

Abstract
Scope
Stress is a known contributor to various forms of disease in humans and animals, although mechanisms are still unknown. In animals, psychosocial stress-induced depression/anxiety phenotypes are coincidental with increased inflammation in both brain and blood. The authors recently showed that a novel treatment with a select bioactive polyphenol preparation promotes resilience to stress-mediated depression/anxiety phenotypes mice. Moreover, selective bioactive phenolic compounds within the polyphenol preparation are identified that are effective in mitigating the behavioral effects of bone marrow transplantation from stressed mice.
Methods and results
Here, an animal model of adult stress and bone marrow transplantation is used to identify an epigenetic signature of repeated social defeat stress (RSDS) that is passed through bone marrow hematopoietic progenitor cells to naïve mice, revealing the maintenance of epigenetic memory following stress both centrally and peripherally. Further, polyphenols are administered to naïve and stress-susceptible mice, demonstrating that polyphenol treatment in mice from both susceptible and naïve donors alters global DNA methylation in the central nervous system and periphery and likewise has an effect on human blood cells after immune challenge.
Conclusions
Findings highlight the enduring molecular memory of stress and the possible mechanism by which select bioactive polyphenols may promote resiliency to stress. Polyphenols may be an efficacious alternative to traditional pharmacological treatments in psychiatry.
Keywords: DNA methylation, epigenetics, polyphenols, social defeat, stress
1. Introduction
Stress during an individual’s lifespan is a known contributor to both physical and mental illness. Although the maladaptive effects of life stress on mental health are generally thought to be caused by malfunctioning of neural processes, there is evidence that the immune system plays a crucial role, which emphasizes the importance of untangling the complex relationship between the brain and immunity. Immune activation resulting from stress renders an individual more susceptible to disease, including psychiatric disorders.[1] This occurs through release of cytokines that mediate inflammatory and anti-inflammatory processes in times of immune activation. Not only are cytokines abundant in the periphery, but both cytokines and their receptors are present in the central nervous system (CNS) and exert effects on monoaminergic neurotransmission and neuronal signaling.[1,2] The manner in which peripheral cytokines reach the CNS is still being investigated, but rodent studies have recapitulated the effects of immune activation on brain function and behavior.[3,4]
There have been great interests in dissecting the potential interactions between environmental stress and depression/anxiety phenotypes. A recent line of preclinical studies has focused on understanding how a select environmental stress, namely repeated social defeat stress (RSDS), may induce the development of depression/anxiety phenotypes in mouse or rat models. This experimental paradigm involves subjecting mice (or rats) to repeated bouts of social defeat by larger and more aggressive mice (or rats), which results in the development of a clear depression-like syndrome characterized by enduring deficits in social interactions.[5,6] Notably, RSDS induces depression phenotypes in some but not all of the animals, and animals are characterized as stress-resilient or stress-susceptible depending on the presence or absence of a depressive-like behavioral phenotype following RSDS.[6–9] In addition to differences in behavioral phenotypes, susceptible and resilient mice are also distinct by different immune phenotypes, with susceptible mice displaying elevated inflammatory cytokines, notably IL-6, which mirrored levels in human patients with treatment-resistant depression.[8] Interestingly, transmission of stress-susceptible phenotypes could be modulated through hematopoietic progenitor cells because transplantation of bone marrows (BM) from susceptible mice (mice that developed depression/anxiety phenotypes following RSDS) into stress-naïve mice promotes susceptibility of naïve mice to develop RSDS-mediated depression/anxiety phenotypes which demonstrates.[8] However, there is little information on how social stress may mechanistically modulate immune activation and depression phenotypes over short and long terms.
