

Abstract
For mice, a maternal vitamin A (VA) deficient diet initiated from mid-gestation (GVAD) produces serum retinol deficiency in mature offspring. We hypothesize that the effects of GVAD arise from pre-weaning developmental changes. We compare the effect of this GVAD protocol in combination with a post-weaning high fat diet (HFD) or high carbohydrate diet (LF12). Each is compared to an equivalent VA sufficient combination. GVAD extensively decreased serum retinol and liver retinol, retinyl esters, and retinoid homeostasis genes (Lrat, Cyp26b1, and Cyp26a1). These suppressions were each more effective with LF12 than with HFD. Post-weaning initiation of VA deficiency with LF12 depleted liver retinoids, but serum retinol was unaffected. Liver retinoid depletion, therefore, precedes serum attenuation. Maternal LF12 decreased the obesity response to the HFD, which was further decreased by GVAD. LF12 fed to the mother and offspring extensively stimulated genes marking stellate activation (Col1a1, Timp2, and Cyp1b1) and novel inflammation markers (Ly6d, Trem2, Nupr1). The GVAD with LF12 diet combination suppressed these responses. GVAD in combination with the HFD increased these same clusters. A further set of expression differences on the HFD when compared to a high carbohydrate diet were prevented when GVAD was combined with HFD. Most of these GVAD gene changes match published effects from deletion of Nr0b2/Shp, a retinoid-responsive, nuclear co-repressor that modulates metabolic homeostasis. The stellate and inflammatory increases seen with the high carbohydrate, LF12 diet may represent postprandial responses. They depend on retinol and Shp, but the regulation reverses with a HFD.
Keywords: Retinoids, vitamin A deficiency, liver, stellate cell activation, small heterodimer partner (Nr0b2/Shp)
1. Introduction
Vitamin A (VA)/retinol is an essential lipophilic micronutrient that regulates development, reproduction and immunity. While a retinaldehyde isomer functions in the retina of the eye as a light sensor, retinol is largely active through conversion to retinoic acid (RA) by a set of selective alcohol and aldehyde dehydrogenases (Rdh1, 10, 15; Aldh1a1-3) [1]. RA activates transcription in the nucleus through a set of receptors, RAR forms (α, β, γ), that, like other non-steroid receptors, function as heterodimers with retinoid X receptors (RXRs) which can be activated by 9-cis retinol [2]. Retinol may scavenge oxygen radicals and may have important functions as an antioxidant [3]. Retinol and RA play key spatiotemporal roles in controlling morphogenic aspects of embryogenesis and later in organogenesis [4].
Fetal development relies on maternal transplacental transfer of retinol [5], which decreases maternal liver retinyl ester (RE) content and disrupts serum retinol homeostasis between gestational day 9.5 and 15.5 in mice [6]. In rats, a post-weaning VA-deficient diet (PWVAD) is sufficient to appreciably lower serum and liver retinoids and to change liver gene expression in accord with altered homeostasis [7, 8]. However, mice exhibit stronger homeostatic control, possibly via coprophagy and recycling of retinoids, as well as conversion of provitamin A carotenoids in the diet [7, 9-11]. Mice only show depletion of serum retinol when VA deficiency is initiated in the maternal diet at mid-gestation and maintained in the post-weaning diet (GVAD). Attenuation of serum retinol concentration becomes extensive as mice approach maturity [10, 12]. A liver decrease in RE stores is likely essential for serum retinol depletion; however, additional gene changes produced during fetal development may be required. We have developed a maternal diet that is particularly effective in causing this serum retinol depletion (VAD diet).
Dietary retinol is absorbed by intestinal epithelia and esterified by lecithin:retinol acyltransferase (LRAT) to RE, which are packaged in chylomicra with triglycerides, and transported through the lymphatic system. Triglycerides are deposited at target tissues and the remaining chylomicron remnants are taken up by liver hepatocytes, which process RE for transport and storage [13-16]. Retinol binding protein 4 (RBP4), produced in the liver and adipose, sequesters retinol in the blood [17]. Holo-RBP4 complexes are internalized by binding with a plasma membrane receptor, STRA6, which then transfers the retinol to the intracellular binding protein, RBP1 [18]. Recent work shows that hepatocytes predominantly express a related receptor, RBPR2, that functions in the same way as STRA6 [19].
The liver is the major storage site for RE, within specialized stellate cells that comprise less than five percent of total liver cells and are located between endothelia and hepatocytes in the sinusoids [20]. The stellate cell is key to the progression of steatosis, wherein endothelia, hepatocytes, macrophages, and platelets produce cytokines, which transform the quiescent stellate cell to an activated myofibroblast morphology [20]. Activation has been characterized by a loss of retinoid storage and increases in proliferation, contractility, matrix degradation and immune cell recruitment [21]. In the absence of stress, the liver contains an intermediate stellate cell type that retains RE in lipid droplets and expresses genes that are characteristic of myofibroblasts, including collagen 1a1 (Col1a1) and cytochrome P450 1b1 (Cyp1b1) [22]. We are particularly interested in Cyp1b1 in the context of the effects of retinoids on obesity because this mRNA increases during adipogenesis [23] and is expressed during early development at sites of high retinol activity, suggesting regulatory crosstalk [24].
The relationship between retinoids and metabolic homeostasis has been extensively studied with respect to catabolism and storage of lipids [8, 15, 25, 26]. Retinoic acid administration can decrease adiposity, improve glucose tolerance [15], and reverse stellate cell activation [21]. Deletion of Aldh1a1 allows accumulation of retinaldehyde, the precursor of RA, and prevents diet-induced obesity (DIO) [27]. While retinoids are largely anti-adipogenic, supplementation of VA in a high lipid content diet increases adipose tissue weight [28]. A mid-gestation VA deficient diet decreases fasting-mediated PPARα activation, concomitant with decreased fatty acid oxidation and increased triglyceride accumulation [25].
We have previously investigated the effects of a post-weaning high fat diet (60 percent kcal from fat, 20 percent kcal carbohydrate; HFD) compared to an approximately isocaloric low fat diet (10 percent kcal fat, 70 percent kcal carbohydrate; LFD). DIO is recognized by increased weight gain and adipose deposition with the HFD relative to the LFD [29, 30]. The DIO changes in males only appear at maturity (approximately diet week 5), at the same time that the GVAD protocol starts to decrease serum retinol.
Mice in which Cyp1b1 is deleted exhibit suppressed DIO [29, 30]. We have identified genes that responded to Cyp1b1 deletion and the difference in diet, HFD versus LFD [30], including gene clusters that did not respond to Cyp1b1 deletion and also those that responded to the diet difference to the same extent in wild type and Cyp1b1-/- mice, which we refer to as HF genes. While the genes that respond to Cyp1b1 deletion are mostly controlled by endocrine mechanisms [30], we show here that HF genes are highly responsive to GVAD.
Similar to Cyp1b1-/- mice, GVAD is effective in blocking DIO. We hypothesize that VA deficiency from the GVAD protocol will affect the fetal/neonatal developmental regulation and imprint changes on liver metabolic homeostasis that are evident in adult offspring.
We have examined the selective responses produced by GVAD combined with a HFD (GVAD-HFD) and, here, provide evidence that the application of GVAD to a novel post-weaning diet with low fat content (LF12) (GVAD-LF12) enhances the extent of serum retinol decreases in the offspring and changes their effects. Additionally, we examine the effects of changing the maternal diet from the standard breeder diet (BD) to the LF12 diet, which have appreciably different fat content. Gene expression was effectively sorted into functionally related groups based on expression ratios between different treatment groups.
The effects of GVAD reveal that several diet-dependent gene clusters rely highly on retinol and show extensive stimulation by the LF12 diet in the maternal and post-weaning periods. Evidence is presented that the high carbohydrate content of the diet activates stellate cells and markers of a novel inflammatory response. We report a remarkable overlap between genes that respond to GVAD and those that are respond to deletion of the co-repressor, Shp/Nr0b2.
2. Materials and Methods
2.1 Animal care and husbandry
Mice were maintained in the AAALAC-accredited University of Wisconsin RARC facility. Mice were provided food and water ad libitum and were maintained in a controlled 12-hour light/dark cycle environment. All protocols were approved by the School of Medicine and Public Health Animal Care and Use Committee (ACUC, Protocol number M00682). Nulliparous C57BL/6J (Jackson Labs, Bar Harbor, ME) females (8-12 weeks of age) were time-mated, such that the presence of a vaginal plug was designated as embryonic day (E)0.5. Prior to mating and until dietary administration, dams were maintained on standard, chow breeder diet (BD, Product Number 2019, Harlan Teklad, Madison, WI).
2.2 Dietary intervention
The initiation of diets occurred at either mid-gestation (E10.5) or weaning [post-natal day (PN)21]. All post-weaning diets were continued for 11 weeks. The detailed composition (and catalog numbers) of the diets is shown in Supplementary Table 1 and all dietary schemes are shown in Figure 1. Mid-gestational initiation and maintenance on a VA deficient diet (GVAD) or administration a VA deficient diet only in the post-weaning period (PWVAD) was used for decreased VA availability. The composition of the VAD diet had less fat (12 percent kcal) and more carbohydrate (70 percent kcal) (LF12) than the standard breeder diet (BD) (Supplementary Table 1, Figure 1).
