

Abstract
Primary congenital glaucoma (PCG) is a severe autosomal recessive ocular disorder associated with considerable clinical and genetic heterogeneity. Recently, rare heterozygous alleles in the angiopoietin receptor-encoding gene TEK were implicated in PCG. We undertook this study to ascertain the second mutant allele in a large cohort (n = 337) of autosomal recessive PCG cases that carried heterozygous TEK mutations. Our investigations revealed 12 rare heterozygous missense mutations in TEK by targeted sequencing. Interestingly, four of these TEK mutations (p.E103D, p.I148T, p.Q214P, and p.G743A) co-occurred with three heterozygous mutations in another major PCG gene CYP1B1 (p.A115P, p.E229K, and p.R368H) in five families. The parents of these probands harbored either of the heterozygous TEK or CYP1B1 alleles and were asymptomatic, indicating a potential digenic mode of inheritance. Furthermore, we ascertained the interactions of TEK and CYP1B1 by co-transfection and pull-down assays in HEK293 cells. Ligand responsiveness of the wild-type and mutant TEK proteins was assessed in HUVECs using immunofluorescence analysis. We observed that recombinant TEK and CYP1B1 proteins interact with each other, while the disease-associated allelic combinations of TEK (p.E103D)∷CYP1B1 (p.A115P), TEK (p.Q214P)∷CYP1B1 (p.E229K), and TEK (p.I148T)∷CYP1B1 (p.R368H) exhibit perturbed interaction. The mutations also diminished the ability of TEK to respond to ligand stimulation, indicating perturbed TEK signaling. Overall, our data suggest that interaction of TEK and CYP1B1 contributes to PCG pathogenesis and argue that TEK-CYP1B1 may perform overlapping as well as distinct functions in manifesting the disease etiology.
Introduction
Autosomal recessive primary congenital glaucoma [PCG (MIM 231300)] is the most common form of glaucoma in infants (<3 years of age). The disease is thought to result from developmental defects in the Schlemm’s canal and trabecular meshwork resulting in raised intraocular pressure (IOP) and optic nerve head damage (deLuise and Anderson 1983; Sarfarazi and Stoilov 2000; Wang and Wiggs 2014). PCG is accompanied with clinical symptoms of buphthalmos (enlarged eye globes), a characteristic Haab’s striae (horizontal breaks in the Descemet membrane), corneal edema, and photophobia, leading to irreversible blindness (Anderson 1981; Chakrabarti et al. 2006). It exhibits a prevalence of 1 in 1250–3300 live births among consanguineous populations (Dandona et al. 1998; Plasilova et al. 1999), and 1 in 10,000–30,000 in outbred populations worldwide (Dimasi et al. 2007; Khan 2011).
PCG is genetically heterogeneous (Libby et al. 2005) with homozygous mutations in CYP1B1 (Li et al. 2011; Stoilov et al. 1997; Vasiliou and Gonzalez 2008) and LTBP2 (Ali et al. 2009) accounting for majority of the cases. In addition, allelic interactions of two unlinked genes, MYOC (Kaur et al. 2005) and FOXC1, with CYP1B1 (Chakrabarti et al. 2009) have been associated in PCG. Recently, rare heterozygous mutations in the angiopoietin receptor TEK (MIM 600221; tunica interna endothelial cell kinase; or TIE2) resulting in haploinsufficiency were implicated in PCG (Souma et al. 2016). No mutations in the known glaucoma genes were identified. These observations were intriguing, since PCG is inherited in an autosomal recessive manner.
Given that the clinical and genetic predisposition to PCG can vary according to ethnicity (Liu and Allingham 2011; Vasiliou and Gonzalez 2008; Wang and Wiggs 2014), we reasoned that heterozygous TEK mutations might co-occur with mutations in another glaucoma-associated gene in autosomal recessive PCG. In this study, we not only identified novel heterozygous TEK mutations but also found evidence for genetic and physical interaction of TEK with CYP1B1 in PCG. We further showed that the disease-associated allelic combinations of TEK and CYP1B1 altered their interaction and affected the ability of TEK to respond to extrinsic signals. Overall, our study implicated TEK and CYP1B1 in common pathways that could affect the disease etiology.
Materials and methods
Study approval
The study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Review Board of the L.V. Prasad Eye Institute (LEC 08-15-097). Written informed consents were obtained from all the adult subjects and guardians of the minors.