One way by which stress and immune activation may alter downstream neural processes and behavior is through epigenetic mechanisms. Epigenetics refers to changes to the DNA structure in chromatins that do not alter the sequence of nucleotide bases. These chemical modifications can alter gene expression patterns and therefore resulting protein expression and behavior. While a multitude of epigenetic modifications exist, DNA methylation is one of the most thoroughly studied and is characterized by the addition of a methyl group to a cytosine nucleotide, which changes chromatin structure but not its base pairing property. DNA methylation represses gene expression via physical blocking of transcription factors or by recruiting transcriptional repressor proteins, although in some cases DNA methylation may activate transcription.[10] DNA methylation can be catalyzed or reversed by environmental factors and persist long after the initial insult, producing stable changes in gene expression during both development and adulthood.[11]
Importantly, stressful experiences can alter DNA methylation patterns in humans and animals, both globally and at gene loci important for brain plasticity and emotion.[12,13] These methylation patterns can affect expression of genes involved in the crosstalk between inflammatory pathways and neural activity, ultimately conferring vulnerability to psychopathology, including depression and anxiety.[2,14] For example, in humans, DNA methylation of FKBP5, which codes for a gene important in hypothalamic–pituitary–adrenal axis activity, was decreased in subjects that experienced childhood trauma, and these subjects likewise had decreased sensitivity of peripheral immune cells to immune challenge in vitro.[15] In another study, Uddin and colleagues found evidence of downstream alterations in immune function following trauma in patients with posttraumatic stress disorder, which they suggest is a result of altered methylation patterns in immune-related genes.[16]
With the known interplay of stress, epigenetics, and immunity, there are now more avenues for possible treatments for stress-related disorders that lie outside of traditional psychiatric medications (most of which do not target inflammation). Plant-derived polyphenols are naturally occurring micronutrients that have demonstrated efficacy in treatment of depressive-like symptoms in animal models, but the mechanism by which this occurs is still unknown.[17] Our group recently identified a polyphenol-rich dietary polyphenol preparation we referred to as BDPP, which is effective in alleviating the effects of RSDS-mediated depression/anxiety phenotypes in mice.[18] Starting from BDPP, we isolated and characterized two select bioactive phenolic compounds, dihydrocaffeic acid (DHCA) and malvidin-3′-O-glucoside (Mal-gluc), that are effective in modulating hematopoietic progenitor cell–mediated transmission of stress-susceptible phenotypes. In particular, we identified a sub-threshold micro-defeat paradigm that is not sufficient to induce depression phenotypes in mice, but that is effective in inducing depression phenotypes in mice with BM transplantation from mice that developed depression/anxiety phenotypes following RSDS.[18] We demonstrated that in mice with BM transplantation, treatment with combined DHCA/mal-gluc was effective promoting resilience against the developing of depression phenotypes in response to sub-threshold micro-defeat.[18]
Dietary polyphenols and many other dietary phytochemicals are becoming increasingly attractive as therapeutic agents because of their safety and efficacy in alleviating psychiatric symptomology.[19,20] The aim of the current study was to investigate long-term effects of a stress-susceptible immune profile on global DNA methylation in the CNS and periphery with or without treatment with DHCA/Mal-gluc. Information gathered from these studies may provide one mechanism by which phenolic compounds can alleviate stress-induced depression/anxiety phenotypes with the hope of becoming an avenue for clinical developments.
2. Experimental Section
2.1. Materials
Mal-gluc was purchased from Extrasynthesis (Genay Cedex, France). DHCA and LPS (from Escherichia coli) were purchased from Sigma–Aldrich. All tested compounds were analyzed by LC–MS and archived in compliance with Product Integrity guides of the U.S. National Institutes of Health National’s Center for Complementary and Integrative Health.
2.2. Animals
CD45.2+ C57BL/6 mice were used for RSDS and as BM recipients. CD45.1+ C57BL/6 mice were used as BM donors. For number of animals per group, see Figure 2. All C57BL/6 mice were purchased from the Jackson Laboratory. Retired breeder CD-1 mice, purchased from Charles River Laboratory, serve as aggressor mice. All animals had access of regular chow ad lib and were maintained on a 12:12-h light/dark cycle with lights on at 07:00 h in a temperature-controlled (20 ± 2 °C) vivarium. All animal procedures were approved by the Institutional Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai.
Figure 2.
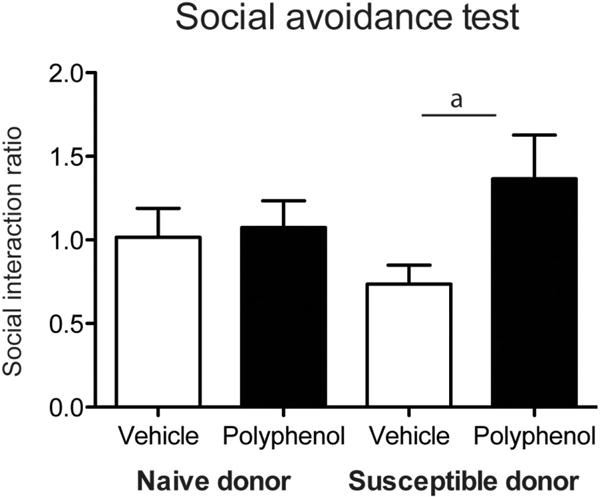
Social avoidance behavioral testing in mice subjected to subthreshold micro-defeat in the presence or absence of DHCA/Mal-gluc treatment. Social avoidance behavioral test in mice treated with DHCA/Mal-gluc or vehicle in mice transplanted with naïve or susceptible donor. (a denotes p < 0.05, n = 9–10 per group).
