

Abstract
Convergent phenotypic evolution is often caused by recurrent changes at particular nodes in the underlying gene regulatory networks (GRNs). The genes at such evolutionary ‘hotspots’ are thought to maximally affect the phenotype with minimal pleiotropic consequences. This has led to the suggestion that if a GRN is understood in sufficient detail, the path of evolution may be predictable. The repeated evolutionary loss of larval trichomes among Drosophila species is caused by the loss of shavenbaby (svb) expression. svb is also required for development of leg trichomes, but the evolutionary gain of trichomes in the ‘naked valley’ on T2 femurs in Drosophila melanogaster is caused by reduced microRNA-92a (miR-92a) expression rather than changes in svb. We compared the expression and function of components between the larval and leg trichome GRNs to investigate why the genetic basis of trichome pattern evolution differs in these developmental contexts. We found key differences between the two networks in both the genes employed, and in the regulation and function of common genes. These differences in the GRNs reveal why mutations in svb are unlikely to contribute to leg trichome evolution and how instead miR-92a represents the key evolutionary switch in this context. Our work shows that variability in GRNs across different developmental contexts, as well as whether a morphological feature is lost versus gained, influence the nodes at which a GRN evolves to cause morphological change. Therefore, our findings have important implications for understanding the pathways and predictability of evolution.
Author summary
A major goal of biology is to identify the genetic causes of organismal diversity. Convergent evolution of traits is often caused by changes in the same genes–evolutionary ‘hotspots’. shavenbaby is a ‘hotspot’ for larval trichome loss in Drosophila, but microRNA-92a underlies the gain of leg trichomes. To understand this difference in the genetics of phenotypic evolution, we compared the expression and function of genes in the underlying regulatory networks. We found that the pathway of evolution is influenced by differences in gene regulatory network architecture in different developmental contexts, as well as by whether a trait is lost or gained. Therefore, hotspots in one context may not readily evolve in a different context. This has important implications for understanding the genetic basis of phenotypic change and the predictability of evolution.
Introduction
A major challenge in biology is to understand the relationship between genotype and phenotype, and how genetic changes modify development to generate phenotypic diversification. The genetic basis of many phenotypic differences within and among species have been identified [e.g. 1–15], and these findings support the generally accepted hypothesis that morphological evolution is predominantly caused by mutations affecting cis-regulatory modules of developmental genes [16]. Moreover, it has been found that changes in the same genes commonly underlie the convergent evolution of traits [reviewed in 17]. This suggests that there are evolutionary ‘hotspots’ in GRNs: changes at particular nodes are repeatedly used during evolution because of the role and position of the gene in the GRN, and hence the limited pleiotropic effect of the change [18–21].
The regulation of trichome patterning is an excellent system for studying the genetic basis of morphological evolution [22]. Trichomes are actin protrusions from epidermal cells that are overlaid by cuticle and form short, non-sensory, hair-like structures. They can be found on various parts of insect bodies during different life stages, and are thought to be involved in, for example, thermo-regulation, aerodynamics, oxygen retention in semi-aquatic insects, grooming, and larval locomotion [23–27] (Fig 1).
Fig 1. The GRN controlling formation of trichomes on larval and leg epidermis differs between these developmental contexts.

(A) Simplified GRN for larval trichome development (see [22,29,81,82]). (B) GRN for leg trichome development. Magenta colour indicates interactions found only during leg trichome development. Dashed lines indicate likely interactions. Expression of svb is controlled by several upstream transcription factors and signalling pathways some of which are not active during leg trichome development. The question mark indicates that there are likely to be other unknown activators of svb in legs. Activation of Svb protein requires proteolytic cleavage involving small peptides encoded by tal [30–32]. Active Svb then regulates the expression of at least 163 target genes in embryos [29,33], the expression of 135 of which is detectable in legs. The products of these downstream genes are involved in actin bundling, cuticle segregation, or changes to the matrix, which lead to the actual formation of trichomes. SoxN and Svb activate each other and act partially redundantly on downstream targets in larvae [34,36] and this interaction probably also occurs in legs based on expression data. miR-92a is only expressed in naked leg cells where it represses sha and probably CG14395 and thereby acts as a short circuit for svb. Its expression is likely controlled by Ubx. (C, D) Trichomes on the ventral side of the larval cuticle form stereotypic bands (denticle belts) separated by trichome-free cuticle. (E, F) A trichome-free region on the posterior of the T2 femur differs in size between different D. melanogaster strains. Shown are OregonR (E) and e4,wo1,ro1 (F).
The GRN underlying trichome formation on the larval cuticle of Drosophila species has been characterised in great detail [reviewed in 21,22,28] (Fig 1). Several upstream transcription factors, signalling pathways, and tarsal-less (tal)-mediated post-translational proteolytic processing, lead to the activation of the key regulatory transcription factor Shavenbaby (Svb), which, with SoxNeuro (SoxN), activates a battery of downstream effector genes [29–37]. These downstream factors modulate cell shape changes, actin polymerisation, or cuticle segregation, which underlie the actual formation of trichomes [29,33]. Importantly, ectopic activation of svb during embryogenesis is sufficient to drive trichome development on otherwise naked larval cuticle, and loss of svb function leads to a loss of larval trichomes [38].
Regions of dorso-lateral larval trichomes have been independently lost at least four times among Drosophila species [39,40]. Recombination mapping and functional studies have shown that in all cases analysed, this phenotypic change is caused by changes in several svb enhancers, resulting in a loss of svb expression [6,10,39–42]. The modular enhancers of svb are thought to allow the accumulation of mutations that facilitate the loss of larval trichomes in certain regions without deleterious pleiotropic consequences. It is thought that evolutionary changes in larval trichome patterns cannot be achieved by mutations in genes upstream of svb because of deleterious pleiotropic effects, while changes in individual svb target genes would only affect trichome morphology rather than their presence or absence [19–21,29,33]. Given the position and function of svb in the larval trichome GRN, these data suggest that svb is a hotspot for the evolution of trichome patterns more generally because it is also required for the formation of trichomes on adult epidermis and can induce ectopic trichomes on wings when over expressed [38,43]. Therefore, one could predict that changes in adult trichome patterns are similarly achieved through changes in svb enhancers [20,21].
The trichome pattern on femurs of second legs also varies within and between Drosophila species [1,44] (Fig 1). In D. melanogaster, an area of trichome-free cuticle or ‘naked valley’ varies in size among strains from small to larger naked regions. Other species of the D. melanogaster species subgroup only exhibit larger naked valleys [1,44]. Therefore, trichomes have been gained at the expense of naked cuticle in some strains of D. melanogaster. Differences in naked valley size between species have previously been associated with differences in the expression of Ultrabithorax (Ubx), which represses the formation of leg trichomes [44]. However, genetic mapping experiments and expression analysis have shown that naked valley size variation among populations of D. melanogaster is caused by cis-regulatory changes in miR-92a [1]. This microRNA represses trichome formation by repressing the svb target gene shavenoid (sha), and D. melanogaster strains with small and large naked valleys exhibit weaker or stronger miR-92a expression, respectively, in developing femurs [1,45]. Therefore, while svb is thought to be a hotspot for the evolutionary loss of patches of larval trichomes, it does not appear to underlie the evolutionary gain of leg trichomes in D. melanogaster.
Differences in GRN architecture among developmental contexts may affect which nodes can evolve to facilitate phenotypic change in different tissues or developmental stages. In addition, an evolutionary gain or loss of a phenotype may also result from changes at different nodes in the underlying GRN, i.e. alteration of a particular gene may allow the loss of a trait but changes in the same gene may not necessarily result in the gain of the same trait. Therefore, a better understanding of the genetic basis of phenotypic change and evaluation of the predictability of evolution require characterising the expression and function of GRN components in different developmental contexts, and studying how the loss versus the gain of a trait is achieved.
Here we report our comparison of the regulation of trichome development in legs versus embryos. Our results reveal differences in expression and function of key components of the GRN between these two developmental contexts. These differences indicate that svb is likely unable to act as a switch for the gain of leg trichomes because it is already expressed throughout the legs in both naked and trichome-producing cells. Instead, regulation of sha by miR-92a appears to act as the switch between naked and trichome-producing cells in the leg. This shows that differences in GRNs between different developmental contexts can affect the pathway used by evolution to generate phenotypic change.