Figure 1. Dietary scheme.
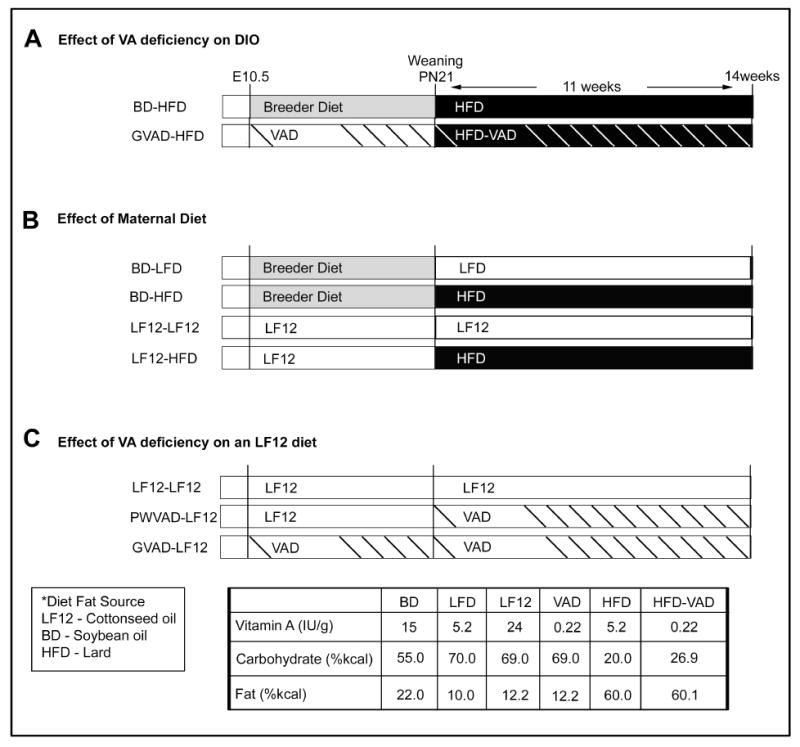
Design for treatment groups showing maternal diet (E10.5-PN21) and post-weaning diet (PN21-week 14). (A) The effect of VA deficiency on diet-induced obesity (DIO), (B) the effect of the maternal diet on DIO, (C) and the effect of VA deficiency on a low fat base diet (LF12). Composition of each diet is shown in Supplementary Table 1.
The GVAD protocol was paired with a post-weaning high fat diet (GVAD-HFD) that otherwise had the same components as the control high fat diet (HFD) protocol (BD-HFD) (Figure 1A). The LF12 maternal diet was also used from mid-gestation to weaning prior to combination with either continued LF12 or HFD post-weaning (LF12-LF12 and LF12-HFD) (Figure 1B). The GVAD and PWVAD protocols were also examined with a post-weaning LF12 diet (GVAD-LF12 and PWVAD-LF12) (Figure 1C).
The VAD diets contained 220IU/kg of retinyl palmitate as the source of VA, compared to the sufficient diet, which contained 24,000IU/kg.
Mice were fasted for 4 hours prior to euthanasia between 1pm and 2pm by CO2 asphyxiation. Body mass is reported at the time of euthanasia. Blood was collected by cardiac puncture and the serum was isolated by centrifugation at 14,000rpm for 10 min and frozen at -80°C. Tissues were weighed, flash frozen, and stored at -80°C.
2.3 Liver RNA isolation
Frozen liver was placed in RNAlater Ice, according to manufacturer’s instructions (Ambion, Foster City, CA). Total RNA was isolated from 20mg tissue using the RNeasy Mini Kit accompanied by Qiashredder columns (Qiagen, Valencia, CA), according to the manufacturer’s instructions. RNA was spectrophotometrically measured for quantity and purity by A260/A280 and A260/A230 on a Nanodrop, followed by visual inspection by denaturing agarose gel electrophoresis.
2.4 Microarray analysis
All microarray analyses were performed with liver mRNA isolated from at least two mice in each treatment group that showed body weight and adipose characteristics close to the mean. Analyses were carried out on the Agilent Technologies 4x44k platforms. Each array includes 43,379 biological features, including replicates of 245 probes.
Samples were prepared according to the manufacturer’s instructions for dual-color or one-color labeling, including sample preparation, hybridization and scanning. mRNA was submitted by J. Bushkofsky to Chris Bradfield’s lab at the University of Wisconsin-Madison for dual color labeling for GVAD-HFD and BD-HFD samples (Bushkofsky, unpublished results), such that competitive binding of each of the treatment samples (Cy3-labeled) was co-hybridized with a pooled control sample, BD-HFD, of three biological replicates (Cy5-labeled). The Cy5 values were used as a measure of binding efficiency and relative expression. The one-color labeling method was employed for a separate set of analyses using BD-HFD, LF12-HFD, LF12-LF12, and GVAD-LF12 samples. All samples were Cy3-labeled. Data was deposited in the NCBI Gene Expression Omnibus and can be accessed through GEO Series Accession Number GSE87845.
Results were analyzed by the EDGE3 software using the Limma analysis, which provides normalized fold change values and assesses significance based on ANOVA statistics [31].
Selections of previously reported liver gene responses to the BD-HFD compared to an approximately isocaloric low fat diet (LFD) were used in comparative analyses [29, 30].
2.5 Real time PCR
RNA (1.5μg) was reverse transcribed with Reverse Transcriptase (Promega, Madison, WI) and amplified by real time PCR (qPCR) in a 10μl reaction volume with Sybr Green (Promega).
Expression was measured using a BioRad CFXConnect real time PCR detection system (Hercules, CA) and quantified using the standard curve method. Separate mRNA from three mice in each treatment group were normalized by comparison to GAPDH expression. Primer sequences:
Cyp26a1
F 5’-ACTTACCTAGGACTCTACCCAC-3’
R 5’-GCTGTTCCAAAGTTTCCATGTC-3’
Col1a1
F 5’-CGCTGGAGAACCTGGAAAG-3’
R 5’-GCGGGACCTTGTTCACCTC-3’
Scd1
F 5’-CCGGAGACCCCTTAGATCGA-3’
R 5’-TAGCCTGTAAAAGATTTCAAA-3’
2.6 Retinoid measurements
Serum and liver retinol and retinyl esters were determined as previously stated [32], with the following modifications. Serum (150μL) was combined with an equal volume ethanol/0.1% butylated hydroxytoluene and extracted three times with 500μL hexane. Liver (0.5g) was ground in sodium sulfate by mortar and pestle. Samples were extracted with dichloromethane to a final volume of 25mL and an aliquot (5mL) was dried under nitrogen. Methanol:dichloroethane (50:50 (v:v), 0.1mL) was used for resuspension, 25μL was analyzed. Retinyl butyrate was added to each sample to measure extraction efficiency. Retinol and retinyl ester peaks were resolved on a Waters HPLC system with a C18 reverse-phase column (Milford, MA). Solvent A consisted of 92.5:7.5 (v:v) acetonitrile:water and solvent B consisted of 85:10:5 (v:v) acetonitrile:methanol:dichloroethane, each with 0.05% triethylamine as a modifier.
2.7 Statistical analysis
Comparisons were analyzed using one-way ANOVA with a Tukey post-test to determine individual p-values or linear regression, where applicable (GraphPad Prism, La Jolla, CA). Data are represented as mean ± SEM.
3. Results
3.1 GVAD diet depletes retinyl ester stores and disrupts circulating retinol
To investigate the effects of VA deficiency on DIO in C57BL/6J mice, we examined the effect of initiation of a VA-deficient diet at mid-gestation (GVAD) in combination with a post-weaning VA-deficient HFD (GVAD-HFD). Each treatment was compared to a previously examined control HFD group (BD-HFD) (Figure 1A). GVAD-HFD decreased serum and liver retinoid content and suppressed DIO. The GVAD protocol, however, used a defined, base maternal diet (LF12) that differed appreciably the control breeder diet (BD) with lower fat and higher carbohydrate (Supplementary Table 1). We compared these two maternal diets in combination with the same post-weaning HFD (LF12-HFD versus BD-HFD) (Figure 2A). The LF12 maternal diet did not affect serum retinol, but increased liver retinol and RE content compared to BD-HFD (Figure 2B).
Figure 2. Maternal diet affects retinoid content and diet-induced obesity, each is further decreased by GVAD-HFD.