Enrolment of patients and controls
Our study cohort consisted of 2057 subjects including index cases of unrelated PCG patients (n = 337) devoid of homozygous mutations in the known PCG-associated genes (CYP1B1, LTBP2, MYOC, and FOXC1), along with their affected sibs (n = 9), unaffected parents and relatives (n = 687), and ethnically matched normal volunteers from the geographic region of habitat of the patients (n = 1024). In addition, we enrolled a cohort of adult glaucoma patients with primary open angle glaucoma (POAG; n = 240) and developmental glaucoma cases comprising Axenfeld–Rieger syndrome (ARS; n = 30), Aniridia (n = 10), and Peter’s anomaly (n 10), to understand the involvement of TEK and CYP1B1 = in these cases.
Clinical evaluation of the subjects
A detailed clinical inclusion and exclusion criteria for the enrolment of PCG (Chakrabarti et al. 2006), POAG (Chakrabarti et al. 2007), and developmental glaucoma cases comprising of ARS (Komatireddy et al. 2003), Aniridia (Dharmaraj et al. 2003), and Peter’s anomaly (Gould and John 2002) have been described earlier. Each case was independently diagnosed by at least two glaucoma specialists. A good inter-observer agreement was seen based on the kappa statistics (κ = 0.94) and all cases with discordant diagnosis were excluded. The normal volunteers (controls) belonged to the same ethnicity of the patients and were matched to their geographical region of habitat. Majority of these subjects belonged to a longitudinal cohort [The Andhra Pradesh Eye Disease Study (APEDS)] who are being followed up for the last 20 years (Khanna et al. 2016). All control subjects, along with the parents and relatives of the PCG patients, underwent a comprehensive ocular examination and did not manifest any signs or symptoms of glaucoma or any other ocular or systemic diseases.
Targeted sequencing and validation
A customized panel containing the entire coding and untranslated regions of 36 genes (Supplementary Table 1) was designed for screening using the Ion Ampliseq Designer (Life Tech). Screening was accomplished by deep sequencing on an Ion Torrent platform using the Ion Ampliseq chemistry following the manufacturer’s guidelines (both from Life Tech). Bar-coded targets were amplified, ligated, and sequenced (in sets of 40 samples pooled in a single P1 HiQ chip) at an average depth of 500 (range from 120× to 1100× coverage). The analysis pipeline included the standard quality control measures followed by alignment with the hg19 sequence. Each sample exhibited 800–900 variants with >98% being on target. Samples that failed in the quality control measures described above were repeated. All TEK variants were validated by Sanger sequencing on an automated 3130 XL DNA sequencer (Applied Biosystems, CA) using the BigDye chemistry and with reported primers (Souma et al. 2016).
Cell culture, transient transfection, GFP pulldown, and immunofluorescence
HEK293 cells were maintained in DMEM/F12 (Thermo Fisher) plus 10% FBS and penicillin/streptomycin. For TEK-CYP1B1 interaction studies, HEK293 cells were transiently transfected with plasmids encoding recombinant HA-TEK and GFP-tagged CYP1B1 proteins. Cells were then subjected to pull down using GFP antibody (Abcam, Cambridge, MA) crosslinked to AminoLink Plus Coupling Resin (Chromotek, Hauppauge, NY), as described earlier (Rao et al. 2016). The pulled-down proteins were washed three times with lysis buffer, and the samples were eluted in glycine lysis buffer (pH 2.8), neutralized using 1 M Tris pH 9.5, and analyzed by SDS-PAGE and immunoblotting.
HUVECs were maintained in EBM-2 media from Lonza (Walkersville, MD) supplemented with EGM-2 SingleQuots (Lonza). After transient transfection, the cells were treated with recombinant human Angiopoietin 1 (ANGPT-1; 600 ng/ml; R&D Systems, Minneapolis, MN), as described earlier (Souma et al. 2016). The cells were then fixed with 4% paraformaldehyde, blocked with 5% goat serum, and incubated with primary antibodies: HA (Abcam) and ZO-1 (Thermo Fisher) overnight. After washing, Alexa 488-conjugated and Alexa 546-conjugated secondary antibodies (Thermo Fisher) were added for 1 h. After washing, nuclear stain Hoechst was added, and the cells were imaged using a Leica microscope (DM5500).