Results
Differences in gene expression between leg and larval trichome development
The embryonic expression, regulation, and function of many genes involved in larval trichome formation is well understood [for example, see 29,32–38,42,46] (Fig 1A). To characterise the regulation of leg trichome development better we first carried out RNA-Seq of T2 pupal legs between 20 and 28 hours after puparium formation (hAPF): the window when leg trichomes are specified [44] (S1–S6 Files). We tested if genes known to play a role during larval trichome formation are also expressed in our samples and used a cut-off of 1 fragment per kilobase per million (FPKM) reads mapped to determine if a gene is most likely expressed or not. Note that we chose not to compare the actual expression levels of trichome genes in our leg data sets with those of previously published expression data for embryos because it is difficult to interpret what any quantitative differences in overall expression levels between these two heterogeneous mixtures of cells might mean with respect to trichome development. We found that key genes known to be involved in larval trichome formation are expressed in legs. These include Ubx, SoxN, tal, svb, and sha, as well as key components of the Delta-Notch, Wnt and EGF signalling pathways (Fig 1, and S1 Table). However, expression of several genes known to regulate larval trichome development [29,33,36] is barely detectable in legs (i.e. below or around 1 FPKM). These include Dichaete, Arrowhead, and abrupt, which are also known to regulate svb expression during larval trichome development [34,42] (Fig 1 and S1 Table). Furthermore, the expression of 28 of the 163 known targets of svb in embryos [29,33] is barely detectable in our dataset (FPKM at or below 1) (S2 Table). In addition, 12 out of the 43 genes thought to be involved in larval trichome formation independently of svb [33,36] are also expressed at levels of less than 1 FPKM in legs (S3 Table). Therefore, our RNA-Seq data evidence key differences in both upstream and downstream components of the leg trichome GRN when comparing it to what is known for the embryonic GRN that specifies larval trichomes.
Our leg RNA-Seq data also allowed us to compare expression between strains of D. melanogaster with different sizes of naked valley: Oregon R (OreR) which has a small naked valley and ebony4, white ocelli1, rough1 (eworo) which has a large naked valley (Fig 1). The size of the naked valley in these two strains is caused by differential expression of miR-92a [1]. We found that none of the known regulators of svb are differentially expressed between these two strains. In addition, we did not detect any significant differences in the expression of svb itself or most of its target genes including sha (S1, S2 and S3 Tables). However, we did find a trend towards higher expression of jing interacting gene regulatory 1 (jigr1) in the large naked valley strain eworo, although this difference is not significant after p value correction for false discovery rate (FDR) (S1 Table). Interestingly, miR-92a is co-expressed with jigr1 during neuroblast self-renewal [47] and it is located in one of its introns. Therefore higher expression of miR-92a may be indirectly detectable in eworo (S1 Table). These results are consistent with miR-92a-mediated post-transcriptional regulation causing differences in naked valley size, and since this only occurs in a small proportion of leg cells, the effect on transcripts is likely to be difficult to detect using RNA-Seq.
miR-92a is sufficient to repress leg trichomes and acts downstream of Ubx
We next further examined the function of specific genes during leg trichome development compared to their roles in the formation of larval trichomes. It was previously shown that mutants of miR-92a have small naked valleys [48], which is consistent with the evolution of this locus underlying natural variation in naked valley size [1]. We confirmed these findings using a double mutant for miR-92a and its paralogue miR-92b [47], which exhibits an even smaller naked valley (Fig 2). Note that we did not detect any changes to the larval trichome pattern in these mutants compared to heterozygotes. We examined the morphology of the proximal leg trichomes gained from the loss of miR-92a compared to the trichomes found more distally. We found that the trichomes gained were indistinguishable from the other leg trichomes (S1 Fig). This suggests that all of the genes required to generate leg trichomes are already transcribed in naked valley cells, but that miR-92a must be sufficient to block their translation. Indeed, we found that the extra trichomes that develop in the naked valley in the absence of miR-92a are dependent on svb because in a svb mutant background no trichomes are gained after loss of miR-92a (Fig 2). Furthermore, these results also show that trichome repression by Ubx in the naked valley requires miR-92a because trichomes in the miR-92a mutant develop in the region where Ubx is expressed [44]. Thus, our data confirm that Ubx plays opposite roles in the larval and leg trichome GRNs: in embryos Ubx activates svb to generate larval trichomes [46], while we show that Ubx-mediated repression of leg trichomes [44,49] depends on miR-92a (Figs 1 and 2).
Fig 2. Leg trichome patterns in miR-92a/miR-92b mutants.
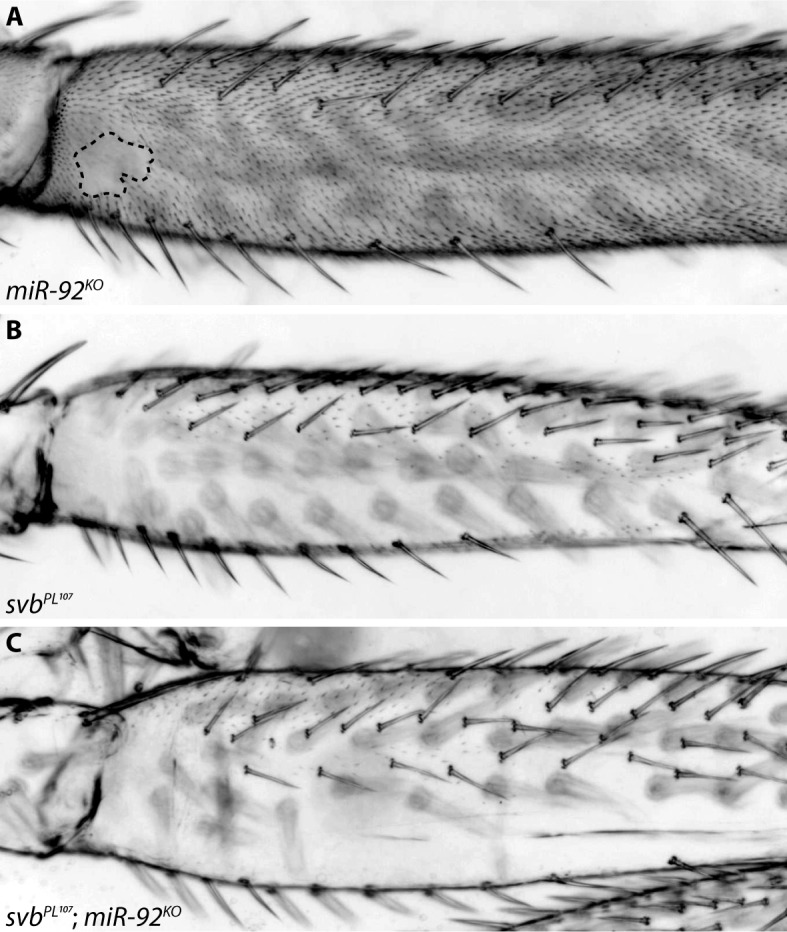
(A) Flies mutant for both miR-92a and miR-92b gain trichomes in the naked valley. (B) Most trichomes on the posterior T2 femur are repressed in svbPL107 flies. (C) No trichomes are gained upon loss of miR-92a and miR-92b in a svbPL107 background.