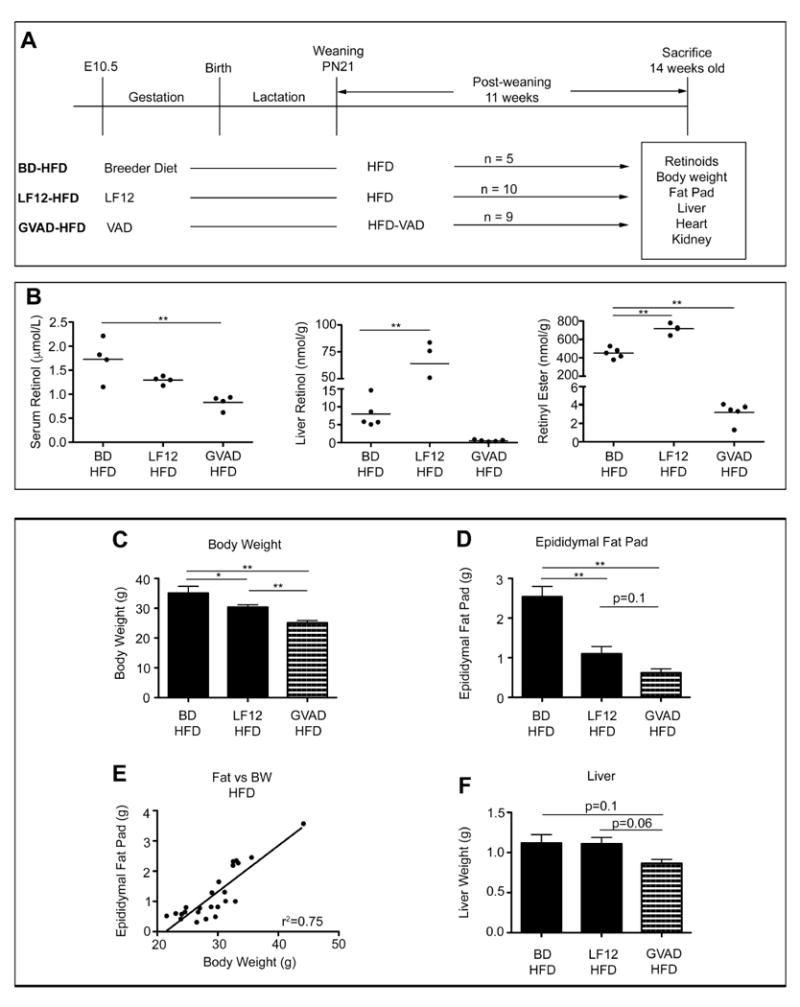
(A) Design for treatment groups showing maternal diet (E10.5-PN21) and post-weaning diet (PN21-week 14). Maternal VAD diet is based on LF12. HFD-VAD is a VA deficient equivalent to HFD. Male mice from at least two litters were used; number (n) is shown per group. (B) Serum retinol, liver retinol, and liver retinyl ester for individual mice in each group. (C) Body weight (D) Epididymal fat pad weight (E) Correlation between fat and body weight among BD-HFD, LF12-HFD, and GVAD-HFD groups (F) Liver weight. Data are mean±SEM *p-value<0.05, **p-value<0.01
3.2 LF12 maternal diet suppressed DIO, which was further decreased by GVAD-HFD
We operationally define DIO as an increase in body weight (BW) and adiposity in mice on the HFD relative to equivalent mice on the LFD [29, 30]. GVAD-HFD decreased DIO and liver weight (Figure 2C, D, and F). Body weight changes correlated with epididymal adiposity for each condition on a post-weaning HFD (Figure 2E). LF12-HFD produced intermediate effects on DIO compared to GVAD-HFD and BD-HFD protocols (Figure 2C and D). Liver weights responded in parallel to body weight and adipose changes (Figure 2F). Heart and kidney weights were unaffected (Supplementary Figure 1). Female GVAD-HFD offspring exhibited a similar BW and liver weight response as males (Bushkofsky, unpublished results).
3.3 Anomalous responses caused by the LF12 diet
GVAD and PWVAD protocols were applied with continuous use of the LF12 diet for dam and offspring (GVAD-LF12, PWVAD-LF12) in comparison to the appropriate control in which LF12 was fed to both dam and post-weaning offspring (LF12-LF12 protocol) (Figure 3A). Normal adult serum retinol content was seen with BD-HFD (Figure 2B) and LF12-LF12 (Figure 3B) protocols and was unaffected by PWVAD-LF12 (Figure 3B). By contrast, the GVAD-LF12 combination produced approximately 90 percent suppression of serum retinol (Figure 3B), which was more effective than GVAD-HFD (Figure 3C) or the original application of this GVAD approach [12]. The GVAD-LF12 combination also removed all detectable retinol and RE from the liver, thus paralleling the greater retinol depletion from serum (Figure 3B and C). Remarkably, the LF12-LF12 protocol increased the capacity for liver RE by more than two-fold compared to the normal documented range (1000-2000nmol/g) [33-35] and by nearly ten-fold compared to the BD-HFD values (Figure 2B).
Figure 3. GVAD lowers serum and liver retinol and attenuates adiposity from LF12 diet.
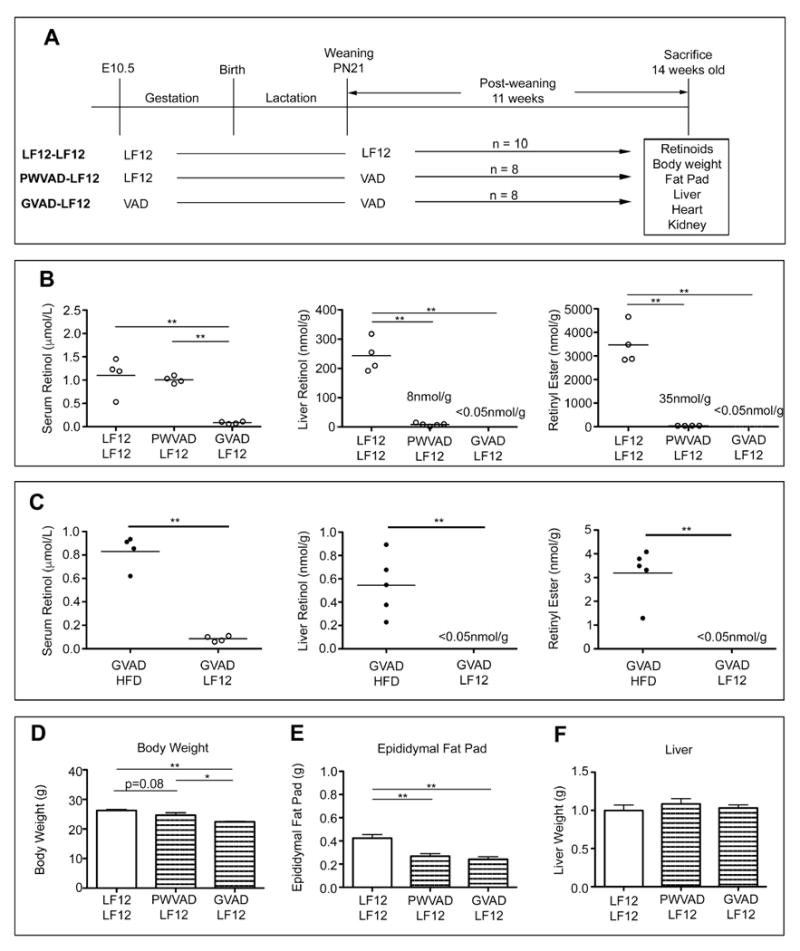
(A) Design for treatment groups showing maternal diet (E10.5-PN21) and post-weaning diet (PN21-week 14). Maternal VAD diet is based on LF12. Male mice from at least two litters were used; number (n) is shown per group. (B and C) Serum retinol, liver retinol, and liver retinyl ester for individual mice in each group. The limit of quantification is <0.05nmol/g (D) Body weight (E) Epididymal fat pad weight (F) Liver weight. Data are mean±SEM *p-value<0.05, **p-value<0.01
The modest deposition of epididymal fat on the low fat diet further decreased two-fold in response to the GVAD-LF12 with a parallel decline in BW (Figure 3D and E). The PWVAD- LF12 protocol also effectively suppressed epididymal fat (Figure 3E). BW changes were proportional to the epididymal adiposity (data not shown). The liver (Figure 3F) and heart (Supplementary Figure 1) weights were unaffected by GVAD-LF12, but the kidney weight was modestly decreased (Supplementary Figure 1). Female littermates showed smaller effects on body and organ weights than males, but retained the kidney responses (Supplementary Figure 2).
3.4 Measuring systemic changes by liver gene expression
The selective effects of the LF12 diet compared to the control breeder diet on DIO (Figure 2) suggest a response in early development that impacts adult endocrine liver regulation. We have used microarray analyses of livers from each of the treatment groups (Figure 1) to identify specific liver mechanisms underlying the role of GVAD in retinoid homeostasis and adiposity and the contribution of a maternal LF12 diet compared to breeder diet. Total numbers of unique detected genes and differentially expressed genes between comparisons (fold change (FC)>2.0, p-value<0.01) are shown in Figure 4. Even with a low threshold (Cy3>50), there were only 42 expression changes when the maternal diet was switched from BD to LF12, despite the large difference in DIO. Nearly 15 times as many differences arose from switching the post-weaning diet or by introducing VA deficiency from mid-gestation to maturity. We show that many of the differences introduced by the post-weaning diet switch are prevented by GVAD (Figure 5).
Figure 4. Agilent microarray feature detection and differentially expressed genes.

The Agilent 4x44k microarray was used to examine liver gene expression of 43,379 features. For each dietary protocol, the number of detected genes with a relative expression of Cy3>50 and genes that exhibited differential expression (fold change (FC)>2.0 and p-value<0.0) are shown.
Figure 5. Effects of maternal diet, post-weaning diet, and GVAD on expression of six genes from different response clusters.