Statistical and bioinformatic analysis
Twenty-seven intragenic variations in TEK were used to generate haplotypes using the Haploview software (version 4.2) (Barrett et al. 2005). The minor allele frequencies, measures of Hardy–Weinberg equilibrium, linkage disequilibrium, and estimated haplotype frequencies were calculated. The associated allele and haplotype frequencies were further analyzed for statistical correction using the permutations tests (n = 10,000 permutations). Allele frequencies of each variant were calculated along with odds ratios and 95% confidence intervals (Supplementary Table 2). The allele frequencies of the individual mutations were compared with the overall frequencies provided at the ExAc (Exome Aggregation Consortium) database (Lek et al. 2016).
Results
Identification of novel TEK mutations in PCG
Screening of TEK mutations in our cohort revealed 12 novel heterozygous changes in 26 unrelated PCG cases (Table 1) that affected highly conserved residues (Fig. 1a–c, Supplementary Fig. 1). The allele frequency of each variant was < 0.01 in the normal controls, indicating that these were rare events (Table 1). Except for the p.I148T allele, the frequencies of 4/12 TEK mutations were significantly lower in the ExAc database compared to our normal controls (Table 1). The overall frequency of TEK mutations in our PCG cohort was 3.26% (95% CI 1.83–5.75%), which was slightly lower than a previous study comprising a mixed Caucasian cohort [5.29%, (95% CI 2.89–9.46%)] (Souma et al. 2016).
Table 1.
Allele frequencies of TEK mutations observed in the PCG cohort
| Chromosomal locations | Genomic position | Protein alteration | Allele frequencies observed in
|
P valuesa | Allele frequencies in the ExAc database (allele count/total alleles) | Pathogenicity scoresb (SIFT; PolyPhen-2; MutationTaster2) | |
|---|---|---|---|---|---|---|---|
| PCG [n = 337] Frequency (Allele count) | Controls [n = 1024] Frequency (Allele count) | ||||||
| 27158085 | g.48947A > C | p.E103D | 0.0089 (6) | 0.0053 (11) | 0.336 | 0.0008978 (109/121404) |
0.26; 0.996; 45 |
| 27158131 | g.48993C > T | p.R119C | 0.0014 (1) | – | – | 0.00001648 (2/121324) |
0.08; 0.997; 180 |
| 27168571 | g.59433T > C | p.I148T | 0.0133 (9) | 0.0161 (33) | 0.579 | 0.01716 (2007/116936) |
0.69; 0; 89 |
| 27172626 | g.63488A > C | p.Q214P | 0.0014 (1) | – | – | Novel | 0.12; 0.032; 76 |
| 27173314 | g.64176T > A | p.Y285X | 0.0014 (1) | – | – | Novel | – |
| 27173358 | g.64220A > G | p.E300G | 0.0014 (1) | – | – | Novel | 0.01; 0.805; 98 |
| 27180247 | g.71109C > T | p.P304L | 0.0014 (1) | 0.0024 (5) | 0.609 | 0.00005 (6/121244) |
0.04; 0.09; 98 |
| 27185499 | g.76361A > G | p.H400R | 0.0029 (2) | 0.0039 (8) | 0.626 | 0.000305 (37/121296) |
0.36; 0; 29 |
| 27185537 | g.76399C > T | p.R413W | 0.0029 (2) | 0.0019 (4) | 0.609 | 0.0001813 (22/121320) |
0.02; 0.987; 101 |
| 27204927 | g.95789G > C | p.G743A | 0.0014 (1) | – | – | 0.0002801 (34/121386) |
0.84; 0.518; 60 |
| 27212870 | g.103732G > A | p.G951D | 0.0014 (1) | – | – | Novel | 0; 1; 94 |
| 27220074 | g.110936G > A | p.C1044Y | 0.0014 (1) | – | – | Novel | 0.19; 0.999; 194 |
Based on test of significance between the PCG cases and controls. The chromosomal position is in accordance with GRCh37/hg19 assembly. The TEK gene reference sequence is NM_000459 and protein reference sequence is NP_000450.2
Pathogenicity scores based on SIFT (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), and MutationTaster2 (www.mutationtaster.org) softwares
Fig. 1.