Regulation of svb during leg trichome patterning
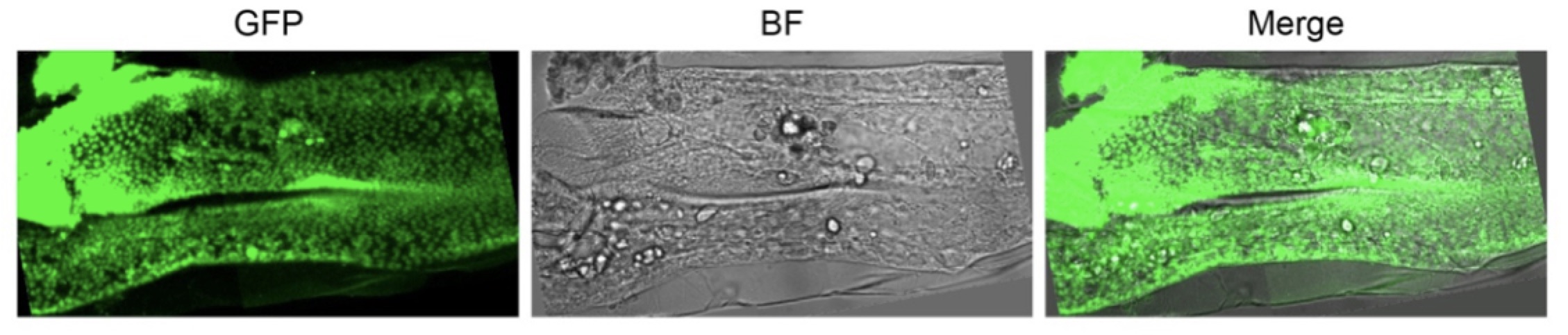
The results above suggest that svb is expressed in the naked valley but is unable to induce the formation of trichomes because of the presence of miR-92a. To test this further we examined the expression of svb transcripts in pupal T2 legs using in situ hybridization. However, this method produced inconsistent results among legs and it was difficult to distinguish between signal and background in the femur. Therefore we examined the expression of a nuclear localising GFP inserted into a BAC containing the entire svb cis-regulatory region, which was previously shown to reliably capture the expression of this gene [43]. We detected GFP throughout T2 legs at 24 hAPF including in the proximal region of the posterior femur (S2 Fig). This indicates that svb is expressed in naked valley cells that do not produce trichomes as well as in more distal trichome-producing cells.
We next investigated the regulatory sequences responsible for svb expression in T2 legs. To do this we carried out ATAC-Seq [50,51] on chromatin from T2 legs during the window of 20 to 28 hAPF when leg trichomes are specified [44]. Embryonic expression of svb underlying larval trichomes is regulated by several enhancers spanning a region of approximately 90 kb upstream of the transcription start site of this gene [5,10] (Fig 3). Several of these larval enhancers also drive reporter gene expression during pupal development [43]. We observed that the embryonic enhancers DG3, E and 7 contained regions of open chromatin according to our T2 leg ATAC-Seq data. However, we found additional accessible chromatin regions that do not overlap with known svb embryonic enhancers (Fig 3).
Fig 3. Enhancers of svb.

(A) Overview of the chromatin accessibility profile (ATAC-seq) at the ovo/svb locus. Indicated are: the deficiency used (dotted line), known larval svb enhancers (black boxes), and tested putative enhancers (grey boxes: no expression in pupal legs, green/orange boxes: expression in pupal legs). Region VT057077 (orange) is able to drive expression during trichome formation (see B-D). The bottom panel shows expressed variants of genes at the locus (black) and genes/variants not expressed (grey). Boxes represent exons, lines represent introns. (B) VT057077 has a naked valley of intermediate size. (C) Expression of sha-ΔUTR under VT057077 control induces trichome formation in the naked valley. (D, D’) Driving miR-92a with VT057077 represses trichome formation on the anterior and posterior of the second leg femur. Small patches of trichomes can sometimes still be observed (arrowhead).
Deletion of a region including the embryonic enhancers DG2 and DG3 [Df(X)svb108] results in a reduction in the number of dorso-lateral larval trichomes when in a sensitized genetic background or at extreme temperatures [5]. Moreover, Preger-Ben Noon and colleagues [43] recently showed that this deletion, as well as a larger deletion that also removes embryonic enhancer A ([Df(X)svb106], see Fig 3), results in the loss of trichomes on abdominal segment A5, specifically in males. We found several peaks of open chromatin in the regions covered by these two deficiencies in our ATAC-seq dataset (Fig 3) and therefore tested the effect of Df(X)svb106 on leg trichome development. We found that deletion of this region and consequently enhancers DG2, DG3, Z and A did not affect the size of the naked valley or the density of trichomes on the femur or other leg segments of flies raised at 17°C, 25°C, or 29°C (compared to the parental lines) (S3 Fig). This suggests that while this region may contribute to svb expression in legs, its removal does not perturb the robustness of leg trichome patterning.
Next, to try to identify enhancer(s) responsible for leg expression, we employed all available GAL4 reporter lines for cis-regulatory regions of svb (S4 Table) that overlap with regions of open chromatin downstream of the above deficiencies (Fig 3). All 10 regions that overlap with open chromatin are able to drive GFP expression to some extent in second legs between 20 and 28 hAPF, as well as in other pupal tissues (S4 Fig). While some of the regions only produce expression in a handful of epidermal cells or particular regions of the T2 legs, none are specific to the presumptive naked valley. Moreover, VT057066, VT057077, VT057081, and VT057083 appear to drive variable levels of GFP expression throughout the leg (S4 Fig). Note that the two regions overlapping with larval enhancers E and 7 (VT057062 and VT057075, respectively) only drive weak expression in a few cells in the tibia and tarsus (S4 Fig).
To further test whether the expression of any of these regions is consistent with a role in trichome formation, we used them to drive expression of the trichome repressor miR-92a and the trichome activator sha-ΔUTR [1]. Intriguingly, driving miR-92a under control of only one of the fragments (VT057077) caused the repression of trichomes on all legs (Fig 3 and S5 Fig) as well as on wings and halteres (S5 Fig). Expressing miR-92a under control of seven fragments (including VT057062 and VT057075) had no noticeable effect, and with two of the other fragments (VT057053, VT057056) only led to repression of trichomes in small patches along the legs consistent with the GFP expression pattern (S4 and S5 Figs).
Driving sha-ΔUTR with VT057077 is sufficient to induce trichome formation in the naked valley (Fig 3) and on the posterior T3 femur (S5 Fig). Driving sha-ΔUTR under control of any of the other nine regions did not produce any ectopic trichomes in the naked valley on T2 or on any other legs. These results indicate that a single enhancer, VT057077, is able to drive svb expression throughout the second leg in both regions which normally produce trichomes and in naked areas.
svb and sha differ in their capacities to induce trichomes in larvae and legs
It was previously shown that miR-92a inhibits leg trichome formation by repressing translation of the svb target sha [1]. However, sha mutants are still able to develop trichomes in larvae, albeit with abnormal morphology [29]. These data suggest that there are differences in the functionality of svb and sha in larval versus leg trichome formation, and therefore we next verified and tested the capacity of svb and sha to produce larval and leg trichomes.
As previously shown [38], ectopic expression of svb is sufficient to induce trichome formation on normally naked larval cuticle (Fig 4). However, we found that ectopic expression of sha in the same cells does not lead to the production of trichomes (Fig 4). svb is also required for posterior leg trichome production [43] (Fig 2 and S6 Fig), but over expression of svb in the naked valley does not produce ectopic trichomes (Fig 4). Over expression of sha on the other hand is sufficient to induce trichome development in the naked valley [1] (Fig 4). These results show that svb and sha differ in their capacities to generate trichomes in larvae versus legs.
Fig 4. Ectopic trichome formation on naked cuticle.

Driving sha-ΔUTR (A) under control of wg-GAL4 does not lead to ectopic trichome formation on otherwise naked larval cuticle. Driving svb (B) or its constitutively active variant ovoB (C) is sufficient to activate trichome development, but expressing only the Svb activator tal (D) is not [32,38]. GFP was co-expressed in each case to indicate the wingless (wg) expression domain (A’-D’). Ectopic activation of sha-ΔUTR in the proximal femur (E) is able to induce trichome formation, but ectopic svb (F) is not. Driving either ovoB (G) or the activator tal (H) leads to ectopic trichome development. Expression of ovoB has additional effects on leg development (e.g. a bending of the proximal femur), while expression of tal also leads to the development of ectopic bristles on the femur (arrowheads in H).
Interestingly, we observed that the ectopic trichomes produced by expression of sha-ΔUTR in the naked valley are significantly shorter than those on the rest of the leg (S1 Fig). This suggests that although sha is able to induce trichome formation in these cells, other genes are also required for their normal morphology. We observed that another characterised svb-target gene, CG14395 [33], is also a high-ranking predicted target of miR-92a [52]: its 3’UTR contains two conserved complete 8-mers corresponding to the binding site for this microRNA. We found that CG14395 is also expressed in pupal second legs according to our leg RNA-Seq data (S2 Table) and furthermore that RNAi against this gene resulted in shorter leg trichomes (S7 Fig). Therefore it appears that miR-92a also represses CG14395 and potentially other svb target genes in addition to sha to block trichome formation.
