKG1202R/alectinib; (D) the distribution of the opening degree of the P-loop between ALKWT/JH-VIII-157-02 and ALKG1202R/JH-VIII-157-02.
Abbreviations: ALK, anaplastic lymphoma kinase; ATP, adenosine triphosphate; P-loop, ATP-binding glycine-rich loop.
Free energy calculations
Conventional MD simulation is commonly used to undertake free energy calculations in the field of computational biophysics. Herein, the free energies were decomposed into the contributions of each energy term for all systems to analyze the energy contributions in determining the receptor–ligand association on the basis of the MM/GBSA methodology. As shown in Table 1, the predicted binding affinities of ALKWT/alectinib, ALKG1202R/alectinib, ALKWT/JH-VIII-157-02, and ALKG1202R/JH-VIII-157-02 are −24.85±7.77, −13.59±4.92, −25.13±7.02, and −25.01±9.01 kcal/mol, respectively. Obviously, it can be observed that the ΔGbind shows significant correlation with experimental data, and various energy terms contribute differently to binding free energies for each system. Herein, we only discussed the van der Waals energy (ΔEvdW) and entropy term (−TΔS). As listed in Table 1, the decrease of van der Waals energy and the increase of entropy both lead to a decrease of the binding free energy for the G1202R solvent front mutation in ALKG1202R/alectinib as compared to ALKWT/alectinib. On comparison, there were no significant differences between ALKWT and ALKG1202R with regard to van der Waals energy and entropy for JH-VIII-157-02. Herein, combining the structural analysis that the P-loop confers a greater degree if opening of the binding pocket for the ALKG1202R/alectinib system and the increase of the entropy calculated from the MM/GBSA method, it can be concluded that the binding of alectinib to the ALK G1202R solvent front mutation with a resultant more flexible P-loop and increased conformational entropy substantially leads to resistance to alectinib.
Table 1.
Binding free energies of alectinib and JH-VIII-157-02 in ALKWT and ALKG1202R (kcal/mol)
| Name | ALKWT/alectinib | ALKG1202R/alectinib | ALKWT/JH-VIII-157-02 | ALKG1202R/JH-VIII-157-02 |
|---|---|---|---|---|
| ΔEvdW | −51.27±2.76 | −46.88±2.79 | −51.05±3.79 | −52.42±3.66 |
| ΔEelec | −6.86±2.36 | −6.58±3.61 | −13.88±5.73 | −15.32±6.73 |
| ΔGGB | 24.07±2.43 | 23.53±3.15 | 32.77±5.38 | 34.97±5.59 |
| ΔGSA | −5.32±0.26 | −6.98±0.21 | −6.01±0.44 | −6.47±0.31 |
| TΔS | −14.53±7.38 | −21.32±4.01 | −13.04±6.29 | −14.23±8.53 |
| ΔEenthalpy | −39.38±2.41 | −34.91±2.86 | −38.17±3.45 | −39.24±2.89 |
| ΔGbind | −24.85±7.77 | −13.59±4.92 | −25.13±7.02 | −25.01±9.01 |
| ΔWPMF | −14.39±0.76 | −9.92±0.42 | −14.48±0.46 | −13.99±1.21 |
| IC50 (nM) | 2 | 207 | 2 | 2 |
Abbreviations: ΔEvdW, van der Waals energy; ΔEelec, electrostatic energy; ΔGGB, electrostatic contribution to solvation; ΔGSA, non-polar contribution to solvation; TΔS, entropic contribution; ΔEenthalpy, binding free energy in the absence of entropic contribution; ΔWPMF, potential of mean force depth based on 16–20 Å along the reaction coordinate; ΔGbind, binding free energy; ALK, anaplastic lymphoma kinase; IC50, half maximal inhibitory concentration.