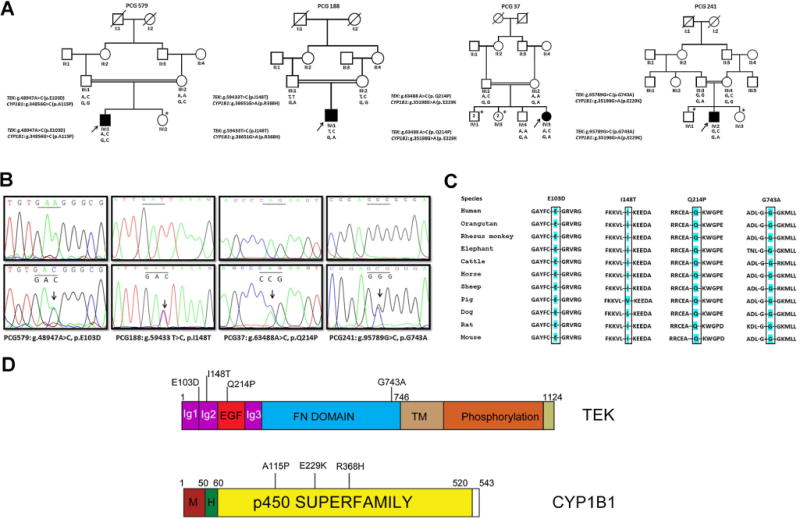
Genetic interaction of TEK and CYP1B1 in PCG patients. a Pedigrees of four PCG families segregating the heterozygous TEK and CYP1B1 alleles. The affected probands are indicated by solid black symbols who harbor both the heterozygous mutant alleles, while their asymptomatic parents carry either of the corresponding heterozygous TEK or CYP1B1 alleles. The genotypes of the individuals for the TEK and CYP1B1 mutations are indicated below the symbols. Asterisk indicates the siblings whose biological samples were unavailable. Co-segregation of TEK p.I148T and CYP1B1 p.R368H was observed in two pedigrees and only a representative pedigree is shown. b Chromatograms of the four probands (lower panel) harboring the four different heterozygous TEK mutations. The site of nucleotide change is indicated by an arrow, compared to the corresponding wild-type sequence (upper panel). c TEK protein sequence conservation across different species for the four mutations (p.E103D, p.I148T, p.Q214P, and p.G743A). The conserved residue for each mutation is highlighted in blue color. d Schematic representation of the TEK and CYP1B1 domains (Ig immunoglobulin, EGF epidermal growth factor, FN fibronectin, TM transmembrane, M membrane, H hinge region) indicating the location of the mutations identified in PCG (color figure online)
As PCG is inherited in an autosomal recessive manner (Dimasi et al. 2007; Sarfarazi and Stoilov 2000; Wang and Wiggs 2014), we suspected the presence of a second mutant allele. Support of this hypothesis comes from the previous reports of digenic inheritance in some forms of glaucoma (Chakrabarti et al. 2009; Kaur et al. 2005; Vincent et al. 2002). Although our present cohort did not carry homozygous changes in any of the known PCG genes, we reanalyzed our samples that harbored heterozygous mutations in any of these genes along with the TEK mutations. We observed that in 5 PCG cases heterozygous CYP1B1 mutations (p.A115P, p.E229 K, and p.R368H) co-occurred with heterozygous TEK mutations (p.E103D, p.I148T, p.Q214P, and p.G743A) indicating a potential digenic inheritance (Fig. 1a). None of the normal controls carried both the heterozygous combinations of CYP1B1 and TEK mutations. The TEK Q214P and G743A alleles were absent in 1024 controls, whereas very low frequencies of heterozygous TEK E103D (0.005) and I148T (0.016) alleles were found in the control population (Table 1). A compound heterozygous TEK mutation (p.E103D and p.E300G) was also observed in 1 family (PCG38). However, the remaining 20 PCG cases harboring a single heterozygous TEK mutation did not carry any additional mutation in the other 35 adult and childhood glaucoma-associated genes (Supplementary Fig. 1; Supplementary Table 1). The co-occurrence of heterozygous TEK and CYP1B1 mutations as seen in our PCG cases were not observed in additional sets of POAG, ARS, Aniridia, and Peter’s Anomaly patients. The CYP1B1 mutations identified in this study have previously been reported in PCG; however, the TEK mutations were novel. We, therefore, analyzed the effect of the observed TEK mutations in silico. Functional predictions by SIFT, Poly-Phen-2, and MutationTaster2 softwares indicated that these mutations were possibly damaging to the protein function (Table 1) and their residues were highly conserved across species (Fig. 1c, Supplementary Fig. 1).