2.3. Generation of BM Chimeras and Treatment
Generation of BM chimera was performed as previously described.[8,21] Briefly, 6 weeks old recipient CD45.2+ C57BL/6 mice were lethally irradiated with 1200 rad delivered in two doses of 600 rad each (10–11 h apart). Following the second irradiation, BM hematopoietic cells from the donor mouse were introduced through a retro-orbital injection. BM cells were obtained from the naïve (SI ratio = 1.81) or susceptible (SI ratio =0.46) CD45.1+ C57BL/6 mice. All mice were treated with ≈3 mg per mouse per day (sulfatrim) for 3 weeks followed by 2-weeks treatment with vehicle or a mixture of DHCA (5 mg kg−1 d−1) and Mal-gluc (500 ng kg−1 d−1) delivered through their drinking water, before subjected to a subthreshhold defeat. This treatment was repeated 4 months later (Figure 1). Stability studies demonstrated that both compounds are stable in aqueous solution for at least 5 days. Therefore, for all the treatment, drinking solutions were freshly made and changed once every 2–3 days.
Figure 1.
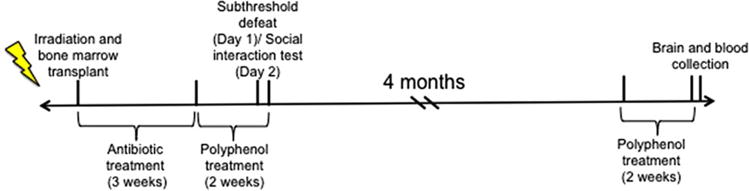
Overall design and timeline of study. Six-week-old male mice were irradiated and received BM transplants from donor mice that experienced RSDS and exhibited a susceptible phenotype or from naïve mice. After antibiotic treatment for 3 weeks and DHCA/Mal-gluc/vehicle treatment for 2 weeks, mice were subjected to subthreshold defeat followed by social interaction testing. After testing, mice were left undisturbed in their home cage for 4 months, and then treated with polyphenols for 2 weeks. The following day, mice were sacrificed and brain/blood was collected for methylation assays.
2.4. Behavioral Testing
2.4.1. Subthreshold Defeat Stress
Subthreshold defeat stress was performed as previously described.[7,22] Briefly, CD-1 mice were screened for aggressive characteristics prior to the start of social defeat experiments based on previously described criteria.[7] During subthreshold defeat, mice were subjected to a novel CD-1 aggressor for three consecutive 5-min defeat bouts, with a 15 min intertrial interval between the defeats. Mice were subjected to social avoidance behavior testing 24 h after the defeat. This protocol does not result in social avoidance behavior in normal mice.
2.4.2. Social Avoidance Test (Social Interaction Test)
Social interaction (SI) testing was performed as previously described.[7] All SI testings were performed under red-light conditions. Mice were placed in a novel interaction open-field arena custom-crafted from opaque Plexiglas (42 × 42 × 42 cm; Nationwide Plastics) with a small animal cage placed at one end. Their movements are then automatically monitored and recorded (Ethovision 3.0; Noldus Information Technology) for 2.5 min in the absence (target absent phase) of a novel CD-1 mouse. This phase is used to determine baseline exploratory behavior. We then immediately measured 2.5 min of exploratory behavior in the presence of a caged CD-1 mouse (target present phase), again recording total distance travelled and duration of time spent in the interaction and corner zones. SI behavior was then calculated as total time spent in each zone, or as a ratio of the time spent in the interaction with target present divided by the time spent in the interaction zone with the target absent. All mice with a ratio above 1.0 were classified as resilient, and all mice below 1.0 were classified as susceptible.
2.5. Collection of Brain Specimens
Mice were rapidly decapitated under baseline conditions and brains were immediately removed, flash frozen in liquid nitrogen, and placed in a −80 °C until further processing. Whole hippocampus was dissected from 250 micron frozen sections and immediately homogenized in buffer RLT for DNA extraction (Qiagen, Valencia, CA).
2.6. Mouse Peripheral Blood Mononuclear Cell Isolation
Mice were sacrificed by decapitation and 300–500 μL trunk blood was immediately collected. Peripheral blood mononuclear cells (PBMCs) were isolated from freshly collected blood using Ficoll-Paque (GE Healthcare, Sweden). In particular, blood samples were diluted with PBS and carefully laid over Ficoll-Paque and centrifuged at 400 × g for 30 min at room temperature in a swinging bucket rotor without brake. The mononuclear cell layer at the interphase was carefully isolated and washed once with PBS at 300 × g for 10 min. The cell pellet was washed once more with PBS at 200 × g for 10 min to remove platelets.
2.7. Human Blood Collection and Isolation of PBMCs
Blood from six healthy male donors (age between 25 and 30) were collected in the heparin-coated tubes and PBMCs were isolated and purified as described above.