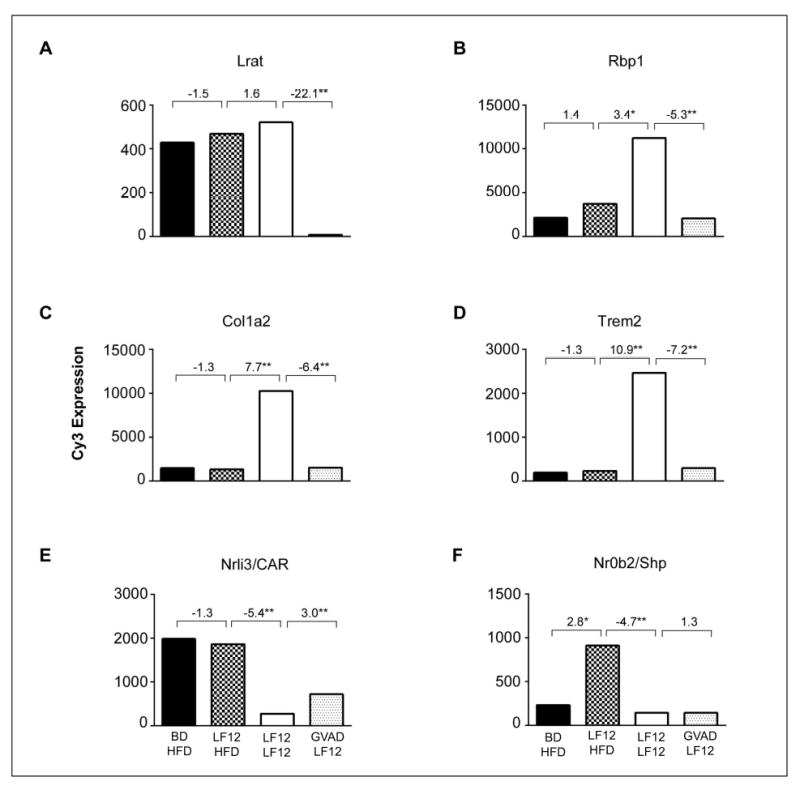
The four treatment groups are shown in Figure 1. Comparison of BD and LF12 maternal diets (bars 1 and 2) with post-weaning HFD shows an impact of maternal diet only for Nr0b2/Shp. Comparison of bars 2 and 3 shows the effect of post-weaning dietary fat and carbohydrate with the LF12 maternal diet. Comparison of bars 3 and 4 shows the effect of depleted retinol by GVAD. *p-value<0.05, **p-value<0.01
Sorting of gene expression across multiple diet and VA deficiency treatment groups organizes individual genes into clusters with similar regulation. Expression changes caused by VA deficiency timing (GVAD versus PWVAD), different maternal diets (BD versus LF12) and post-weaning diets (LFD, LF12, HFD) arise from associations between maternal diet, retinol activity, and development.
Figure 5 shows expression differences for six genes for these four treatment protocols. We address the effects of maternal diet, BD versus LF12 each with a post weaning HFD, dietary fat/carbohydrate by comparing post-weaning LF12 and HFD diets each with a maternal LF12 diet, and the impact of GVAD by applying this to the LF12-LF12 combination. Lecithin:retinol acyltransferase (Lrat), which converts retinol to RE, and cellular retinol binding protein (Rbp1/Crbp1), which mediates the transfer of retinol into hepatocytes, each exhibited a large selective suppression by GVAD in parallel with the decline in liver retinoids (Figure 3B). The stellate marker, collagen 1a2 (Col1a2), and the myeloid cell marker, Trem2, were increased by the change in post-weaning diet from HFD to LF12 and decreased by depletion of serum and liver retinoids. The lipid responsive nuclear receptor (Nr1i3/Car) showed minimal impact of maternal diet, but a large suppression by post-weaning LF12. By contrast, the small heterodimer partner (Nr0b2/Shp) showed extensive dependence on both the maternal and post-weaning high carbohydrate, LF12 diet, but was insensitive to GVAD.
Each of these genes will be addressed more specifically in the context of other liver mechanisms in Tables 1-5. For each table, we used the EDGE3 software to process expression ratios between groups: upper condition versus lower condition. EDGE3 produced a normalized FC between two treatment groups and calculated the p-value, with a negative ratio indicating proportionately higher expression by the lower condition.
Table 1.
Genes associated with retinoid homeostasis and stellate cell activation
| Fold change upper versus | GVAD-HFD | GVAD-LF12 |
|---|---|---|
| lower | BD-HFD | LF12-LF12 |
| Retinoid Homeostasis | ||
| Cyp26a1 | -26.5** | -28.4** |
| Cyp26b1 | -4.8** | -78.1** |
| Lrat | -2.2** | -22.1** |
| Rbpr2 | -1.4** | n.s. |
| Stra6 | n.s | n.s. |
| Rbp4 | 1.8** | n.s. |
| Rbp1 (Crbp1) | 3.2** | -5.3** |
| Stellate Cell Activation | ||
| Acta2 (Sma) | 1.7* | -2.7* |
| Col1a1 | 4.6** | -14.0** |
| Col1a2 | 1.9** | -6.4** |
| Col3a1 | 2.8** | -9.9** |
| Col4a1 | 2.1** | -1.7 |
| Col4a5 | 2.1** | n.s. |
| Col5a1 | 1.6** | -2.8** |
| Col5a2 | 2.9** | -3.8** |
| Col6a1 | 2.0** | -3.6** |
| Mmp13 | 1.9** | -3.8** |
| Mmp3 | 4.0** | n.s. |
| Smpd3 | 3.1** | -3.3** |
| Timp2 | 1.7** | -1.8** |
| Timp3 | 2.6** | -2.1* |
| Cyp1b1 | 2.9** | -2.9** |
p-value ≤ 0.05
p-value ≤ 0.01
n.s. Non-significant values where the net FC < 1.5 and p-value > 0.05
Table 5.
Shp-/- effects on gene expression overlap with GVAD effects in a diet dependent manner
| Fold change upper versus | Shp-/- WD | GVAD-HFD | GVAD-LF12 |
|---|---|---|---|
| lower | WT WD | BD-HFD | LF12-LF12 |
| Stellate | |||
| Col1a1 | -6.8** | 4.6** | -14.0** |
| Col1a2 | -3.9** | 1.9** | -6.4** |
| Col3a1 | -4.5** | 2.8** | -9.9** |
| Col4a1 | -2.2** | 2.1** | -1.7 |
| Col6a1 | -1.9 | 2.0** | -3.6** |
| Fbn1 | -5.9* | 1.5** | -2.2** |
| Lum | -3.0 | 1.5** | -3.3** |
| Timp1 | -6.7 | n.s. | -2.6** |
| Mmp12 | -15.2** | 3.6** | -4.3** |
| Inflammation | |||
| Cd63 | -5.7** | 1.8** | -5.0** |
| Ly6d | -15.7** | 2.8* | -39.8** |
| Nupr1 | -5.0** | 2.8** | -3.3** |
| Trem2 | -12.4 | 3.7** | -7.2** |
| Osbpl3 | -2.7 | 5.5** | -4.3** |
| Ms4a7 | -6.7** | 2.5* | -6.9** |
| Atf3 | -10.9** | 1.8 | -5.9** |
| Phlda3 | -3.5** | 1.9* | -5.0** |
| Lyz2 | -5.6** | 1.5* | -3.6** |
| Cxcl10 | -3.5** | -2.3** | -3.7** |
| Spp1 | -2.2** | n.s. | -3.6** |
| Ubd | -8.5* | n.s. | -22.1** |
| Ntrk2 | -11.5** | 2.8* | -29.0** |
| HF Genes | |||
| Rbp1 (Crbp1) | -2.9 | 3.2** | -5.3** |
| Lpl | -6.2* | 2.7** | -4.2** |
| Nr1i3/Car | 1.7 | -1.8** | 3.0** |
| Hsd17b7 | 1.9** | -1.5* | 2.2* |
| Gadd45g | 1.6 | 5.1** | 4.0* |
Fold change values obtained from S.C. Kim, 2014 [39] wherein Shp-/- mice on a 45% kcal fat western diet (WD) liver gene expression is compared to WT on the same diet
p-value ≤ 0.05
p-value ≤ 0.01
n.s. Non-significant values where the net FC < 1.5 and p-value > 0.05
3.5 VA depletion alters expression of genes associated with retinoid homeostasis
We have examined expression changes produced by the GVAD-HFD protocol, which substantially suppressed DIO and serum retinol. Genes that respond directly to RA (Lrat, Cyp26a1, Cyp26b1) were extensively suppressed (Table 1). The GVAD protocol has a base maternal diet that is different from the BD. The gene expression changes produced by GVAD with a matched LF12 base diet (GVAD-LF12 versus LF12-LF12) showed a similar suppression for Cyp26a1, but larger suppressions for Cyp26b1 and Lrat expression (Table 1). This extra suppression matches the greater depletion of serum and liver retinoids (Figure 3C). qPCR confirmed extensive depletion of Cyp26a1 expression by GVAD-LF12 and PWVAD-LF12 (Figure 6A), which matches liver retinol and RE content (Figure 3B).
Figure 6. Effects on PWVAD and GVAD on liver gene expression determined by qPCR.