To test if the observed mutations originated from a common founder in our cohort, we generated haplotypes with 27 polymorphic intragenic variants in TEK. Six variants that were not in Hardy–Weinberg equilibrium in the controls (rs3824410, g.64042C > T, rs682632, rs2252883, rs681754, and rs681707) were excluded. The remaining 21 variants did not exhibit any significant association to the PCG cases following Bonferroni correction (Supplementary Table 2). Overall, there was weak linkage disequilibrium across these TEK variants (Supplemental Fig. 2), and there were no significant haplotypes associated with PCG. In addition, the TEK mutations were not present on any uniform haplotype background, as was earlier observed with respect to the CYP1B1 mutations globally (Chakrabarti et al. 2006).
Clinical presentation of PCG cases harboring heterozygous TEK and CYP1B1 mutations
The probands of all 5 PCG cases (Fig. 1) exhibited poor visual prognosis based on long-term follow-up after surgical intervention. While a specific genotype–phenotype correlation could not be drawn based on the combination of TEK and CYP1B1 mutant alleles, the IOP remained on the higher side (> 28 mmHg) in their worst affected eye along with total cupping of their optic discs (0.9:1) and poor visual acuity (ranging from mild perception of light to no light perception). Even the probands of the PCG188 and PCG200 families harboring the same TEK (p.I148T)∷CYP1B1 (p.R368H) allelic combinations (Fig. 1a) had variable manifestations of IOP, corneal diameter, cup-to-disc ratio, and visual acuity at presentation. Following surgery, although the visual acuity remained poor in PCG188 and PCG200, the latter had a slight reduction in the IOP in one eye at the end of 4 years. Similar situation was observed with the proband (PCG38) harboring the compound heterozygous TEK mutations, who exhibited reduced IOP post-surgery but with poor visual acuity. Intriguingly, parents of the PCG-affected children who carried a single copy of the TEK or CYP1B1 mutant allele were asymptomatic, suggesting that the heterozygous variations by themselves were not pathogenic.
Interaction of TEK and CYP1B1
As the identified allelic combinations were extremely rare in our cohort, we carried out in vitro analysis to determine biochemical interaction between these proteins. First, we tested if TEK and CYP1B1 could physically interact with each other. Co-transfection of HEK293 (human embryonic kidney) cells with plasmids encoding recombinant HA-TEK (hemagglutinin-tagged TEK) and GFP-CYP1B1 followed by co-immunoprecipitation with anti-GFP-conjugated beads demonstrated that HA-TEK and GFP-CYP1B1 are part of the same complex. As negative control, no interaction was detected between the GFP tag and HA-TEK proteins (Fig. 2). Next, we asked whether the mutant combinations identified in patients can associate in the same assay. Compared to WT (wild-type) proteins, we found that the ability of GFP-CYP1B1 A115P and GFP-CYP1B1 E229K to immunoprecipitate HA-TEK E103D and HA-TEK Q214P, respectively, was significantly diminished. GFP-CYP1B1 R368H also exhibited relatively reduced ability to immunoprecipitate HA-TEK I148T (~70%). No significant change was observed with HA-TEK G743A with GFP-CYP1B1 E229 K as compared to WT proteins (Fig. 2). The WT and mutant TEK proteins expressed at similar levels in the cells, indicating that the mutations did not affect the expression or stability of the proteins (Fig. 2). We also tested the potential of the mutant TEK and CYP1B1 proteins to associate with wild-type CYP1B1 and TEK, respectively. As shown in Supplementary Fig. 3a, the mutant HA-TEK proteins E103D and I148T exhibited diminished interaction with wild-type GFP-CYP1B1. On the other hand, mutant GFP-CYP1B1 A115P and R368H showed perturbed interaction with HA-TEK.
Fig. 2.