2.8. PBMC Stimulation by LPS
Human PBMCs were seeded at 2 × 106 cells per well in six-well plate in culture medium RPMI-1640 (Sigma) supplemented with 20% horse serum, 10% FBS, 2.05 mm l-glutamine, 25 mm Hepes, and 100 U mL−1 penicillin/streptomycin. PBMCs were treated with vehicle or DHCA (final concentration 50 μm) for 16 h before stimulating with 100 ng mL−1 LPS for 24 h. The cell pellets were washed once with ice-cold PBS and snap frozen for DNA isolation and methylation analysis.
2.9. DNA Extraction
For mouse hippocampus, DNA and RNA were simultaneously extracted from homogenized tissue using the Qiagen AllPrep DNA/RNA kit (Qiagen) according to the manufacturer’s instructions. For mouse and human PBMCs, DNA and RNA were simultaneously extracted using the Zymo Duet DNA/RNA kit (Zymo, Irvine, CA) according to the manufacturer’s instructions. DNA concentration was measured with Qubit fluorometer (Life technologies) and stored at −20 °C until further processing.
2.10. Global DNA Methylation Assays
Global DNA methylation was assessed using the 5-methylcytosine (5-mC) DNA ELISA kit according to the manufacturer’s instructions (Zymo, Irvine, CA). Briefly, 100ng of DNA and 5-mC coating buffer was added to PCR tubes and denatured at 98 °C for 5 min. Samples were then transferred to 5-mC plate wells and incubated at 37 °C for 1 h. Wells were washed three times with buffer and incubated again for 30 min at 37 °C. Antibody mix containing 5-mC antibody and a secondary antibody were then added to the wells and incubated for 1 h at 37 °C. After three more washes with buffer, color developer solution was added to each well and allowed to sit at room temperature on the benchtop for ≈ 20 min. Absorbance was read at 405 nm using an ELISA plate reader. All samples were run in duplicate and optical density (OD) values were averaged across both reads. A standard curve of methylation ranging from 0 to 100% CpG methylation was run on each plate (positive and negative controls provided by manufacturer). Methylation percentages for samples was calculated by subtracting the y-intercept of the standard curve from the average sample OD and dividing by the slope of the standard curve. Values were then converted into percent cytosine methylation for human or mouse respectively.
2.11. Statistical Analyses
Mouse global methylation levels across treatment groups were analyzed first with two-way analysis of variances and followed with unpaired t-tests. For the social avoidance test data, we used two-tailed Student’s t-test because we only conducted comparisons between treatment groups and already had pilot data suggesting that there would be a difference between groups. For human PBMC global methylation, methylation levels after LPS stimulaiton and BDPP treatment for each individual were calculated as a percentage of unstimulated control. Means were then compared to a theoretical value of 100% using one-sample t-tests. For all tests, outliers were excluded if they fell more than two SDs above or below the mean. Significance was denoted by p < 0.05.
3. Results
3.1. DHCA/Mal-Gluc Treatment Alleviate Social Defeat-Mediated Depression/Anxiety Behavioral Phenotypes
Male mice irradiated and transplanted with BM from naïve donors or susceptible donors (mice that developed depression/anxiety phenotypes following RSDS) were treated with either vehicle or DHCA/Mal-gluc for 2 weeks. Thereafter, mice subjected to a subthreshold defeat, followed by assessment for depression-like behavioral phenotypes using the social avoidance test. Results from the test was summarized into SI ratios, whereby an SI ratio ≥ 1 and < 1 reflect, respectively, the presence or the absence of depression-like behavioral phenotypes. As expected among mice that were transplanted with BM from naïve donors, subthreshold defeat did not induce depression-like phenotypes, regardless of treatment with vehicle or DHCA/Mal-gluc (Figure 2). In contrast, among mice that were transplanted with BM from susceptible donor, subthreshold defeat resulted in depression-like phenotypes (Figure 2, vehicle treated mice, mean SI ratio = 0.74). We also found that DHCA/Mal-gluc treatment significantly alleviated the sensitivity of mice with BM transplantation from susceptible donor to develop depression-like phenotypes in response to subthreshold defeat (Figure 2, DHCA/Mal-gluc-treated mice, SI ratio = 1.36).