To further understand the receptor–ligand recognition patterns and emphasize the important energetic distributions to drug resistance, the decomposition of the receptor–ligand interaction was also generated by the MM/GBSA method. As shown in Figure 5A and B, the interactions between ALKWT and alectinib are mainly mediated by Leu-1122, Met-1199, Leu-1198, Leu-1256, Val-1130, Gly-1202, Ala-1200, Leu-1196, Ala-1148, and Val-1180. The major favorable energetic contributions for ALKG1202R/alectinib originate predominantly from the residues Met-1199, Val-1180, Val-1130, Leu-1198, Leu-1256, Ala-1200, Leu-1196, Leu-1198, Ala-1148, Arg-1202, Leu-1122, and Val-1130 (Figure 5C and D). Meanwhile, the major favorable energy contributions for ALKWT/JH-VIII-157-02 are Leu-1122, Leu-1256, Met-1199, Val-1130, Leu-1198, Gly-1202, Leu-1196, Ala-1148, Val-1180, and Lys-1150 (Figure 5E and F). The 10 most contributed key residues for the ALKG1202R/JH-VIII-157-02 are Leu-1122, Arg-1202, Met-1199, Leu-1198, Val-1130, Leu-1256, Ala-1200, Leu-1196, Ala-1148, and Val-1180 (Figure 5G and H). It can be seen that the predominant residues for all simulated systems are hydrophobic amino acids, and these systems share mostly identical key residues, located mainly in the hinge region and P-loop or near them. These results not only show the most contributed key residues, but also indicate that the enthalpy cannot be ignored and that entropy is also one of the major variations that govern binding free energy (Table 1).
Figure 5.

The 10 most contributed residues of alectinib and JH-VIII-157-02 to ALKWT and ALKG1202R.
Notes: The most contributed Per-residue interaction spectra of ALKWT/alectinib (A), ALKG1202R/alectinib (C), ALKWT/JH-VIII-157-02 (E), and ALKG1202R/JH-VIII-157-02 (G); structural analysis of ALKWT/alectinib (B), ALKG1202R/alectinib (D), ALKWT/JH-VIII-157-02 (F), and ALKG1202R/JH-VIII-157-02 (H).
Abbreviation: ALK, anaplastic lymphoma kinase.
Assessing G1202R resistance mechanisms by US simulations
Conventional MD simulations are not appropriate to simulate biological processes due to the fact that these processes usually occur on the microsecond to millisecond, or even longer, timescales.39 Therefore, biasing simulation methods have been found useful in enhanced MD sampling simulation techniques and improve the efficiency of conformational sampling, such as through accelerated MD simulation, replica exchange MD simulation, steered MD simulation, and the US simulation method.40–43 In this study, US simulation – a method which overcomes the problem of sampling trap into local minima by adding a bias to the potential function along certain RCs – was carried out to characterize the unbinding processes of alectinib and JH-VIII-157-02 in ALKWT and ALKG1202R to further clarify the resistance mechanisms.
In this study, the equilibrated snapshots extracted from the 100-ns conventional MD simulations trajectories were used as initial structures for a total of 1,312-ns US simulations. Guaranteeing the sampling convergence of the US simulations is inevitable; therefore, 8-ns US simulations for each window were conducted for all systems, and the convergence of PMF was checked after each nanosecond. As shown in Figure 6, all systems are convergent after roughly 5-ns US simulations, suggesting that 5-ns simulations for each window are sufficient to achieve convergent sampling (difference of PMFs <0.5 kcal/mol).
Figure 6.

Convergence of the PMFs calculated for four simulated systems by US simulations. The PMFs gradually decrease with the increase in the simulation time. When the simulation time was lengthened to 5–8 ns for each window, the PMFs became convergent.
Note: Converged PMFs of ALKWT/alectinib (A), ALKG1202R/alectinib (B), ALKWT/JH-VIII-157-02 (C), and ALKG1202R/JH-VIII-157-02 (D).
Abbreviations: ALK, anaplastic lymphoma kinase; PMF, potential of mean force.