Over expression of tal or ovoB can induce trichomes
Svb acts as a transcriptional repressor and requires cleavage by the proteasome to become a transcriptional activator. This cleavage is induced by small proteins encoded by the tal locus [30–32]. We therefore tested if svb is unable to promote trichome development in the naked valley because it is not activated in these cells. We found that expressing the constitutively active form ovoB or tal in naked leg cells is sufficient to induce trichome formation (Fig 4), which is consistent with loss of trichomes in tal mutant clones of leg cells (S6 Fig). Furthermore, it appears that tal, like svb, is expressed throughout the leg (S6 Fig). It follows that svb and tal are expressed in naked cells but are unable to induce trichome formation under normal conditions because of repression of sha, CG14395 and possibly other genes by miR-92a. We hypothesise that over expression of tal on the other hand must be able to produce enough active Svb to result in an increase of sha transcription to overwhelm miR-92a repression.
Discussion
The GRNs for larval and leg trichome patterning differ in composition and evolution
The causative genes and even nucleotide changes that underlie the evolution of an increasing number and range of phenotypic traits have been identified [17]. An important theme that has emerged from these studies is that the convergent evolution of traits is often explained by changes in the same genes–so called evolutionary ‘hotspots’ [17,53]. This suggests that the architecture of GRNs may influence or bias the genetic changes that underlie phenotypic changes [18,19,21]. However, relatively little is known about the genetic basis of changes in traits in different developmental contexts and when features are gained versus lost [18].
It was shown previously that changes in the enhancers of svb alone underlie the convergent evolution of the loss of larval trichomes, while the gain of leg trichomes in D. melanogaster is instead mainly explained by evolutionary changes in cis-regulatory regions of miR-92a [1,6,10,39–41]. We investigated this further by comparing the GRNs involved in both developmental contexts and by examining the regulation and function of key genes.
Our results show that there are differences between the GRNs underlying the formation of larval and leg trichomes in terms of the expression of components and their functionality. These changes are found both in upstream genes of the GRN that help to determine where trichomes are made, and in downstream genes whose products are directly involved in trichome formation (Fig 1). The latter may also determine the differences in the fine-scale morphology of these structures on larval and leg cuticle (Fig 1) [29].
Furthermore, while the key evolutionary switch in embryos, the gene svb, is also necessary for trichome production on the posterior leg, over expression of this gene is not sufficient to produce leg trichomes in the naked proximal region of the T2 femur. This is because the leg trichome GRN employs miR-92a, which inhibits trichome production by blocking the translation of the svb target gene sha and probably other target genes including CG14395. In the legs of D. melanogaster, miR-92a therefore acts as the evolutionary switch for trichome production, and consequently the size of the naked valley depends on the expression of this gene (Fig 5) [1].
Fig 5. The size of the naked valley differs between and within species and is dependent on miR-92a expression.

Reduction of miR-92a expression in D. melanogaster T2 legs has led to a derived (d) smaller naked valley in some populations while the ancestral state (a) is thought to be a large naked valley like in other D. melanogaster group species and other species (e.g. D. pseudoobscura). The absence of a naked valley in D. virilis is possibly due to absence of miR-92a expression, while the presence of small naked valleys in other species of the virilis group (e.g. D. americana) could be explained by a gain of microRNA expression. The coloured bars represent the spatial expression of each gene in the femur with lighter orange indicating where sha expression is post-transcriptionally repressed by miR-92a.
Interestingly, we observed that the ectopic trichomes produced by over expression of sha-ΔUTR in the naked valley are significantly shorter than those on the rest of the leg (S1 Fig). Therefore, while sha is able to induce trichome formation in these cells, other genes, including CG14395, are also required for normal trichome morphology. This suggests that GRNs may be able to co-opt regulators, in this case possibly miR-92a, that can act in trans to regulate existing components. Such changes can facilitate phenotypic evolution by phenocopying the effects of ‘hotspot’ genes in contexts where their evolution may be constrained. While trichomes can be lost as a result of the loss of svb expression but not loss of sha alone, interestingly, over expression of miR-92a is also able to suppress trichomes on other structures, including wings [1,45], presumably through repression of sha and other genes like CG14395.
Other genetic bases for the evolution of leg trichome patterns?
In contrast to larvae, it is unlikely that mutations in svb can lead to evolutionary changes in legs to gain trichomes and decrease the size of the naked valley. This is because this gene (and likely all the other genes necessary for trichome production) is already transcribed in naked valley cells. In addition, a single svb enhancer is able to drive expression throughout the legs including the naked valley. Although other enhancer regions of this gene are able to drive some expression in patches of leg cells, none of these is naked valley-specific. This suggests that evolutionary changes to svb enhancers would be unlikely to only affect expression of this gene in the naked valley. It remains possible that binding sites could evolve in this global leg enhancer to increase the Svb concentration specifically in naked valley cells. This could overcome miR-92a-mediated repression of trichomes similar to our experiments where tal and ovoB are over expressed in these cells, or when molecular sponges are used to phenocopy the loss of microRNAs [54]. However, this does not seem to have been the preferred evolutionary route in D. melanogaster [1] (Fig 5).
Our study also corroborates that Ubx represses leg trichomes [44] whereas it promotes larval trichome development through activation of svb [46]. Moreover, our results indicate that Ubx acts upstream of miR-92a in legs because it is unable to repress leg trichomes in the absence of this microRNA. It is possible that Ubx even directly activates miR-92a since ChIP-chip data indicate that there are Ubx binding sites within the jigr1/miR-92a locus [55]. Intriguingly, there is no naked valley in D. virilis, and Ubx does not appear to be expressed in the second legs of this species during trichome development [44] (Fig 5). However naked valleys are evident in other species of the virilis and montana groups and it would be interesting to determine if these differences were caused by changes in Ubx, miR-92a, or even other loci (Fig 5).
Evolutionary hotspots and developmental context
To the best of our knowledge, our study is the first to directly compare the expression and function of components of the GRNs underlying the formation of similar structures that have evolved in different developmental contexts. Our results show that the GRNs for trichome production in larval versus leg contexts retain a core set of genes but also exhibit differences in the components used and in their wiring. These differences likely reflect changes that accumulate in GRNs during processes such as co-option [e.g. 56] and developmental systems drift [57–59], although it remains possible that the changes have been selected for unknown reasons.
Importantly, we show that the differences in these GRNs may help to explain why they have evolved at different nodes to lead to the gain or loss of trichomes. This supports the suggestion that GRN architecture can influence the pathway of evolution and lead to hotspots for the convergent evolution of traits [17–19,21]. Indeed, such hotspots can also underlie phenotypic changes in different developmental contexts. For example, yellow underlies differences in abdominal pigmentation and wing spot pigmentation among Drosophila species [7,11,60,61]. However, we demonstrate that it cannot be assumed that evolutionary hotspots in one development context represent the nodes of evolution in a different context as a consequence of differences in GRN architecture.
Our findings also highlight that the genes that underlie the loss of features might not have the capacity to lead to the gain of the same feature. Therefore, while evolution may be predictable in a particular context, it is very important to consider different developmental contexts and whether a trait is lost versus gained. Indeed, even when we map the genetic basis of phenotypic change to the causative genes it is important to understand the changes in the context of the wider GRN to fully appreciate how the developmental program functions and evolves. Since evolution is thought to favour changes with low pleiotropy [19,62–65], the effects of genetic changes underlying phenotypic change should be tested more widely during development. Such an approach recently revealed that svb enhancers underlying differences in larval trichomes are actually also used in other contexts [43]. Interestingly, miR-92a is employed in several roles, including self-renewal of neuroblasts [47], germline specification [48], and circadian rhythms [66]. It remains to be seen if the evolutionary changes in this microRNA underlying naked valley differences also have pleiotropic consequences, and therefore if natural variation in naked valley size is actually a pleiotropic outcome of selection on another aspect of miR-92a function.
Materials and methods
Fly strains, husbandry and crosses
Fly strains used in this study are listed in S4 Table. GAL4 lines for analysis of svb expression and RNAi lines for analysis of CG14395 were obtained from the Vienna Drosophila Resource Center (VDRC) [67, 68]. Flies were reared on standard food at 25°C unless otherwise indicated.