3.2. Mouse Brain Global Methylation
We measured global 5mC levels in the hippocampus of mice with the immune profiles of naïve or susceptible donors following two bouts of treatment with DHCA/Mal-gluc over a 4 month period. A two-way analysis of variance revealed an interaction between donor condition and DHCA/Mal-gluc treatment (Figure 3; F(1,25) = 25.68, p < 0.001). DHCA/Mal-gluc -treated mice with the BM chimeras of naïve donors had significantly lower global cytosine methylation compared to vehicle-treated controls (t(16) = 3.381, p < 0.01). Conversely, DHCA/Mal-gluc -treated mice that received the immune profile of susceptible donors had significantly higher methylation compared to vehicle-treated controls (t(9) = 3.49, p < 0.01) as well as DHCA/Mal-gluc -treated mice from naïve donors (t(15) = 3.025, p < 0.01). Further, vehicle treated mice with susceptible donor chimeras had a significant decrease in methylation compared to mice with naïve donor chimeras (t(10) = 3.906, p < 0.01).
Figure 3.
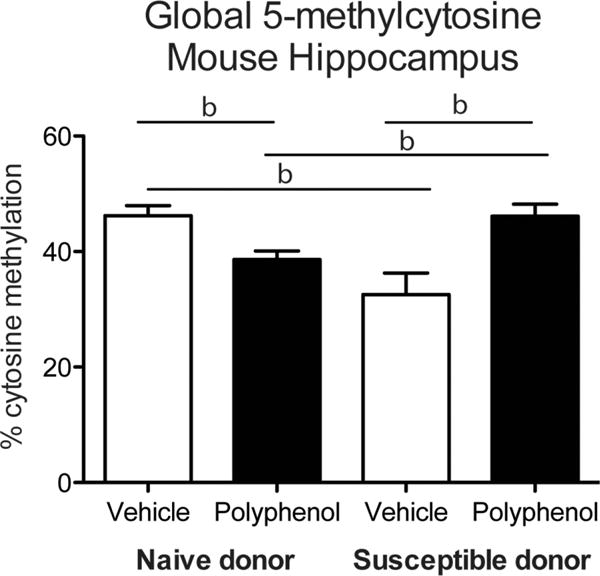
Global 5-methylcytosine levels in the mouse hippocampus. Global DNA methylation was measured in mouse hippocampal tissue following transplantation of BM from naïve or susceptible donors and polyphenol treatment (b denotes p < 0.01; error bars represent SEM).
3.3. Mouse Blood Global Methylation
We isolated PBMCs from mouse blood collected during brain removal and measured global 5-mC levels. There was a significant effect of DHCA/Mal-gluc treatment on methylation (Figure 4; F(1,28) = 10.12, p < 0.01) and a marginal effect of donor condition (F(1,28) = 4.157, p = 0.051). Post hoc tests revealed that DHCA/Mal-gluc-treated mice from naïve donors had decreased methylation compared to vehicle-treated controls from naïve donors (t(16) = 2.889, p < 0.05). Additionally, DHCA/Mal-gluc -treated mice from susceptible donors had decreased methylation compared to vehicle-treated controls, although this effect was only marginally significant (t(12) = 1.835, p = 0.091).
Figure 4.
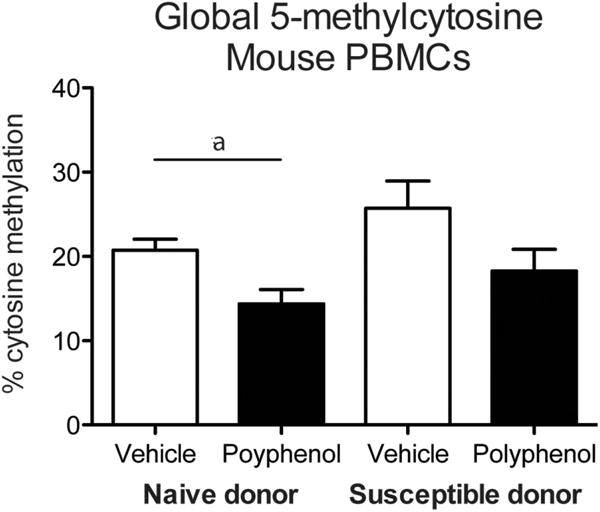
Global 5-methylcytosine levels in mouse PBMCs. Global DNA methylation was measured in mouse PBMCs following transplantation of BM from naïve or susceptible donors and polyphenol treatment (a denotes p < 0.05; error bars represent SEM).
3.4. Human Blood Global Methylation
Previously, we reported that DHCA-mediated anti-inflammatory responses in the periphery and Mal-gluc-mediated normalization of synaptic plasticity in the CNS contributed to the preclinical efficacy of DHCA/Mal-gluc on attenuation of depression-like phenotype.[18] To further test the potential epigenetic mechanisms of this DHCA on peripheral mechanisms, we iso lated PBMCs from healthy human subjects and pretreated with vehicle or DHCA for 16 h before stimulating with LPS for 24 h before conducting global DNA methylation analysis. Each individual’s methylation percentage was calculated as a percent of his control (before stimulation). Although no statistically significant differences were found, there was a trending increase in global methylation after LPS stimulation, and this effect was not present after treatment with DHCA (Figure 5).