As listed in Table 1, it can be seen that the binding free energies of ALKWT/alectinib, ALKG1202R/alectinib, ALKWT/JH-VIII-157-02, and ALKG1202R/JH-VIII-157-02 predicted by US simulations are −14.39±0.76, −9.92±0.42, −14.48±0.46, and −13.99±1.21 kcal/mol, respectively. These results can be correctly ranked and show good consistency with the experimental data. It should be noted that the binding free energies based on the PMF depth (ΔWPMF) and standard binding free energy (ΔGbind) from the MM/GBSA method are the same with respect to the corresponding systems – that is, not much effort should be taken to distinguish the ΔWPMF and ΔGbind in our US simulation strategy. The conventional MD simulations and binding free energy calculations make it hard to give the detailed free energies of a small-molecule dissociated along the unbinding pathways, therefore the PMF curves were used to clarify the different dissociative processes of alectinib and JH-VIII-157-02 in ALKWT and ALKG1202R. As shown in Figure 7, it is obvious that the PMF curves for alectinib in ALKWT are different from that for ALKG1202R. There are various peaks and valleys for ALKWT/alectinib than for ALKG1202R/alectinib. This finding indicated alectinib dissociates from ALKWT and ALKG1202R in a significantly different manner. Dissociating from ALKWT needs to overcome a much higher free energy barrier than that from ALKG1202R. Compared with alectinib, both JH-VIII-157-02 in ALKWT and ALKG1202R need to overcome a considerable free energy barrier.
Figure 7.
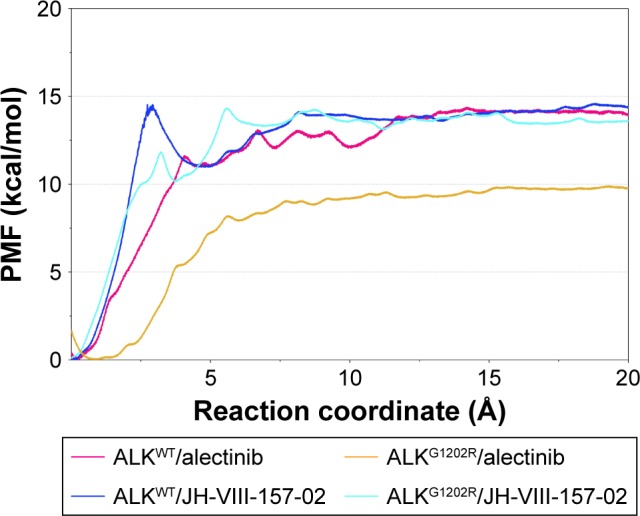
Comparison of the converged PMFs based on different complexes by US simulations.
Abbreviations: ALK, anaplastic lymphoma kinase; PMF, potential of mean force.
Furthermore, the alignment of all converged PMFs shows no increase at ∼12 Å along the RCs (Figure 7). It is well known that the higher the curve, the more the energy released during ligand binding to the receptor. Similar ΔWPMF were clearly observed for ALKWT/alectinib, ALKWT JH-VIII-157-02, and ALKG1202R/JH-VIII-157-02, whereas the ΔWPMF of ALKG1202R/alectinib is much lower than for the other three systems. In other words, alectinib forms a higher affinity to ALKWT, and the G1202R are able to cause resistance. In summary, drug resistance may be caused by the mutation-induced conformational change of the energetic distributions and dissociative processes between the inhibitors and the binding pocket.
MD simulations and drug discovery
MD simulations can offer insights into protein motion and often play important roles in drug discovery. For instance, nuclear magnetic resonance and X-ray crystallographic structures often show well-established binding pockets. However, sometimes, the models produced by these experimental techniques obscure other potentially drug-able sites. MD simulations is an excellent tool for identifying these cryptic sites. As an example, Schames et al44 conducted an MD simulation of HIV integrase in 2004, and the simulation revealed a previously unidentified binding site that was not evident from any of the available crystal structures.44 As predicted, this crystal structure later displayed that known inhibitors do, in fact, bind to this binding site. Further development was subsequently applied to the discovery of a highly effective antiretroviral drug, raltegravir – the first FDA-approved HIV integrase inhibitor.45
Despite the significant advances made in clinical trials for novel ALK agents, improvements are still required.46 The dynamic and energetic aspects obtained in this study are crucial and helpful to undertake structure-based virtual screening to rationally develop next-generation inhibitors to overcome the drug resistance caused by G1202R in ALK.