Replacement of the P{lacW}l(3)S011041 element, which is inserted 5’ of the tal gene, by a P{GaWB} transposable element was carried out by mobilization in omb-GAL4; +/CyO Δ2–3; l(3)S011041/TM3,Sb flies as described in [31]. Replacements were screened by following UAS-GFP expression in the progeny. The P{GaWB} element is inserted in the same nucleotide position as P{lacW}S011041. Clonal analysis of tal S18.1 and svbR9 alleles were performed as previously described [69].
A transgenic line that contains the cis-regulatory region of svb upstream of a GFP reporter (svbBAC-GFP) [43] was used to monitor svb expression. Legs of pupae were dissected 24 h hAPF, fixed and stained following the protocol of Halachmi et al. [70], using a chicken anti-GFP as primary antibody (Aves Labs, 1:250) and an anti-chicken as secondary (AlexaFluor 488, 1:400). Images were obtained on a confocal microscope with a 60X objective. SUM projections of the z-stacks were generated after background subtraction. A filter median implemented in ImageJ software [71] was applied. The proximal femur image was reconstructed from two SUM projections using Adobe Photoshop.
Measurement of trichome length
For trichome length measurements, T2 legs were dissected, mounted in Hoyer’s medium/lactic acid 1:1 and imaged under a Zeiss Axioplan microscope using ProgRes MF cool camera (Jenaoptik, Germany). Trichomes on distal and proximal femurs were measured and analysed using ImageJ software [71]. Statistical analyses were done in R version 3.4.2 [72].
RNA-Seq
Pupae were collected within 1 hAPF and allowed to develop for another 20 to 28 h at 25°C. Second legs were dissected in PBS from approximately 80 pupae per replicate and kept in RNAlater. RNA was isolated using phenol-chloroform extraction. This was done in three replicates for two different strains (e4,wo1,ro1 and OregonR). Library preparation and sequencing (75 bp paired end) were carried out by Edinburgh Genomics. Reads were aligned to D. melanogaster genome version 6.12 [73] using TopHat 2.1.1. [74]. Transcripts were quantified using Cufflinks 2.2.1 and differential expression analysis conducted using Cuffdiff [75] (S1–S7 Files). Genes expressed below or around 1 FPKM were considered not expressed. Raw sequencing reads are deposited in the Gene Expression Omnibus with accession number GSE113240.
ATAC-seq
Pupae were reared and dissected as described above. Dissected legs were kept in ice cold PBS. Leg cells were lysed in 50 μl Lysis Buffer (10 mM Tris-HCl, pH = 7.5; 10 mM NaCl; 3 mM MgCl2; 0.1% IGEPAL). Nuclei were collected by centrifugation at 500 g for 5 min. Approximately 60,000 nuclei were suspended in 50 μl Tagmentation Mix [25 μl Buffer (20 mM Tris-CH3COO-, pH = 7.6; 10 mM MgCl2; 20% Dimethylformamide); 2.5 μl Tn5 Transposase; 22.5 μl H2O] and incubated at 37°C for 30 min. After addition of 3 μl 2 M NaAC, pH = 5.2 DNA was purified using a QIAGEN MinElute Kit. PCR amplification for library preparation was done for 15 cycles with NEBNext High Fidelity Kit; primers were used according to [50]. This procedure was carried out for three replicates for each of two strains (e4,wo1,ro1 and OregonR). Paired end 50 bp sequencing was carried out by the Transcriptome and Genome Analysis Laboratory Göttingen, Germany. Reads were end-to-end aligned to D. melanogaster genome version 6.12 (FlyBase) [73] using bowtie2 [76]. After filtering of low quality reads and removal of duplicates using SAMtools [77,78], reads were re-centered according to [50]. Peaks were called with MACS2 [79] and visualisation was done using Sushi [80] (S8 and S9 Files). The reads have been deposited in the Gene Expression Omnibus with accession number GSE113240.
Supporting information
(A) Trichomes gained in the naked valley after loss of miR-92a and miR-92b have a similar morphology as trichomes on the more distal femur. Trichomes gained after ectopic expression of sha-ΔUTR (B) are significantly shorter, while trichomes developing after expression of ovoB (C) are significantly longer than on the remaining femur. (D) Trichomes on the more distal femur have a similar length as in the driver line (VT42733) regardless of whether ovoB or sha are expressed under its control, but trichomes gained in the naked valley are significantly longer or shorter, respectively (p<0.001). Tukey’s multiple comparison test was used to test for significance.
(JPG)
{kind=link}
GFP is expressed throughout the posterior femur of a T2 leg at 24 hAPF.
(JPG)
{kind=link}
The control line still contains both pBac insertions used to generate the deficiency [5,43]. There is no detectable difference in naked valley size or trichome density between deficiency and control flies at 25°C, 29°C, or 17°C.
(JPG)
{kind=link}
All tested drivers show some expression in T2 legs as well as in other pupal tissues.
(JPG)
{kind=link}
(A, A’, B, B’) Trichomes on the wing are largely repressed upon expression of miR-92a under control of VT057077. Note that trichomes on the alula (arrowhead in B) develop normally. Also trichomes on T1 and T3 legs (C, C’ D, F, F’, G) and on the halteres (E, E’, H, H’) are repressed when miR-92a is driven by VT057077. (I) Driving sha-ΔUTR under control of VT057077 leads to ectopic formation of trichomes on the posterior T3 leg (compare to D’). (J, J’) Trichomes on the ventral side of the femur are partially repressed when miR-92a is expressed under control of VT057053. Trichomes are repressed in a patch on the dorsal side of the distal T2 femur (K) and around the rim of the distal wing (L) after expression of miR-92a under control of VT057056.
(JPG)
{kind=link}
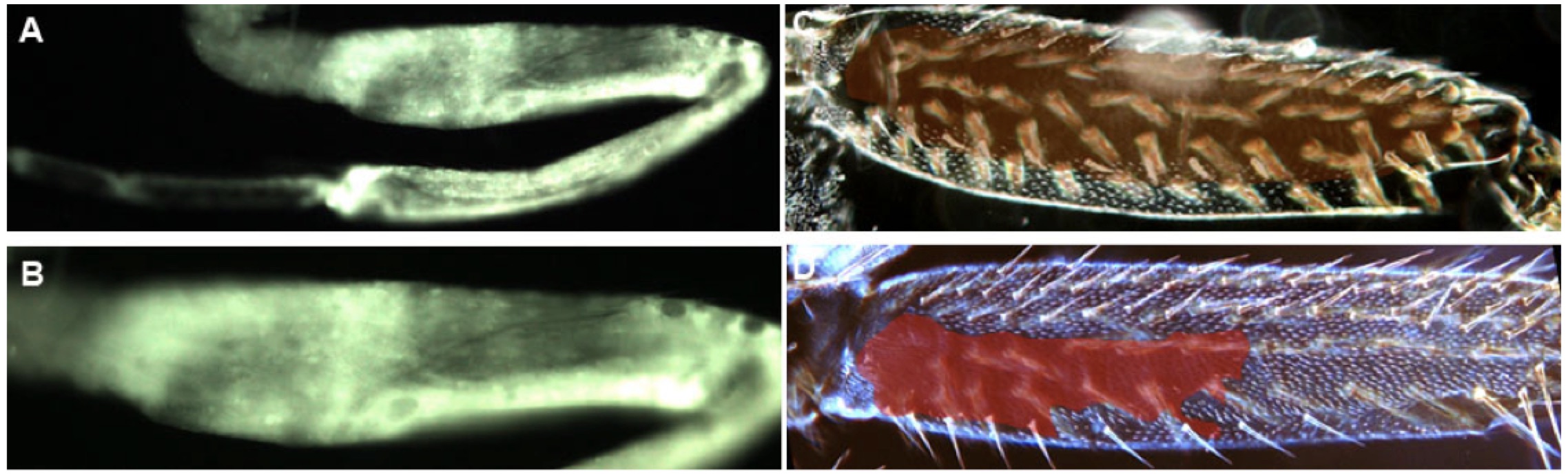
GFP is expressed throughout all the leg segments (A) including the femur (B) of the second leg. Mutant clones of tals18 (C) (brown shaded area) and svbR9 (D) (red shaded area) lack trichomes on the femur of a second leg.