Figure 5.
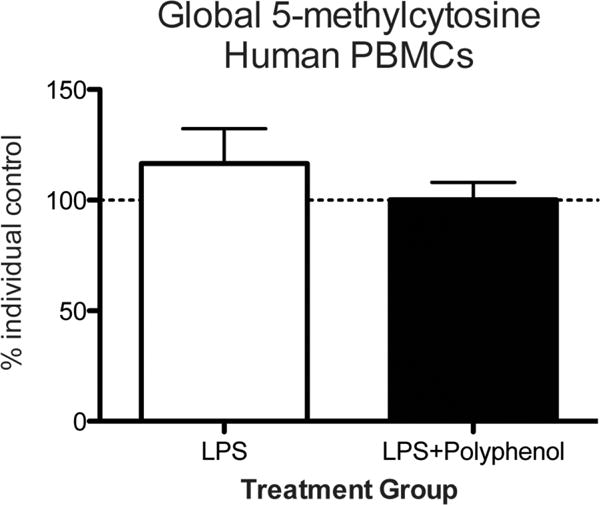
Global 5-methylcytosine levels in LPS-stimulated PBMCs. Global DNA methylation was measured in human PBMCs at baseline, after LPS stimulation, or when LPS stimulation was preceded by polyphenol treatment (error bars represent SEM).
4. Discussion
Here, we investigated the global DNA methylation patterns in brain and blood of DHCA/Mal-gluc-treated mice that were transplanted with the immune profile of a stress-susceptible or non-stressed naïve donor. We found DHCA/Mal-gluc treatment-and donor-specific changes in global methylation patterns in both the mouse hippocampus and PBMCs that were maintained months distal to the BM transplant. Further, we investigated the effect of immune challenge on global methylation of healthy human PBMCs and found a trending increase in methylation after immune challenge that was attenuated after treatment with DHCA, the component of DHCA/Mal-gluc that is known to attenuate peripheral inflammation.[18] These findings highlight DNA methylation as a possible mechanism by which stress-induced immune changes, in combination with polyphenol treatment, can be maintained in molecular memory long after the initial stressor.
Previous work has shown the ability of hematopoietic progenitor cell transplants to transfer stress-susceptible phenotypes across individual mice.[8,18] Although we have identified IL-6 and its role in synaptic plasticity as a key player in the mechanistic action of polyphenols in this model, mechanisms upstream of protein expression had not yet been identified. We have now shown that DNA methylation may be a way in which the peripheral immune system interacts with the CNS to produce stress susceptibility.
The opposing effects of DHCA/Mal-gluc treatment on global DNA methylation levels depending on donor condition was an unexpected but interesting finding. In the hippocampus of mice with a stress-susceptible bone marrow chimera, DHCA/Mal-gluc increased methylation to levels of the vehicle-treated mice from naïve donors, suggesting that these compounds can rescue changes induced by the immune profile receieved from a stress-susceptible donor. Conversely, DHCA/Mal-gluc decreased methylation in mice from naïve donors (to levels of vehicle-treated mice from susceptible donors). This highlights the variability among individuals when assessing the therapeutic efficacy of drug treatments.[23] These findings suggest that future studies should consider individual immune profiles when assessing the most effective pharmacological treatment strategy.
In mouse PBMCs, DHCA/Mal-gluc treatment decreased global methylation in mice from both naïve donors and susceptible donors. The concomittant decrease of methylation in both the hippocampus and PBMCs of mice from naïve donors demonstrates the conserved effect of DHCA/Mal-gluc on global DNA demethylation across healthy tissues. There are currently conflicting ideas about the correlation between blood and brain DNA methylation,[24,25] but identifying a similar pattern of methylation in both tissues after DHCA/Mal-gluc treatment suggests that this treatment may be useful in the clinic to reflect therapeutic effects on the brain.
Another piece of evidence suggesting that this treatment has clinical relevance is our human PBMC findings. Although findings were nonsignificant, LPS treatment increased global methylation in human PBMCs while subsequent DHCA treatment returned methylation to baseline levels. Inflammation is associated with increased global DNA methylation levels,[26] which is in line with our findings of an increase in methylation after LPS treatment. Furthermore, the demethylating effects of DHCA in human PBMCs are similar to our findings in the mouse. The choice of DHCA or Mal-gluc was based on the observation that DHCA can reduce peripheral inflammation while Mal-gluc can modulate synaptic plasticity in the brain. Therefore, for all the ex vivo studies with PBMCs, the main source of peripheral inflammation, the treatment was always DHCA.