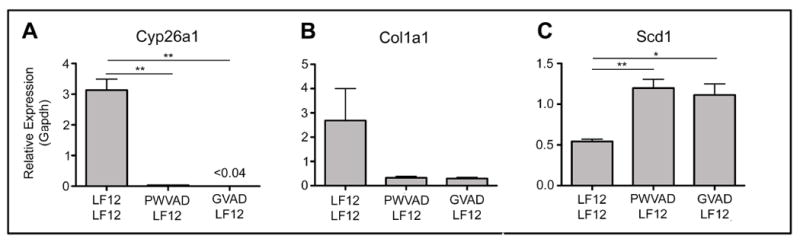
Key genes associated with retinoid signaling (Cyp26a1), stellate cell activation (Col1a1), and hepatic energy metabolism (Scd1) were examined by qPCR using mRNA from at least three mice from two or more litters. Data are mean±SEM *p-value<0.05, **p-value<0.01
Other genes involved in retinoid homeostasis showed different responses to each GVAD protocol. Rbp4, which sequesters serum retinol, showed stimulation with GVAD-HFD that matches the elevated serum retinol compared to GVAD-LF12. Stra6, the holo-RBP4 receptor, did not respond to these retinol changes. The recently identified liver RBP4 receptor, Rbpr2, was modestly suppressed by GVAD-HFD. Rbp1 showed opposing expression by GVAD-HFD and GVAD-LF12 (Figure 7A).
Figure 7. Adult retinoid deficiency induced by GVAD reverses the diet dependence of gene expression.
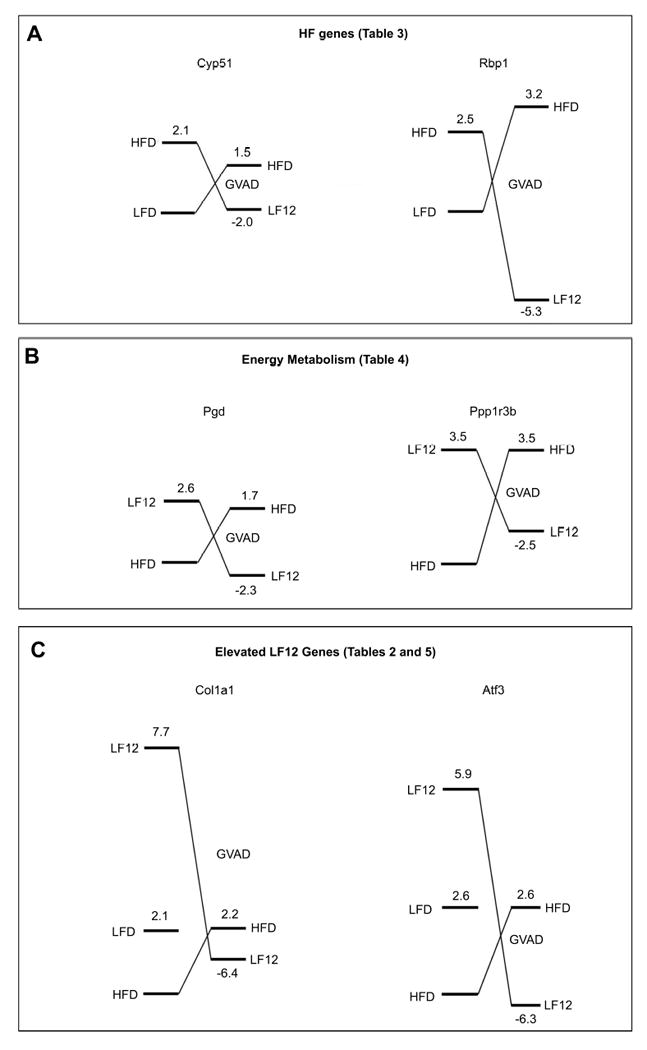
Each diagram presents EDGE3 processed expression ratios relative to a standard diet set to 1.0 (HFD or LFD). The right hand values represent changes induced by GVAD on a HFD or LF12 base diet, scaled proportionately. The levels approximate to the absolute relative Cy3 values for the treatments. (A) HF genes are represented by Cyp51 and Rbp1 (Table 3). (B) Energy metabolism genes are represented by Pgd and Ppp1r3b (Table 4). (C) Elevated LF12 genes are represented by Col1a2 and Atf3 (Tables 2 and 5).
3.6 Stellate cell markers depend on maternal diet and timing of VA deficiency
GVAD-HFD stimulated markers of stellate cell activation, including α-smooth muscle actin (Sma/Acta2), and several matrix-associated genes, such as multiple collagen family members (Col genes), matrix metalloproteinases (Mmp genes) and tissue inhibitor of metalloproteinase (Timp genes) (Table 1). Basal stellate cells include a population with an intermediate activation state, which also retain RE droplets and express Cyp1b1 [22]. The GVAD-HFD-specific increase in matrix genes and Cyp1b1 suggests an expansion of this semi-activated population.
Surprisingly, GVAD-LF12 decreased expression of the same set of stellate markers (Table 1). This cluster of genes was stimulated by the carbohydrate-rich base diet (LF12-LF12 protocol) (Figure 5, Table 2). qPCR confirmed suppression of Col1a1 by GVAD-LF12. However PWVAD-LF12 was equally effective in reversing this stellate marker (Figure 6B), despite suppression of serum retinol by GVAD, but not PWVAD (Figure 3B).
Table 2.
Elevated LF12 genes are retinol-sensitive and include stellate and inflammation markers
| Fold change upper versus | BD-HFD | LF12-HFD | GVAD-LF12 |
|---|---|---|---|
| lower | BD-LFD | LF12-LF12 | LF12-LF12 |
| Stellate Cell Activation | |||
| Acta2 | n.s. | -3.7* | -2.7* |
| Col1a1 | -2.7 | -20.8** | -14.0** |
| Col1a2 | -1.8 | -7.7** | -6.4** |
| Col3a1 | -1.8 | -10.3** | -9.9** |
| Col4a1 | -1.5 | -3.8** | -1.7 |
| Col6a1 | -1.6 | -5.0** | -3.6** |
| Col6a2 | -1.7 | -3.9** | -3.1** |
| Smpd3 | -2.8* | n.s. | -3.3** |
| Lum | -1.9 | -9.9** | -3.3** |
| Fbn1 | n.s. | -3.7** | -2.2** |
| Timp1 | n.s. | -2.5** | -2.6** |
| Cyp1b1 | n.s. | -4.8** | -2.9** |
| Inflammation | |||
| Lyz2 | -1.8 | -5.3** | -3.6** |
| Cd63 | -2.6* | -10.0** | -5.0** |
| Spp1 | -2.0 | -3.4** | -3.6** |
| Ly6d | -4.3** | -62.4** | -39.8** |
| Nupr1 | -5.5** | -4.2** | -3.3** |
| Trem2 | -2.2 | -10.9** | -7.2** |
| Osbpl3 | -3.3** | -8.6** | -4.3** |
| Ms4a7 | -2.7* | -9.9** | -6.9** |
| Atf3 | -3.1* | -6.7** | -5.9** |
| Phlda3 | -1.8 | -8.2** | -5.0** |
| Ubd | -1.7 | -23.1** | -22.1** |
| Ntrk2 | -14.7** | -14.5** | -29.0** |
| Cxcl10 | -1.9 | -3.6** | -3.7** |
p-value ≤ 0.05
p-value ≤ 0.01
n.s. Non-significant values where the net FC < 1.5 and p-value > 0.05
3.7 LF12-LF12 diet activates both stellate and inflammatory marker genes
Stellate genes were extensively stimulated by the LF12-LF12 protocol compared to the LF12- HFD protocol, as shown for Col1a2 in Figure 5C and the stellate markers in Table 2. The increased fold change response by LF12-HFD/LF12-LF12 (center column) compared to the control BD-HFD/BD-LFD ratio (left column) derives from this high LF12-LF12 expression. GVAD-LF12 prevented these increases in stellate cell markers on the LF12-LF12 protocol. This stellate activation is, therefore, nearly completely retinol dependent (Table 2). These changes in stellate cell markers for the LF12-LF12 protocol occur without the loss of RE storage, which is typical of full stellate activation [21]. Indeed the liver retinoid levels increase suggesting elevated stellate storage (Figure 3B).
The response by stellate markers was matched by the macrophage gene, Trem2, which increased with the LF12-LF12 protocol and was reversed by GVAD-LF12 (Figure 5D). Table 2 provides 12 additional responses termed elevated LF12 genes, which represent less than half of this cluster. Remarkably, these genes are largely markers for inflammatory cells, such as Lyz2, Cd63, Ly6d, and Osbpl3. These responses also include stress related transcription factors that control these responses, Nupr1 and Atf3, and chemokines, Cxcl10.