Conclusion
In this study, molecular docking, conventional MD simulations, and MM/GBSA free energy calculations successfully modeled the G1202R solvent front mutation systems. Moreover, the binding free energies of alectinib and JH-VIII-157-02 in ALKWT and ALKG1202R were well pre dicted. The analysis of individual energy terms suggests that the major variations of alectinib binding between ALKWT and ALKG1202R are van der Waals energy and entropy term, compared to JH-VIII-157-02. Further structural analysis revealed that the conformational change of the P-loop plays an important role for alectinib in the resistance of the G1202R solvent front mutation. Additionally, US simulations were used to prove the principle of the binding preference of alectinib and JH-VIII-157-02 toward ALKWT and ALKG1202R. The ΔWPMF derived from US simulations were consistent with the experimental data, and obvious energy barriers were observed from the PMF profile of alectinib toward ALKWT. However, no energy barrier was observed toward ALKG1202R, suggesting alectinib is more easily dissociated from ALKG1202R than from ALKWT, thus indicating lesser residence time. In contrast, the more flexible chemical structure of JH-VIII-157-02 has similar dissociative processes in both ALKWT and ALKG1202R. Our results unambiguously suggested that both the conformational change of the P-loop and the variance of the van der Waals interactions are responsible for the resistance of the ALK G1202R mutation due to the inherent planar and rigid chemical structure of alectinib. Overall, our study not only revealed the determinants of drug resistance but also provided a feasible way to predict the unbinding kinetics of protein–ligand systems that can potentially facilitate the rational design of highly selective inhibitors with longer residence time for the ALK G1202R mutation.
Acknowledgments
Financial support was received from the Natural Science Foundation of Fujian Province, People’s Republic of China (grant no 2017J01831).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 2.Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6(12):3314–3322. doi: 10.1158/1535-7163.MCT-07-0365. Pt 1. [DOI] [PubMed] [Google Scholar]
- 3.Cui JJ, Tran-Dubé M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54(18):6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 4.Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56(14):5675–5690. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 5.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menichincheri M, Ardini E, Magnaghi P, et al. Discovery of entrectinib: a new 3-aminoindazole as a potent anaplastic lymphoma kinase (ALK), c-ros oncogene 1 kinase (ROS1), and pan-tropomyosin receptor kinases (Pan-TRKs) inhibitor. J Med Chem. 2016;59(7):3392–3408. doi: 10.1021/acs.jmedchem.6b00064. [DOI] [PubMed] [Google Scholar]
- 8.Choi YL, Soda M, Yamashita Y, et al. ALK Lung Cancer Study Group EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki T, Okuda K, Zheng W, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70(24):10038–10043. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54–61. doi: 10.1056/NEJMoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iragavarapu C, Mustafa M, Akinleye A, et al. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17. doi: 10.1186/s13045-015-0122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. 2016;9:19. doi: 10.1186/s13045-016-0251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CC, Jia Y, Li N, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010;430(3):425–437. doi: 10.1042/BJ20100609. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Sun R, Wu Y. L1198F mutation resensitizes crizotinib to ALK by altering the conformation of inhibitor and ATP binding sites. Int J Mol Sci. 2017;18(3):E482. doi: 10.3390/ijms18030482. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagasundaram N, Wilson Alphonse CR, Samuel Gnana PV, Rajaretinam RK. Molecular dynamics validation of crizotinib resistance to ALK mutations (L1196M and G1269A) and identification of specific inhibitors. J Cell Biochem. 2017;118(10):3462–3471. doi: 10.1002/jcb.26004. [DOI] [PubMed] [Google Scholar]
- 17.Sun H, Li Y, Li D, Hou T. Insight into crizotinib resistance mechanisms caused by three mutations in ALK tyrosine kinase using free energy calculation approaches. J Chem Inf Model. 2013;53(9):2376–2389. doi: 10.1021/ci400188q. [DOI] [PubMed] [Google Scholar]