(JPG)
{kind=link}
Expression of the RNAi construct and UAS-Dicer was under control of GAL4 driver lines VT042733 (drives in the proximal femur) and VT057077 (drives in the whole leg). Box plots show the length of trichomes in the distal part of the posterior femur and around the naked valley (NV). Parents (UAS-Dcr/CyO;VT042733/TM6B or UAS-Dcr/CyO;VT057077/TM6B females, VDRC CG14395-RNAi males) and siblings without knockdown effect were used as controls (Ctrl). (A) Trichomes developing after knockdown of CG14395 in the proximal femur are significantly shorter around the naked valley area than on the remaining femur (distal part) and on femurs of the controls (p < 0.001). Data are normally distributed (Shapiro-Wilk normality test). Tukey’s multiple comparison test was used to test for significance. (B) After knockdown of CG14395 in the whole leg, trichomes are significantly shorter both around the naked valley area and on the remaining femur (p < 0.001 and p < 0.01). Note that some controls show significantly different trichome lengths. Data are not normally distributed (Shapiro-Wilk normality test). Kruskal-Wallis and pairwise comparisons using Wilcoxon rank sum test were used to test for significance.
(PDF)
Genes are sorted by gene name. Two rows for a gene indicate alternative transcription start sites. Expression level in fragments per kilobase per million (FPKM) with low and high confidence values, base 2 log of the fold change, and p and q values are given after mapping with TopHat 2.1.1, transcriptome assembly with Cufflinks 2.2.1 and Cuffmerge, and comparison with Cuffdiff 2.2.1 [74,75]. q values are false discovery rate (FDR)-corrected p values.
(XLSX)
Genes are sorted by gene name. Two rows for a gene indicate alternative transcription start sites. Expression level in fragments per kilobase per million (FPKM) with low and high confidence values, base 2 log of the fold change, and p and q values are given after mapping with TopHat 2.1.1, transcriptome assembly with Cufflinks 2.2.1 and Cuffmerge, and comparison with Cuffdiff 2.2.1 [74,75]. q values are false discovery rate (FDR)-corrected p values.
(XLSX)
Genes are sorted by gene name. Two rows for a gene indicate alternative transcription start sites. Expression level in fragments per kilobase per million (FPKM) with low and high confidence values, base 2 log of the fold change, and p and q values are given after mapping with TopHat 2.1.1, transcriptome assembly with Cufflinks 2.2.1 and Cuffmerge, and comparison with Cuffdiff 2.2.1 [74,75]. q values are FDR-corrected p values.
(XLSX)
(DOCX)
(CSV)
(CSV)
(CSV)
(CSV)
(CSV)
(CSV)
(CSV)
(TXT)
(TXT)
Acknowledgments
We thank Georgina Haines-Woodhouse for technical assistance, Daniel Leite for help with bioinformatics, and members of the McGregor lab and Maike Kittelmann for comments and suggestions throughout the project. We also thank Francois Payre (University of Toulouse), David Stern (Janelia Farm), Fen-Biao Gao (UMass Med School) and the Vienna Drosophila Resource Center for providing fly stocks. We also thank three anonymous reviewers for their valuable comments and suggestions to improve the manuscript.
Data Availability
All RNA-Seq and ATAC-Seq data files are available from the Gene Expression Omnibus database (accession number GSE113240).
Funding Statement
This work was funded by DFG Research Fellowships to SK (Ki 1831/1-1) and FAF (FR 3929/1-1), a BBSRC DTP studentship to ADB, grants from Ministerio de Economía y Competitividad (BFU2016-74961-P) and the Andalusian Government (BIO-396) to JLGS, and an Austrian Science Fund (FWF) Fellowship to APM (M1059-B09). RNA library preparation and sequencing were carried out by Edinburgh Genomics, The University of Edinburgh. Edinburgh Genomics is partly supported through core grants from NERC (R8/H10/56), MRC (MR/K001744/1) and BBSRC (BB/J004243/1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Arif S, Murat S, Almudi I, Nunes MD, Bortolamiol-Becet D, et al. (2013) Evolution of mir-92a Underlies Natural Morphological Variation in Drosophila melanogaster. Curr Biol 23: 523–528. doi: 10.1016/j.cub.2013.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnoult L, Su KF, Manoel D, Minervino C, Magrina J, et al. (2013) Emergence and diversification of fly pigmentation through evolution of a gene regulatory module. Science 339: 1423–1426. doi: 10.1126/science.1233749 [DOI] [PubMed] [Google Scholar]
- 3.Chan YF, Marks ME, Jones FC, Villarreal G Jr., Shapiro MD, et al. (2010) Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327: 302–305. doi: 10.1126/science.1182213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colosimo PF, Peichel CL, Nereng K, Blackman BK, Shapiro MD, et al. (2004) The genetic architecture of parallel armor plate reduction in threespine sticklebacks. PLoS Biol 2: E109 doi: 10.1371/journal.pbio.0020109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frankel N, Davis GK, Vargas D, Wang S, Payre F, et al. (2010) Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466: 490–493. doi: 10.1038/nature09158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frankel N, Erezyilmaz DF, McGregor AP, Wang S, Payre F, et al. (2011) Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature 474: 598–603. doi: 10.1038/nature10200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gompel N, Prud'homme B, Wittkopp PJ, Kassner VA, Carroll SB (2005) Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature 433: 481–487. doi: 10.1038/nature03235 [DOI] [PubMed] [Google Scholar]
- 8.Guerreiro I, Gitto S, Novoa A, Codourey J, Nguyen Huynh TH, et al. (2016) Reorganisation of Hoxd regulatory landscapes during the evolution of a snake-like body plan. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lang M, Murat S, Clark AG, Gouppil G, Blais C, et al. (2012) Mutations in the neverland gene turned Drosophila pachea into an obligate specialist species. Science 337:1658–1661. doi: 10.1126/science.1224829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGregor AP, Orgogozo V, Delon I, Zanet J, Srinivasan DG, et al. (2007) Morphological evolution through multiple cis-regulatory mutations at a single gene. Nature 448: 587–590. doi: 10.1038/nature05988 [DOI] [PubMed] [Google Scholar]
- 11.Prud'homme B, Gompel N, Rokas A, Kassner VA, Williams TM, et al. (2006) Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature 440: 1050–1053. doi: 10.1038/nature04597 [DOI] [PubMed] [Google Scholar]
- 12.Reed RD, Papa R, Martin A, Hines HM, Counterman BA, et al. (2011) optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 333: 1137–1141. doi: 10.1126/science.1208227 [DOI] [PubMed] [Google Scholar]
- 13.Santos ME, Le Bouquin A, Crumière AJJ, Khila A (2017) Taxon-restricted genes at the origin of a novel trait allowing access to a new environment. Science 358: 386–390. doi: 10.1126/science.aan2748 [DOI] [PubMed] [Google Scholar]
- 14.Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, et al. (2004) Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 428: 717–723. doi: 10.1038/nature02415 [DOI] [PubMed] [Google Scholar]
- 15.Martin A, McCulloch KJ, Patel NH, Briscoe AD, Gilbert LE, et al. (2014) Multiple recent co-options of Optix associated with novel traits in adaptive butterfly wing radiations. Evodevo 5: 7 doi: 10.1186/2041-9139-5-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll SB (2008) Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134: 25–36. doi: 10.1016/j.cell.2008.06.030 [DOI] [PubMed] [Google Scholar]
- 17.Martin A, Orgogozo V (2013) The Loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution 67: 1235–1250. doi: 10.1111/evo.12081 [DOI] [PubMed] [Google Scholar]
- 18.Kopp A (2009) Metamodels and phylogenetic replication: a systematic approach to the evolution of developmental pathways. Evolution 63: 2771–2789. doi: 10.1111/j.1558-5646.2009.00761.x [DOI] [PubMed] [Google Scholar]
- 19.Stern DL (2011) Evolution, Development and the Predictable Genome. Greenwood Village: Roberts and Company. [Google Scholar]