3.8 Genes exhibit opposite GVAD-dependent effects for high fat and high carbohydrate diets
Obesity suppression is a hallmark of both the GVAD-HFD protocol and Cyp1b1 gene deletion [30]. We identified 166 liver genes that exhibited similar expression differences between HFD and LFD in WT mice and Cyp1b1-/- mice (termed HF genes) [30]. The HF genes were relatively insensitive to the breeder diet (LF12-LF12 versus BD-LFD protocols) (Table 3, columns 1 and 3). However, nearly all the HF gene responses were reversed by GVAD-HFD (98 genes, FC>2.0 or p-value<0.05) (Supplementary Table 2). Thus, the expression of HF genes on the HFD depends on retinol. For nine of the 14 HF genes in Table 3, the LF12 expression is sensitive to GVAD, but opposite in direction to expression on the HFD.
Table 3.
HF genes respond to GVAD-LF12
| Fold change upper versus | BD-HFD | GVAD-HFD | LF12-HFD | GVAD-LF12 |
|---|---|---|---|---|
| lower | BD-LFD | BD-HFD | LF12-LF12 | LF12-LF12 |
| HF genes | ||||
| Cyp2c54 | 7.4** | -3.6* | 15.3** | 8.7** |
| Nr0b2 | 2.2** | -2.9** | 4.7** | n.s. |
| Hmgcs1 | 2.2* | -2.7** | n.s. | n.s. |
| Apol7a | 2.1* | -2.0** | 1.8 | n.s. |
| Cyp51 | 2.1** | -2.0* | 2.3* | 1.5 |
| Nr1i3 | 3.0* | -1.8** | 5.4** | 3.0** |
| Hsd17b7 | 2.0** | -1.5* | 1.9 | 2.2* |
| Acacb | -3.2* | 1.3** | n.s. | 1.6* |
| Scd2 | -4.5* | 1.4** | -6.8** | -4.5** |
| Pck2 | -2.4** | 2.5** | -2.5** | -1.8* |
| Lpl | -3.3* | 2.7** | -5.1** | -4.2** |
| Rbp1 | -2.5* | 3.2** | -3.4* | -5.3** |
| Igh-2 | -7.9** | 3.9** | -2.6 | -2.0 |
| Gadd45g | -4.8** | 5.1** | -4.9 | 4.0* |
p-value ≤ 0.05
p-value ≤ 0.01
n.s. Non-significant values where the net FC < 1.5 and p-value > 0.05
This reversal effect is shared for HF genes, the stellate and inflammatory markers, and metabolic regulators. In Figure 7, we diagram how these reversal effects of GVAD change the sensitivity of gene expression to high carbohydrate (LF12 or LFD) and high fat (HFD). This reversal by GVAD is shown for HF genes Cyp51 and Rbp1, for Col1a1, a stellate activation marker, and for Atf3, an inflammatory response marker (Tables 2 and 3).
For most of the genes linked to energy homeostasis, GVAD responses again reversed between post-weaning dietary fat/carbohydrate (HFD and LF12) (Table 4). This reversal is shown in Figure 7B for phosphogluconate dehydrogenase (Pgd) and a regulator of glycogen synthesis, Ppp1r3b [36]. Additional genes include steroyl CoA desaturase (Scd1), malic enzyme (Me1), fatty acid synthase (Fasn), another regulator of glycogen synthesis, Ppp1r3g, and Agpat9/Gpat3, a limiting enzyme in triglyceride synthesis [37].
Table 4.
GVAD-LF12 activates energy metabolism genes
| Fold change upper versus | GVAD-LF12 | GVAD-HFD |
|---|---|---|
| lower | LF12-LF12 | BD-HFD |
| Scd1 | 2.1** | -1.6 |
| Me1 | 2.0** | -1.7* |
| Fasn | 1.7 | -1.3 |
| Mup3 | 2.1** | n.s. |
| Slc2a4 | 2.1* | 1.9* |
| Pdk1 | 3.5** | -3.1** |
| Ppp1r3b | 3.5** | -2.5** |
| Lpin1 | 5.6** | 4.4** |
| Ppp1r3g | -6.4* | 3.3** |
| Lgals1+ | -4.8** | n.s. |
| Pdk4 | -2.6* | -1.7* |
| Agpat9 | -2.6 | 3.5** |
| Pgd | -2.3** | 1.5* |
Additional genes within the same family respond similarly: Lgals3
p-value ≤ 0.05
p-value ≤ 0.01
n.s. Non-significant values where the net FC < 1.5 and p-value > 0.05
qPCR analysis confirmed the increase of Scd1 by GVAD-LF12 and demonstrated an equal response to PWVAD-LF12 (Figure 6C), which also corresponds to the equal adiposity effects by these treatments (Figure 3).
3.9 Shp-/- mice display overlapping gene expression
The HF gene Nr0b2/Shp is a transcriptional co-repressor that plays key roles in development and hormonal activity through heterodimerization with several nuclear receptors [38-40]. We discovered a remarkable overlap between the liver gene responses to GVAD and expression changes reported for deletion of Shp [39]. Shp has previously been shown to be retinoid responsive [40], as confirmed here by the GVAD-HFD suppression (Table 3). Shp-/- mice, like GVAD mice, exhibit decreased DIO [39, 41].
The reported effects of Shp deletion [39] were obtained with a defined Western Diet, which has the same base components as the HFD and LFD diets, except with 40 percent kcal from each carbohydrate and fat. The reported study [39] used a 12-hour fast rather than the 4-hour fast used here. GVAD produced opposite effects on the stellate marker, Col1a1, and on the inflammatory marker, Atf3, with the LF12 and HFD diets (Figure 7). Table 5 shows that the GVAD-HFD combination stimulated the inflammatory markers and produced opposite responses to those for the GVAD-LF12 combination. Shp-/- effects match the responses produced by GVAD-LF12 for 27 genes that are found in the stellate and inflammation clusters (Table 5).
Out of 138 genes that are affected by both GVAD-LF12 and Shp-/-, 90 percent showed this match (Supplementary Table 3). This relationship was seen for stellate, inflammatory, and HF genes (Table 5). Shp-/-, like GVAD, functions in an anti-inflammatory role for genes induced by the high carbohydrate LF12-LF12 combination. Shp is induced by LRH-1, a major stimulant of glucose metabolism that is subject to feedback inhibition through dimerization with Shp [42].
4. Discussion
In mice, the initiation of VA deficiency at mid-gestation (GVAD) is necessary to cause a substantial adult decline in serum retinol [9, 12]. This contrasts with rats where post-weaning initiation (PWVAD) is sufficient to reset retinol homeostasis [7, 8]. We show that livers from mice on the carbohydrate-rich, LF12 diet are similarly and extensively depleted of liver retinol, RE, and the retinoic acid marker, Cyp26a1, by PWVAD and GVAD. Serum retinol is, however, only suppressed by GVAD (Figure 3B). Thus, additional effects of the maternal GVAD diet function beyond liver retinoid content to overcome the normal serum retinol homeostasis.
GVAD is also more effective at suppressing serum retinol with the LF12 post-weaning diet than with the post-weaning HFD (Figure 3C). We show here that GVAD prevents many effects with the high carbohydrate LF12 diet, including partial activation of stellate cells and a set of inflammatory responses that we suggest arises from mild postprandial glucose-induced stress [43]. For mice fed a HFD, the effect of GVAD on these same genes is typically opposite.
Use of the LF12 diet for both the dam and offspring diets (LF12-LF12 protocol) produced remarkably high stimulation of adult gene expression markers for both stellate cell activation and inflammation. Their suppression by GVAD suggests a dependence on retinol or RA. The match of these expression changes with those in Shp-/- mice [39] also suggests dependence on this nuclear co-suppressor. This close relationship between the stellate cell and inflammation in liver hepatitis has been well documented [20, 21].
The overlap of retinol and Shp regulation also extends to the HF gene cluster, which is defined by appreciable expression differences between LFD and HFD and relative insensitivity to the maternal diet (BD versus LF12) (Table 3). Notable contributors include cellular retinol binding protein (Rbp1) and lipoprotein lipase (Lpl). These genes exhibit closely correlated expression in individual mice across multiple treatments. RBP1 and LPL function together to mediate the uptake of lipids and RE through LRP-1-mediated processing of chylomicron remnants [44]. This HF cluster includes several lipid metabolism genes, which may participate in this process, such as nuclear receptors Nr1i3/Car and Nr4a1 [2, 45].
Shp is induced by the nuclear factor, LRH-1, a major regulator of cell glucose levels through gluconeogenesis and glucokinase [42]. Shp exerts feedback control of LRH-1 through dimerization [46]. Shp expression is induced by retinoic acid [40], circadian clock regulatory genes [47] and numerous nuclear receptors [48]. Shp-/- mice are protected from DIO, although with diminished glucose tolerance [39]. Over-expression of SHP causes dysregulation of lipid metabolism through indirect activation of PPARγ and SREBP-1c, and inhibition of HNF4α signaling [38, 49]. Shp also inhibits many other receptors involved in lipid metabolism (HNF4α, LXR, PXR) [38, 49, 50]. GVAD-HFD and LF12-LF12 protocols, which stimulated the stellate and inflammatory clusters, appreciably decreased Shp expression (Table 3). Thus, under these ad libitum conditions, the high carbohydrate LF12-LF12 condition may generate these unusual stimuli by removing the constraint provided by Shp.