- 18.Kumar A, Shanthi V, Ramanathan K. Computational investigation and experimental validation of crizotinib resistance conferred by C1156Y mutant anaplastic lymphoma kinase. Mol Inform. 2015;34(2–3):105–114. doi: 10.1002/minf.201400070. [DOI] [PubMed] [Google Scholar]
- 19.Raju TN. The Nobel chronicles. 1988: James Whyte Black, (b 1924), Gertrude Elion (1918–1999), and George H Hitchings (1905–1998) Lancet. 2000;355(9208):1022. doi: 10.1016/s0140-6736(05)74775-9. [DOI] [PubMed] [Google Scholar]
- 20.McKeage K. Alectinib: a review of its use in advanced ALK-rearranged non-small cell lung cancer. Drugs. 2015;75(1):75–82. doi: 10.1007/s40265-014-0329-y. [DOI] [PubMed] [Google Scholar]
- 21.Hatcher JM, Bahcall M, Choi HG, et al. Discovery of inhibitors that overcome the G1202R anaplastic lymphoma kinase resistance mutation. J Med Chem. 2015;58(23):9296–9308. doi: 10.1021/acs.jmedchem.5b01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 23.Kuntal BK, Aparoy P, Reddanna P. EasyModeller: a graphical interface to MODELLER. BMC Res Notes. 2010;3:226. doi: 10.1186/1756-0500-3-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDock-Tools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 26.Lindorff-Larsen K, Piana S, Palmo K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 28.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. [Google Scholar]
- 29.Kräutler V, Van Gunsteren WF, Hünenberger PH. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem. 2001;22(5):501–508. [Google Scholar]
- 30.Izaguirre JA, Catarello DP, Wozniak JM, Skeel RD. Langevin stabilization of molecular dynamics. J Chem Phys. 2001;114(5):2090–2098. [Google Scholar]
- 31.Xue W, Pan D, Yang Y, Liu H, Yao X. Molecular modeling study on the resistance mechanism of HCV NS3/4A serine protease mutants R155K, A156V and D168A to TMC435. Antiviral Res. 2012;93(1):126–137. doi: 10.1016/j.antiviral.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Liang D, Chen Q, Guo Y, Zhang T, Guo W. Insight into resistance mechanisms of AZD4547 and E3810 to FGFR1 gatekeeper mutation via theoretical study. Drug Des Devel Ther. 2017;11:451–461. doi: 10.2147/DDDT.S129991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srivastava HK, Sastry GN. Molecular dynamics investigation on a series of HIV protease inhibitors: assessing the performance of MM-PBSA and MM-GBSA approaches. J Chem Inf Model. 2012;52(11):3088–3098. doi: 10.1021/ci300385h. [DOI] [PubMed] [Google Scholar]
- 34.Miller BR, 3rd, McGee TD, Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8(9):3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 35.Onufriev A, Bashford D, Case DA. Modification of the generalized Born model suitable for macromolecules. J Phys Chem B. 2000;104(15):3712–3720. [Google Scholar]
- 36.Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO) J Comput Chem. 1999;20(2):217–230. [Google Scholar]
- 37.Kongsted J, Ryde U. An improved method to predict the entropy term with the MM/PBSA approach. J Comput Aided Mol Des. 2009;23(2):63–71. doi: 10.1007/s10822-008-9238-z. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem. 1992;13(8):1011–1021. [Google Scholar]
- 39.Bernardi RC, Melo MC, Schulten K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim Biophys Acta. 2015;1850(5):872–877. doi: 10.1016/j.bbagen.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys. 2004;120(24):11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 41.Zhou R. Replica exchange molecular dynamics method for protein folding simulation. Methods Mol Biol. 2007;350:205–223. doi: 10.1385/1-59745-189-4:205. [DOI] [PubMed] [Google Scholar]
- 42.Park S, Schulten K. Calculating potentials of mean force from steered molecular dynamics simulations. J Chem Phys. 2004;120(13):5946–5961. doi: 10.1063/1.1651473. [DOI] [PubMed] [Google Scholar]
- 43.Okamoto Y. Generalized-ensemble algorithms: enhanced sampling techniques for Monte Carlo and molecular dynamics simulations. J Mol Graph Model. 2004;22(5):425–439. doi: 10.1016/j.jmgm.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Schames JR, Henchman RH, Siegel JS, Sotriffer CA, Ni H, McCammon JA. Discovery of a novel binding trench in HIV integrase. J Med Chem. 2004;47(8):1879–1881. doi: 10.1021/jm0341913. [DOI] [PubMed] [Google Scholar]
- 45.Steigbigel RT, Cooper DA, Kumar PN, et al. BENCHMRK Study Teams Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med. 2008;359(4):339–354. doi: 10.1056/NEJMoa0708975. [DOI] [PubMed] [Google Scholar]
- 46.Su S, Wu YL. Clinical trials of tyrosine kinase inhibitors for lung cancer in China: a review. J Hematol Oncol. 2017;10(1):147. doi: 10.1186/s13045-017-0514-z. [DOI] [PMC free article] [PubMed] [Google Scholar]