Stress-susceptible mice display increased inflammation that can be compared to experimentally induced inflammation in human PBMCs. Importantly, DHCA/Mal-gluc has the same demethylating effect on both humans and mice, as well as conservation between tissue types in mice. It is important to note that our human methylation data has a rather small sample size due to the preliminary nature of the study, providing only trending results. Due to possible batch effects of the LPS-stimulation, we were unable to add subjects to the current study, but are planning on replicating this part of the study with a higher number in the future.
A key aspect of our model is the delay between BM transplant and time of brain/blood collection. Although previous studies investigated molecular markers and behavior weeks following transfer of hematopoietic progenitor cells,[8,18] our study design allowed over 4 months to pass following transplant, during which animals were left undisturbed. Intriguingly, the molecular memory of brain and immune cells remained after the lengthy delay. Changes in DNA methylation elicited by stress are known to be long lasting and can even span generations in both the CNS and periphery,[11] but to our knowledge, there are no studies of the long-term effects of BM transplants on DNA methylation in the brain. Immune cells received from the donor during hematopoietic progenitor cell transplant develop into the host immune system but also exhibit neuronal phenotypes,[27,28] which may be the reason that hippocampal DNA methylation levels exhibit long-term changes depending on stress phenotype of the donor.
Global changes in methylation have the capacity to alleviate depressive-like behavior in rodents,[29] which suggests that DHCA/Mal-gluc treatment, which elicits large changes in global methylation in the brain and blood, may do so as well. Future studies will assess the behavioral effects of DHCA/Mal-gluc treatment and investigate the correlation between methylation and behavior. The studies described here are of a preliminary nature and give rise to many further questions that must be addressed before DHCA/Mal-gluc can be used in a clinical setting. Limitations to the current study will be addressed in the future, including assessing sex differences, gene expression and immune profiles, and the applicability of polyphenols to be used in other realms of medicine and psychiatry. In conclusion, we have shown here that treatment with a polyphenolic compound alters global DNA methylation across human and mouse tissues following inflammation, although this is just a beginning step in discovering the therapeutic potential of polyphenols.
Acknowledgments
F.H. and G.M.P. contributed equally to this work. J.B. and F.H. conceptually designed molecular experiments. J.W. and L.H. conceptually designed and performed all behavioral experiments and BM transfers. J.B. and N.M. performed all molecular assays, and J.B. analyzed all resulting data. J.B. drafted the manuscript, and F.H., G.M.P., and J.W. critically reviewed the manuscript before submission. G.M.P is a VA Senior Career Scientist at the James J. Peters Veterans Affairs Medical Center. We would like to thank Tiffany Doherty of the University of Delaware for her assistance with global methylation assays. This work was supported by the James J. Peters VA Medical Center under Grants 1I01RX001705 and 1I01CX001395-01 to Dr. Fatemeh Haghighi. Dr. Pasinetti was supported by the P50 AT008661-01 to G.M.P. from the National Center for Complementary and Integrative Health (NCCIH) and the Office of Dietary Supplements (ODS) and support from Altschul Foundation. Following initial online publication, the Acknowledgements section was updated.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Dr. Jennifer Blaze, Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029
Dr. Jun Wang, Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY 10029; Research and Development Service, James J. Peters Veterans Affairs Medical Center, Bronx, NY 10468
Dr. Lap Ho, Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY 10029
Natalia Mendelev, Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029.
Dr. Fatemeh Haghighi, Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029; Research and Development Service, James J. Peters Veterans Affairs Medical Center, Bronx, NY 10468.