- 20.Stern DL, Orgogozo V (2008) The loci of evolution: how predictable is genetic evolution? Evolution 62: 2155–2177. doi: 10.1111/j.1558-5646.2008.00450.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stern DL, Orgogozo V (2009) Is genetic evolution predictable? Science 323: 746–751. doi: 10.1126/science.1158997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arif S, Kittelmann S, McGregor AP (2015) From shavenbaby to the naked valley: trichome formation as a model for evolutionary developmental biology. Evol Dev 17: 120–126. doi: 10.1111/ede.12113 [DOI] [PubMed] [Google Scholar]
- 23.Balmert A, Florian Bohn H, Ditsche-Kuru P, Barthlott W (2011) Dry under water: comparative morphology and functional aspects of air-retaining insect surfaces. J Morphol 272: 442–451. doi: 10.1002/jmor.10921 [DOI] [PubMed] [Google Scholar]
- 24.Ditsche-Kuru P, Schneider ES, Melskotte JE, Brede M, Leder A, et al. (2011) Superhydrophobic surfaces of the water bug Notonecta glauca: a model for friction reduction and air retention. Beilstein J Nanotechnol 2: 137–144. doi: 10.3762/bjnano.2.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodwyn PJ, Voigt D, Fujisaki K (2008) Skating and diving: Changes in functional morphology of the setal and microtrichial cover during ontogenesis in Aquarius paludum fabricius (Heteroptera, Gerridae). J Morphol 269: 734–744. doi: 10.1002/jmor.10619 [DOI] [PubMed] [Google Scholar]
- 26.Goodwyn PP, De Souza E, Fujisaki K, Gorb S (2008) Moulding technique demonstrates the contribution of surface geometry to the super-hydrophobic properties of the surface of a water strider. Acta Biomater 4: 766–770. doi: 10.1016/j.actbio.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 27.Inestrosa NC, Sunkel CE, Arriagada J, Garrido J, Godoy-Herrera R (1996) Abnormal development of the locomotor activity in yellow larvae of Drosophila: a cuticular defect? Genetica 97: 205–210. [DOI] [PubMed] [Google Scholar]
- 28.Stern DL, Frankel N (2013) The structure and evolution of cis-regulatory regions: the shavenbaby story. Philos Trans R Soc Lond B Biol Sci 368: 20130028 doi: 10.1098/rstb.2013.0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chanut-Delalande H, Fernandes I, Roch F, Payre F, Plaza S (2006) Shavenbaby couples patterning to epidermal cell shape control. PLoS Biol 4: e290 doi: 10.1371/journal.pbio.0040290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chanut-Delalande H, Hashimoto Y, Pelissier-Monier A, Spokony R, Dib A, et al. (2014) Pri peptides are mediators of ecdysone for the temporal control of development. Nat Cell Biol 16: 1035–1044. doi: 10.1038/ncb3052 [DOI] [PubMed] [Google Scholar]
- 31.Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP (2007) Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol 5: e106 doi: 10.1371/journal.pbio.0050106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, et al. (2010) Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science 329: 336–339. doi: 10.1126/science.1188158 [DOI] [PubMed] [Google Scholar]
- 33.Menoret D, Santolini M, Fernandes I, Spokony R, Zanet J, et al. (2013) Genome-wide analyses of Shavenbaby target genes reveals distinct features of enhancer organization. Genome Biol 14: R86 doi: 10.1186/gb-2013-14-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overton PM, Chia W, Buescher M (2007) The Drosophila HMG-domain proteins SoxNeuro and Dichaete direct trichome formation via the activation of shavenbaby and the restriction of Wingless pathway activity. Development 134: 2807–2813. doi: 10.1242/dev.02878 [DOI] [PubMed] [Google Scholar]
- 35.Ren N, He B, Stone D, Kirakodu S, Adler PN (2006) The shavenoid gene of Drosophila encodes a novel actin cytoskeleton interacting protein that promotes wing hair morphogenesis. Genetics 172: 1643–1653. doi: 10.1534/genetics.105.051433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rizzo NP, Bejsovec A (2017) SoxNeuro and Shavenbaby act cooperatively to shape denticles in the embryonic epidermis of Drosophila. Development 144: 2248–2258. doi: 10.1242/dev.150169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanet J, Benrabah E, Li T, Pelissier-Monier A, Chanut-Delalande H, et al. (2015) Pri sORF peptides induce selective proteasome-mediated protein processing. Science 349: 1356–1358. doi: 10.1126/science.aac5677 [DOI] [PubMed] [Google Scholar]
- 38.Delon I, Chanut-Delalande H, Payre F (2003) The Ovo/Shavenbaby transcription factor specifies actin remodelling during epidermal differentiation in Drosophila. Mech Dev 120: 747–758. [DOI] [PubMed] [Google Scholar]
- 39.Sucena E, Delon I, Jones I, Payre F, Stern DL (2003) Regulatory evolution of shavenbaby/ovo underlies multiple cases of morphological parallelism. Nature 424: 935–938. doi: 10.1038/nature01768 [DOI] [PubMed] [Google Scholar]
- 40.Sucena E, Stern DL (2000) Divergence of larval morphology between Drosophila sechellia and its sibling species caused by cis-regulatory evolution of ovo/shaven-baby. Proc Natl Acad Sci U S A 97: 4530–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frankel N, Wang S, Stern DL (2012) Conserved regulatory architecture underlies parallel genetic changes and convergent phenotypic evolution. Proc Natl Acad Sci U S A 109: 20975–20979. doi: 10.1073/pnas.1207715109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Preger-Ben Noon E, Davis FP, Stern DL (2016) Evolved Repression Overcomes Enhancer Robustness. Dev Cell 39: 572–584. doi: 10.1016/j.devcel.2016.10.010 [DOI] [PubMed] [Google Scholar]
- 43.Preger-Ben Noon E, Sabaris G, Ortiz DM, Sager J, Liebowitz A, et al. (2018) Comprehensive Analysis of a cis-Regulatory Region Reveals Pleiotropy in Enhancer Function. Cell Rep 22: 3021–3031. doi: 10.1016/j.celrep.2018.02.073 [DOI] [PubMed] [Google Scholar]
- 44.Stern DL (1998) A role of Ultrabithorax in morphological differences between Drosophila species. Nature 396: 463–466. doi: 10.1038/24863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schertel C, Rutishauser T, Forstemann K, Basler K (2012) Functional characterization of Drosophila microRNAs by a novel in vivo library. Genetics 192: 1543–1552. doi: 10.1534/genetics.112.145383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, et al. (2015) Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 160: 191–203. doi: 10.1016/j.cell.2014.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuva-Aydemir Y, Xu XL, Aydemir O, Gascon E, Sayin S, et al. (2015) Downregulation of the Host Gene jigr1 by miR-92 Is Essential for Neuroblast Self-Renewal in Drosophila. PLoS Genet 11: e1005264 doi: 10.1371/journal.pgen.1005264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen YW, Song S, Weng R, Verma P, Kugler JM, et al. (2014) Systematic study of Drosophila microRNA functions using a collection of targeted knockout mutations. Dev Cell 31: 784–800. doi: 10.1016/j.devcel.2014.11.029 [DOI] [PubMed] [Google Scholar]
- 49.Davis GK, Srinivasan DG, Wittkopp PJ, Stern DL (2007) The function and regulation of Ultrabithorax in the legs of Drosophila melanogaster. Dev Biol 308: 621–631. doi: 10.1016/j.ydbio.2007.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. doi: 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109: 21.29.21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, et al. (2007) Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res 17: 1850–1864. doi: 10.1101/gr.6597907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richardson MK, Brakefield PM (2003) Developmental biology: hotspots for evolution. Nature 424: 894–895. doi: 10.1038/424894a [DOI] [PubMed] [Google Scholar]
- 54.Cohen SM (2009) Use of microRNA sponges to explore tissue-specific microRNA functions in vivo. Nat Methods 6: 873–874. doi: 10.1038/nmeth1209-873 [DOI] [PubMed] [Google Scholar]
- 55.Agrawal P, Habib F, Yelagandula R, Shashidhara LS (2011) Genome-level identification of targets of Hox protein Ultrabithorax in Drosophila: novel mechanisms for target selection. Sci Rep 1: 205 doi: 10.1038/srep00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glassford WJ, Johnson WC, Dall NR, Smith SJ, Liu Y, et al. (2015) Co-option of an Ancestral Hox-Regulated Network Underlies a Recently Evolved Morphological Novelty. Dev Cell 34: 520–531. doi: 10.1016/j.devcel.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buffry AD, Mendes CC, McGregor AP (2016) The Functionality and Evolution of Eukaryotic Transcriptional Enhancers. Adv Genet 96: 143–206. doi: 10.1016/bs.adgen.2016.08.004 [DOI] [PubMed] [Google Scholar]