In C57Bl/6J mice, the diet induced increase in obesity in males starts at maturity [29, 30], which also corresponds to the time at which serum retinol responds to GVAD [9]. Key components of the liver participation in retinol homeostasis are Rbp4, which binds serum retinol, Rbp1 and Lpl, which mediate dietary uptake, Lrat, which generates RE, and Cyp26 forms, which metabolize retinoic acid. Differences between post-weaning HFD and LF12 diets partly explain the greater effectiveness in retinoid depletion in GVAD mice fed the LF12 diet. Rbp4 was elevated by GVAD in combination with the HFD, but not by GVAD in combination with the LF12 diet (Table 1). Rbp1 and Lpl were elevated by GVAD with the HFD, but suppressed by GVAD with the LF12 diet (Table 3). Lrat was more suppressed in GVAD mice fed the LF12 diet. Cyp26a1, Cyp26b1, and Lrat were equally were equally suppressed by GVAD and PWVAD, consistent with equal liver retinoid depletions, but in contrast to differences in serum retinol (Figure 6, Bushkofsky, unpublished). The match between serum retinol and adipose levels (PWVAD>GVAD) suggests a possible link when liver retinoid levels are depleted. On VA-sufficient diets, hepatocyte Rbp4 controls serum retinol, but recent work with adipose-specific Rbp4 indicates more complex crosstalk with the liver, which may be affected by extreme retinol depletion [51, 52].
Stellate cells store RE and modulate liver inflammatory and stress processes [21]. The GVAD-HFD and LF12-LF12 activations target at least eight collagen genes, including Col1a1, 1a2, 3a1, 4a1, 5a1 and 6a1, other matrix genes [fibulin (Fbln1) and lumican (Lum)] (Table 5), several forms of metalloproteinases (Mmp) and their inhibitors (Timp) and, notably, Cyp1b1. Complete stellate activation to myofibroblasts releases stored retinyl esters [21]. A novel, intermediate population of stellate cells that retains these lipid droplets also exhibits increased expression of these markers [22]. An increase in this population seems more appropriate to account for changes produced by the LF12-LF12 activation, which substantially increased retinoid storage (Figure 3B).
Gene expression has been measured under non-fasting conditions to avoid the extensive metabolic changes associated with this process. Notably, responses mediated by glucagon and glucocorticoids include CREB-mediated gluconeogenesis and PPARα activation of fatty acid oxidation [53]. We suggest that the appearance of inflammation markers arises from the 70 percent carbohydrate content of LFD and LF12 diets, which generates postprandial leukocyte infiltration during ad libitum feeding [54]. Stellate activation is closely associated with hepatic inflammation in steatotic hepatitis [21]. The inflammation and stress markers that we attribute to postprandial carbohydrate metabolism include nuclear factors (Atf3, Nupr1, Ubd), macrophage markers (Trem2 and Osbp13), other inflammatory cells (Ly6d and Lyz2) and chemokines (Cxcl10) (Tables 2 and 5). The LF12-LF12 protocol generates a combination of low adipose with high inflammation that may represent a retinol-mediated lipodystrophy [55].
The connections between maternal and adult diet, retinol, and Shp are summarized in Figure 8. We distinguish HF genes from LF12-LF12 genes based on their enhanced expression. Each is dependent on retinol (reversed by GVAD) on the post-weaning LF12 diet, but suppressed by retinol on the HFD, thus causing a reversal of diet sensitivity. The close correlation with Shp activity across all gene groups suggests that this reversal derives from Shp signaling. For this diversity of mechanisms, we propose that Shp suppression of retinol/RA activity provides a core regulatory partnership around which other pathways (metabolism, inflammation, diurnal rhythm) function. The retinol contribution may arise from direct stimulation by RA through interaction with RARα [39], the Rbp4/Stra6/Rbrp2 pathway, or inflammatory Rbp4 activation of toll-like receptor 4 (TLR4) [52].
Figure 8. Representation of dietary impacts on three groups of genes that share regulation from three sources.
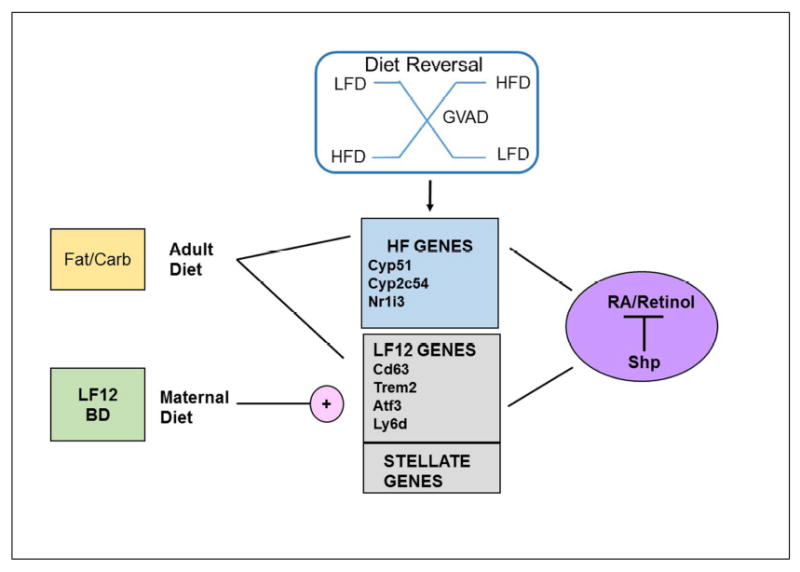
Each group of genes (HF genes, elevated LF12 genes, and stellate cell markers) functions coordinately in response to three perturbations (maternal diet, fat/carbohydrate content in the post-weaning diet, and VA deficiency), shows diet-dependent reversals with GVAD, and correlates responses with Shp deletion.
Supplementary Material
Highlights.
High carbohydrate, LF12 diet stimulates stellate activation and inflammation genes
Retinol and Shp modulate gene expression associated with a high carbohydrate diet
Retinol restrains stellate and inflammation genes in mice fed a high fat diet
Gestational VA deficient diet lowers serum retinol, liver retinoids, and Cyp26a1
Maternal and post-weaning LF12 enhances VA deficiency-induced retinol suppression
Acknowledgments
This work was supported by the National Institutes of Health [R01 DK090249, T32 HD041921, T32 ES007015].
Abbreviations
- VA
Vitamin A
- RA
retinoic acid
- RE
retinyl ester
- PWVAD
post-weaning initiation of vitamin A deficient diet
- GVAD
gestational initiation of vitamin A deficient diet
- VAD diet
vitamin A deficient diet
- DIO
diet-induced obesity
- HFD
high fat diet
- LFD
low fat diet
- LF12
novel low fat diet with 12 percent kcal from fat
- BD
standard breeder diet
- E
embryonic day
- PN
post-natal day
- BW
body weight
References
- 1.Napoli JL. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim Biophys Acta. 2012;1821:152–67. doi: 10.1016/j.bbalip.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–66. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meerza D, Iqbal S, Zaheer S, Naseem I. Retinoids have therapeutic action in type 2 diabetes. Nutrition. 2016;32:898–903. doi: 10.1016/j.nut.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–31. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiegler E, Kim YK, Wassef L, Shete V, Quadro L. Maternal-fetal transfer and metabolism of vitamin A and its precursor beta-carotene in the developing tissues. Biochim Biophys Acta. 2012;1821:88–98. doi: 10.1016/j.bbalip.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satre MA, Ugen KE, Kochhar DM. Developmental changes in endogenous retinoids during pregnancy and embryogenesis in the mouse. Biol Reprod. 1992;46:802–10. doi: 10.1095/biolreprod46.5.802. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy PT, Cerecedo LR. Vitamin A deficiency in the mouse. J Nutr. 1952;46:361–76. doi: 10.1093/jn/46.3.361. [DOI] [PubMed] [Google Scholar]
- 8.Oliveros LB, Domeniconi MA, Vega VA, Gatica LV, Brigada AM, Gimenez MS. Vitamin A deficiency modifies lipid metabolism in rat liver. Br J Nutr. 2007;97:263–72. doi: 10.1017/S0007114507182659. [DOI] [PubMed] [Google Scholar]
- 9.Smith JE. Preparation of vitamin A-deficient rats and mice. 1990;190:229–36. doi: 10.1016/0076-6879(90)90027-x. [DOI] [PubMed] [Google Scholar]
- 10.Ross AC. Diet in vitamin A research. Methods Mol Biol. 2010;652:295–313. doi: 10.1007/978-1-60327-325-1_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wassef L, Spiegler E, Quadro L. Embryonic phenotype, beta-carotene and retinoid metabolism upon maternal supplementation of beta-carotene in a mouse model of severe vitamin A deficiency. Arch Biochem Biophys. 2013;539:223–9. doi: 10.1016/j.abb.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith SM, Levy NS, Hayes CE. Impaired immunity in vitamin A-deficient mice. J Nutr. 1987;117:857–65. doi: 10.1093/jn/117.5.857. [DOI] [PubMed] [Google Scholar]
- 13.Zolfaghari R, Ross AC. Lecithin:retinol acyltransferase from mouse and rat liver. CDNA cloning and liver-specific regulation by dietary vitamin a and retinoic acid. J Lipid Res. 2000;41:2024–34. [PubMed] [Google Scholar]
- 14.Quadro L, Hamberger L, Colantuoni V, Gottesman ME, Blaner WS. Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Molecular Aspects of Medicine. 2003;24:421–30. doi: 10.1016/s0098-2997(03)00038-4. [DOI] [PubMed] [Google Scholar]