Dr. Giulio Maria Pasinetti, Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY 10029; Research and Development Service, James J. Peters Veterans Affairs Medical Center, Bronx, NY 10468; Veterans Affairs Medical Center, Geriatric Research Education and Clinical Center, Bronx, NY 10468; James J. Peters Veterans Affairs Medical Center, Geriatric Research Education and Clinical Center, Bronx, NY 10468
References
- 1.Connor TJ, Leonard BE. Life Sci. 1998;62:583. doi: 10.1016/s0024-3205(97)00990-9. [DOI] [PubMed] [Google Scholar]
- 2.Miller AH, Haroon E, Raison CL, Felger JC. Depress Anxiety. 2013;30:297. doi: 10.1002/da.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raison CL, Capuron L, Miller AH. Trends Immunol. 2006;27:24. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prinz M, Priller J. Nat Rev Neurosci. 2014;15:300. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 5.Berton O, McClung CA, DiLeone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M. Science. 2006;311:864. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 6.Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, LaPlant Q, Graham A, Lutter M, Lagace DC. Cell. 2007;131:391. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Golden SA, Covington HE, III, Berton O, Russo SJ. Nat Protoc. 2011;6:1183. doi: 10.1038/nprot.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, Rebusi N, Heshmati M, Aleyasin H, Warren BL, Labonté B, Horn S, Lapidus KA, Stelzhammer V, Wong EHF, Bahn S, Krishnan V, Bolaños-Guzman CA, Murrough JW, Merad M, Russo SJ. Proc Natl Acad Sci USA. 2014;111:16136. doi: 10.1073/pnas.1415191111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo SJ, Nestler EJ. Nat Rev Neurosci. 2013;14:609. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. Science. 2008;320:1224. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaze J, Roth TL. Semin Cell Dev Biol. 2015;43:76. doi: 10.1016/j.semcdb.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bagot RC, Labonté B, Pe˜na CJ, Nestler EJ. Dialogues Clin Neurosci. 2014;16:281. doi: 10.31887/DCNS.2014.16.3/rbagot. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vialou V, Feng J, Robison AJ, Nestler EJ. Annu Rev Pharmacol Toxicol. 2013;53:59–87. doi: 10.1146/annurev-pharmtox-010611-134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller AH, Raison CL. Nat Rev Immunol. 2016;16:22. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS, Bradley B, Nemeroff CB, Holsboer F, Heim CM, Ressler KJ, Rein T, Binder EB. Nat Neurosci. 2013;16:33. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de los Santos R, Goldmann E, Galea S. Proc Natl Acad Sci USA. 2010;107:9470. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pathak L, Agrawal Y, Dhir A. Expert Opin Investig Drugs. 2013;22:863. doi: 10.1517/13543784.2013.794783. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Hodes GE, Zhang H, Zhang S, Zhao W, Golden SA, Bi W, Menard C, Kana V, Leboeuf M, Xie M, Bregman D, Pfau ML, Flanigan ME, Esteban-Fernández A, Yemul S, Sharma A, Ho L, Dixon R, Merad M, Han M-H, Russo SJ. Pasinetti GM: Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nature Communications. 2018 doi: 10.1038/s41467-017-02794-5. https://doi.org/10.1038/s41467-017-02794-5. [DOI] [PMC free article] [PubMed]
- 19.Naveen S, Siddalingaswamy M, Singsit D, Khanum F. Psychiatry Clin Neurosci. 2013;67:501. doi: 10.1111/pcn.12100. [DOI] [PubMed] [Google Scholar]
- 20.Dias GP, Cavegn N, Nix A, do Nascimento Bevilaqua MC, Stangl D, Zainuddin MSA, Nardi AE, Gardino PF, Thuret S. Oxidative medicine and cellular longevity. 2012;2012:541971. doi: 10.1155/2012/541971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogunovic M, Ginhoux F, Wagers A, Loubeau M, Isola LM, Lubrano L, Najfeld V, Phelps RG, Grosskreutz C, Scigliano E, Frenette PS, Merad M. J Exp Med. 2006;203:2627. doi: 10.1084/jem.20060667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K, Cahill ME, Dias C, Ribeiro E, Ables JL, Kennedy PJ, Robison AJ, Gonzalez-Maeso J, Neve RL, Turecki G, Ghose S, Tamminga CA, Russo SJ. Nat Med. 2013;19:337. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uher R. Harv Rev Psychiatry. 2011;19:109. doi: 10.3109/10673229.2011.586551. [DOI] [PubMed] [Google Scholar]
- 24.Walton E, Hass J, Liu J, Roffman JL, Bernardoni F, Roessner V, Kirsch M, Schackert G, Calhoun V, Ehrlich S. Schizophr Bull. 2015 doi: 10.1093/schbul/sbv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hannon E, Lunnon K, Schalkwyk L, Mill J. Epigenetics. 2015;10:1024. doi: 10.1080/15592294.2015.1100786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stenvinkel P, Karimi M, Johansson S, Axelsson J, Suliman M, Lindholm B, Heimbürger O, Barany P, Alvestrand A, Nordfors L. J Intern Med. 2007;261:488. doi: 10.1111/j.1365-2796.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- 27.Brazelton TR, Rossi FMV, Keshet GI, Blau HM. Science. 2000;290:1775. doi: 10.1126/science.290.5497.1775. [DOI] [PubMed] [Google Scholar]
- 28.Mezey É, Chandross KJ, Harta G, Maki RA, McKercher SR. Science. 2000;290:1779. doi: 10.1126/science.290.5497.1779. [DOI] [PubMed] [Google Scholar]
- 29.Sales AJ, Biojone C, Terceti MS, Guimaraes FS, Gomes MV, Joca SR. Br J Pharmacol. 2011;164:1711. doi: 10.1111/j.1476-5381.2011.01489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]