- 58.Halfon MS (2017) Perspectives on Gene Regulatory Network Evolution. Trends Genet 33: 436–447. doi: 10.1016/j.tig.2017.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.True JR, Haag ES (2001) Developmental system drift and flexibility in evolutionary trajectories. Evol Dev 3: 109–119. [DOI] [PubMed] [Google Scholar]
- 60.Jeong S, Rokas A, Carroll SB (2006) Regulation of body pigmentation by the Abdominal-B Hox protein and its gain and loss in Drosophila evolution. Cell 125: 1387–1399. doi: 10.1016/j.cell.2006.04.043 [DOI] [PubMed] [Google Scholar]
- 61.Wittkopp PJ, Vaccaro K, Carroll SB (2002) Evolution of yellow gene regulation and pigmentation in Drosophila. Curr Biol 12: 1547–1556. [DOI] [PubMed] [Google Scholar]
- 62.Nunes MD, Arif S, Schlotterer C, McGregor AP (2013) A perspective on micro-evo-devo: progress and potential. Genetics 195: 625–634. doi: 10.1534/genetics.113.156463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orr HA (2000) Adaptation and the cost of complexity. Evolution 54: 13–20. [DOI] [PubMed] [Google Scholar]
- 64.True J (2003) Insect melanism: the molecules matter. Trends in Ecology and Evolution 18: 640–647. [Google Scholar]
- 65.Waxman D, Peck JR (1998) Pleiotropy and the preservation of perfection. Science 279: 1210–1213. [PubMed] [Google Scholar]
- 66.Chen X, Rosbash M (2017) MicroRNA-92a is a circadian modulator of neuronal excitability in Drosophila. Nat Commun 8: 14707 doi: 10.1038/ncomms14707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448: 151–156. doi: 10.1038/nature05954 [DOI] [PubMed] [Google Scholar]
- 68.Kvon EZ, Kazmar T, Stampfel G, Yanez-Cuna JO, Pagani M, et al. (2014) Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature 512: 91–95. doi: 10.1038/nature13395 [DOI] [PubMed] [Google Scholar]
- 69.Pueyo JI, Couso JP (2008) The 11-aminoacid long Tarsal-less peptides trigger a cell signal in Drosophila leg development. Dev Biol 324: 192–201. doi: 10.1016/j.ydbio.2008.08.025 [DOI] [PubMed] [Google Scholar]
- 70.Halachmi N, Nachman A, Salzberg A (2012) Visualization of proprioceptors in Drosophila larvae and pupae. J Vis Exp: e3846 doi: 10.3791/3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.R Core Team (2017) R: A language and environment for statistical computing. R Foundation for Statistical Computing. [Google Scholar]
- 73.Gramates LS, Marygold SJ, Santos GD, Urbano JM, Antonazzo G, et al. (2017) FlyBase at 25: looking to the future. Nucleic Acids Res 45: D663–D671. doi: 10.1093/nar/gkw1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. doi: 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. doi: 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993. doi: 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137 doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Phanstiel DH, Boyle AP, Araya CL, Snyder MP (2014) Sushi.R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics 30: 2808–2810. doi: 10.1093/bioinformatics/btu379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Payre F (2004) Genetic control of epidermis differentiation in Drosophila. Int J Dev Biol 48: 207–215. doi: 10.1387/ijdb.041828fp [PubMed] [Google Scholar]
- 82.Stern DL (2013) The genetic causes of convergent evolution. Nat Rev Genet 14: 751–764. doi: 10.1038/nrg3483 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Trichomes gained in the naked valley after loss of miR-92a and miR-92b have a similar morphology as trichomes on the more distal femur. Trichomes gained after ectopic expression of sha-ΔUTR (B) are significantly shorter, while trichomes developing after expression of ovoB (C) are significantly longer than on the remaining femur. (D) Trichomes on the more distal femur have a similar length as in the driver line (VT42733) regardless of whether ovoB or sha are expressed under its control, but trichomes gained in the naked valley are significantly longer or shorter, respectively (p<0.001). Tukey’s multiple comparison test was used to test for significance.
(JPG)
GFP is expressed throughout the posterior femur of a T2 leg at 24 hAPF.
(JPG)
The control line still contains both pBac insertions used to generate the deficiency [5,43]. There is no detectable difference in naked valley size or trichome density between deficiency and control flies at 25°C, 29°C, or 17°C.
(JPG)
All tested drivers show some expression in T2 legs as well as in other pupal tissues.
(JPG)
(A, A’, B, B’) Trichomes on the wing are largely repressed upon expression of miR-92a under control of VT057077. Note that trichomes on the alula (arrowhead in B) develop normally. Also trichomes on T1 and T3 legs (C, C’ D, F, F’, G) and on the halteres (E, E’, H, H’) are repressed when miR-92a is driven by VT057077. (I) Driving sha-ΔUTR under control of VT057077 leads to ectopic formation of trichomes on the posterior T3 leg (compare to D’). (J, J’) Trichomes on the ventral side of the femur are partially repressed when miR-92a is expressed under control of VT057053. Trichomes are repressed in a patch on the dorsal side of the distal T2 femur (K) and around the rim of the distal wing (L) after expression of miR-92a under control of VT057056.
(JPG)
GFP is expressed throughout all the leg segments (A) including the femur (B) of the second leg. Mutant clones of tals18 (C) (brown shaded area) and svbR9 (D) (red shaded area) lack trichomes on the femur of a second leg.
(JPG)
Expression of the RNAi construct and UAS-Dicer was under control of GAL4 driver lines VT042733 (drives in the proximal femur) and VT057077 (drives in the whole leg). Box plots show the length of trichomes in the distal part of the posterior femur and around the naked valley (NV). Parents (UAS-Dcr/CyO;VT042733/TM6B or UAS-Dcr/CyO;VT057077/TM6B females, VDRC CG14395-RNAi males) and siblings without knockdown effect were used as controls (Ctrl). (A) Trichomes developing after knockdown of CG14395 in the proximal femur are significantly shorter around the naked valley area than on the remaining femur (distal part) and on femurs of the controls (p < 0.001). Data are normally distributed (Shapiro-Wilk normality test). Tukey’s multiple comparison test was used to test for significance. (B) After knockdown of CG14395 in the whole leg, trichomes are significantly shorter both around the naked valley area and on the remaining femur (p < 0.001 and p < 0.01). Note that some controls show significantly different trichome lengths. Data are not normally distributed (Shapiro-Wilk normality test). Kruskal-Wallis and pairwise comparisons using Wilcoxon rank sum test were used to test for significance.
(PDF)
Genes are sorted by gene name. Two rows for a gene indicate alternative transcription start sites. Expression level in fragments per kilobase per million (FPKM) with low and high confidence values, base 2 log of the fold change, and p and q values are given after mapping with TopHat 2.1.1, transcriptome assembly with Cufflinks 2.2.1 and Cuffmerge, and comparison with Cuffdiff 2.2.1 [74,75]. q values are false discovery rate (FDR)-corrected p values.
(XLSX)
Genes are sorted by gene name. Two rows for a gene indicate alternative transcription start sites. Expression level in fragments per kilobase per million (FPKM) with low and high confidence values, base 2 log of the fold change, and p and q values are given after mapping with TopHat 2.1.1, transcriptome assembly with Cufflinks 2.2.1 and Cuffmerge, and comparison with Cuffdiff 2.2.1 [74,75]. q values are false discovery rate (FDR)-corrected p values.
(XLSX)
Genes are sorted by gene name. Two rows for a gene indicate alternative transcription start sites. Expression level in fragments per kilobase per million (FPKM) with low and high confidence values, base 2 log of the fold change, and p and q values are given after mapping with TopHat 2.1.1, transcriptome assembly with Cufflinks 2.2.1 and Cuffmerge, and comparison with Cuffdiff 2.2.1 [74,75]. q values are FDR-corrected p values.
(XLSX)
(DOCX)
(CSV)
(CSV)
(CSV)
(CSV)
(CSV)
(CSV)
(CSV)
(TXT)
(TXT)
Data Availability Statement
All RNA-Seq and ATAC-Seq data files are available from the Gene Expression Omnibus database (accession number GSE113240).