- 15.Bonet ML, Ribot J, Palou A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim Biophys Acta. 2012;1821:177–89. doi: 10.1016/j.bbalip.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Harrison EH. Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochim Biophys Acta. 2012;1821:70–7. doi: 10.1016/j.bbalip.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quadro L, Blaner WS, Hamberger L, Novikoff PM, Vogel S, Piantedosi R, et al. The role of extrahepatic retinol binding protein in the mobilization of retinoid stores. J Lipid Res. 2004;45:1975–82. doi: 10.1194/jlr.M400137-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Zhong M, Kawaguchi R, Ter-Stepanian M, Kassai M, Sun H. Vitamin A transport and the transmembrane pore in the cell-surface receptor for plasma retinol binding protein. PLoS One. 2013;8:e73838. doi: 10.1371/journal.pone.0073838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alapatt P, Guo F, Komanetsky SM, Wang S, Cai J, Sargsyan A, et al. Liver retinol transporter and receptor for serum retinol-binding protein (RBP4) J Biol Chem. 2013;288:1250–65. doi: 10.1074/jbc.M112.369132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell. 2010;18:175–89. doi: 10.1016/j.devcel.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Wallace MC, Friedman SL, Mann DA. Emerging and disease-specific mechanisms of hepatic stellate cell activation. Semin Liver Dis. 2015;35:107–18. doi: 10.1055/s-0035-1550060. [DOI] [PubMed] [Google Scholar]
- 22.D’Ambrosio DN, Walewski JL, Clugston RD, Berk PD, Rippe RA, Blaner WS. Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS One. 2011;6:e24993. doi: 10.1371/journal.pone.0024993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho YC, Zheng W, Yamamoto M, Liu X, Hanlon PR, Jefcoate CR. Differentiation of pluripotent C3H10T1/2 cells rapidly elevates CYP1B1 through a novel process that overcomes a loss of Ah Receptor. Arch Biochem Biophys. 2005;439:139–53. doi: 10.1016/j.abb.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 24.Chambers D, Wilson L, Maden M, Lumsden A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development. 2007;134:1369–83. doi: 10.1242/dev.02815. [DOI] [PubMed] [Google Scholar]
- 25.Kang HW, Bhimidi GR, Odom DP, Brun PJ, Fernandez ML, McGrane MM. Altered lipid catabolism in the vitamin A deficient liver. Mol Cell Endocrinol. 2007;271:18–27. doi: 10.1016/j.mce.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Amengual J, Ribot J, Bonet ML, Palou A. Retinoic acid treatment enhances lipid oxidation and inhibits lipid biosynthesis capacities in the liver of mice. Cell Physiol Biochem. 2010;25:657–66. doi: 10.1159/000315085. [DOI] [PubMed] [Google Scholar]
- 27.Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Redonnet A, Ferrand C, Bairras C, Higueret P, Noel-Suberville C, Cassand P, et al. Synergic effect of vitamin A and high-fat diet in adipose tissue development and nuclear receptor expression in young rats. Br J Nutr. 2008;100:722–30. doi: 10.1017/S0007114508967568. [DOI] [PubMed] [Google Scholar]
- 29.Larsen MC, Bushkofsky JR, Gorman T, Adhami V, Mukhtar H, Wang S, et al. Cytochrome P450 1B1: An unexpected modulator of liver fatty acid homeostasis. Arch Biochem Biophys. 2015;571:21–39. doi: 10.1016/j.abb.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bushkofsky JR, Maguire M, Larsen MC, Fong YH, Jefcoate CR. Cyp1b1 affects external control of mouse hepatocytes, fatty acid homeostasis and signaling involving HNF4alpha and PPARalpha. Arch Biochem Biophys. 2016;597:30–47. doi: 10.1016/j.abb.2016.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vollrath AL, Smith AA, Craven M, Bradfield CA. EDGE(3): a web-based solution for management and analysis of Agilent two color microarray experiments. BMC Bioinformatics. 2009;10:280. doi: 10.1186/1471-2105-10-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riabroy N, Tanumihardjo SA. Oral doses of alpha-retinyl ester track chylomicron uptake and distribution of vitamin A in a male piglet model for newborn infants. J Nutr. 2014;144:1188–95. doi: 10.3945/jn.114.191668. [DOI] [PubMed] [Google Scholar]
- 33.Clugston RD, Jiang H, Lee MX, Berk PD, Goldberg IJ, Huang LS, et al. Altered hepatic retinyl ester concentration and acyl composition in response to alcohol consumption. Biochim Biophys Acta. 2012;1831:1276–86. doi: 10.1016/j.bbalip.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Obrochta KM, Kane MA, Napoli JL. Effects of diet and strain on mouse serum and tissue retinoid concentrations. PLoS One. 2014;9:e99435. doi: 10.1371/journal.pone.0099435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim YK, Zuccaro MV, Costabile BK, Rodas R, Quadro L. Tissue- and sex-specific effects of beta-carotene 15,15’ oxygenase (BCO1) on retinoid and lipid metabolism in adult and developing mice. Arch Biochem Biophys. 2015;572:11–8. doi: 10.1016/j.abb.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo X, Zhang Y, Ruan X, Jiang X, Zhu L, Wang X, et al. Fasting-induced protein phosphatase 1 regulatory subunit contributes to postprandial blood glucose homeostasis via regulation of hepatic glycogenesis. Diabetes. 2011;60:1435–45. doi: 10.2337/db10-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khatun I, Clark RW, Vera NB, Kou K, Erion DM, Coskran T, et al. Characterization of a novel intestinal glycerol-3-phosphate acyltransferase pathway and its role in lipid homeostasis. J Biol Chem. 2015 doi: 10.1074/jbc.M115.683359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boulias K, Katrakili N, Bamberg K, Underhill P, Greenfield A, Talianidis I. Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. EMBO J. 2005;24:2624–33. doi: 10.1038/sj.emboj.7600728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SC, Kim CK, Axe D, Cook A, Lee M, Li T, et al. All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade. Hepatology. 2014;59:1750–60. doi: 10.1002/hep.26699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mamoon A, Subauste A, Subauste MC, Subauste J. Retinoic acid regulates several genes in bile acid and lipid metabolism via upregulation of small heterodimer partner in hepatocytes. Gene. 2014;550:165–70. doi: 10.1016/j.gene.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 41.Park YJ, Kim SC, Kim J, Anakk S, Lee JM, Tseng HT, et al. Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J Lipid Res. 2011;52:2234–44. doi: 10.1194/jlr.M016048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oosterveer MH, Mataki C, Yamamoto H, Harach T, Moullan N, van Dijk TH, et al. LRH-1-dependent glucose sensing determines intermediary metabolism in liver. J Clin Invest. 2012;122:2817–26. doi: 10.1172/JCI62368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oliveira MC, Menezes-Garcia Z, Henriques MC, Soriani FM, Pinho V, Faria AM, et al. Acute and sustained inflammation and metabolic dysfunction induced by high refined carbohydrate-containing diet in mice. Obesity (Silver Spring) 2013;21:E396–406. doi: 10.1002/oby.20230. [DOI] [PubMed] [Google Scholar]
- 44.Wassef L, Quadro L. Uptake of dietary retinoids at the maternal-fetal barrier: in vivo evidence for the role of lipoprotein lipase and alternative pathways. J Biol Chem. 2011;286:32198–207. doi: 10.1074/jbc.M111.253070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Close AF, Rouillard C, Buteau J. NR4A orphan nuclear receptors in glucose homeostasis: a minireview. Diabetes Metab. 2013;39:478–84. doi: 10.1016/j.diabet.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Bavner A, Sanyal S, Gustafsson JA, Treuter E. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol Metab. 2005;16:478–88. doi: 10.1016/j.tem.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 47.Lee SM, Zhang Y, Tsuchiya H, Smalling R, Jetten AM, Wang L. Small heterodimer partner/neuronal PAS domain protein 2 axis regulates the oscillation of liver lipid metabolism. Hepatology. 2015;61:497–505. doi: 10.1002/hep.27437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta. 2011;1812:893–908. doi: 10.1016/j.bbadis.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang J, Iqbal J, Saha PK, Liu J, Chan L, Hussain MM, et al. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46:147–57. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- 50.Zou A, Lehn S, Magee N, Zhang Y. New insights into orphan nuclear receptor SHP in liver cancer. Nucl Receptor Res. 2015;2 doi: 10.11131/2015/101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SA, Yuen JJ, Jiang H, Kahn BB, Blaner WS. Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice. Hepatology. 2016;64:1534–46. doi: 10.1002/hep.28659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Norseen J, Hosooka T, Hammarstedt A, Yore MM, Kant S, Aryal P, et al. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol- independent mechanism. Mol Cell Biol. 2012;32:2010–9. doi: 10.1128/MCB.06193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56:952–64. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 54.Yin X, Zheng F, Pan Q, Zhang S, Yu D, Xu Z, et al. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J Mol Endocrinol. 2015;55:169–81. doi: 10.1530/JME-15-0101. [DOI] [PubMed] [Google Scholar]
- 55.Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–97. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.