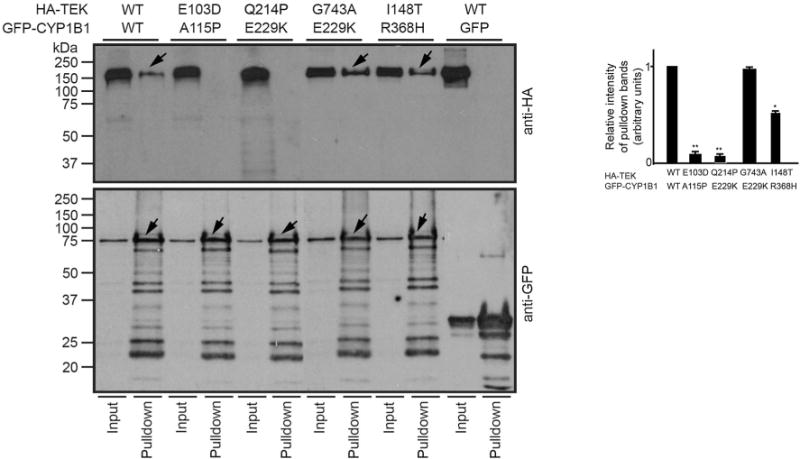
TEK and CYP1B1 interact in cells. HEK293 were transiently transfected with indicated GFP-CYP1B1 and HA-TEK plasmids. Negative controls utilized cells expressing GFP-alone and HA-TEK or untransfected cells. The cell extracts were subjected to pull down using the GFP antibody, and the precipitated proteins (arrows) were analyzed by SDS-PAGE and immunoblotting using indicated antibodies. Lower panel shows positive control for IP. Input lane represents 5% of the protein used for the pull-down assay. Right panel shows quantification of the relative intensity (in arbitrary units) of the HA-immunoreactive bands in the pull-down lane relative to the input lane for each combination. Intensity of the ratio of the pull-down bands to the input of WT (wild-type) proteins was set to 1
The residues E103, I148, and Q214 lie in the N-terminal extracellular domain of TEK (Fig. 1d). This suggested that either the N-terminal TEK domain was involved in the interaction with CYP1B1 or that the mutations altered the conformation of the TEK protein, which affected a secondary CYP1B1-binding site. To directly test this, we determined the CYP1B1-binding domain of TEK. We generated two TEK variants encoding HA-tagged N-terminal extracellular domain (amino acids 1-815; HA-TEKN) and C-terminal intracellular domain (816-1124; HA-TEKC). Co-immunoprecipitation with GFP-CYP1B1 showed that CYP1B1 associated predominantly with HA-TEKC; however, although low yet detectable HA-TEKN could also be pulled down with GFP-CYP1B1 (Supplementary Fig. 3B; arrows). These results suggested that although the intracellular domain of TEK-mediated most association with CYP1B1, the extracellular domain was likely involved in modulating this interaction. Mutations in the N-terminal domain of TEK may alter the conformation of the C-terminal domain, which could perturb the interactions.
Effect of mutations on Angiopoietin-TEK signaling
TEK is a receptor tyrosine kinase with an extracellular ligand-binding domain consisting of three immunoglobulin-like Ig domains and EGF domains (Fig. 1) (Barton et al. 2006). The E103 residue of TEK is part of the first Ig domain (Ig1), the I148 residue lies in the Ig2 domain, and the Q214 and E300 residues in the EGF domain. We, therefore, hypothesized that mutations in these residues would alter the ligand-mediated localization of TEK to cell–cell junctions. We expressed HA-tagged WT or mutant TEK proteins in HUVECs (human umbilical vein endothelial cells) and assessed their subcellular localization after ligand stimulation using the agonist human Angiopoietin 1 (ANGPT-1; 600 ng/ml; R&D Systems, Minneapolis, MN), as described earlier (Souma et al. 2016). ANGPT1 treatment resulted in discrete localization of WT TEK to ZO-1 positive cellular junctions as compared to diffuse membrane and cytosolic association in the absence of ANGPT1. While the TEK-E103D and TEK-Q214P mutants exhibited severely compromised ability to respond to ANGPT1 stimulus, TEK-I148T showed partial response to ANGPT1. The G743A mutant on the other hand, followed the WT TEK localization pattern (Fig. 3). Although this residue lies close to the transmembrane domain, the G743A change may not significantly alter the localization and interaction of TEK with CYP1B1. Further studies are needed to delineate the effect of this mutation on TEK function. We also tested the localization of the TEK-E300G mutant in the same assay. We found that this mutant also exhibited a reduced but not eliminated ability to respond to the ANGPT1 stimulus (Supplementary Fig. 4).
Fig. 3.
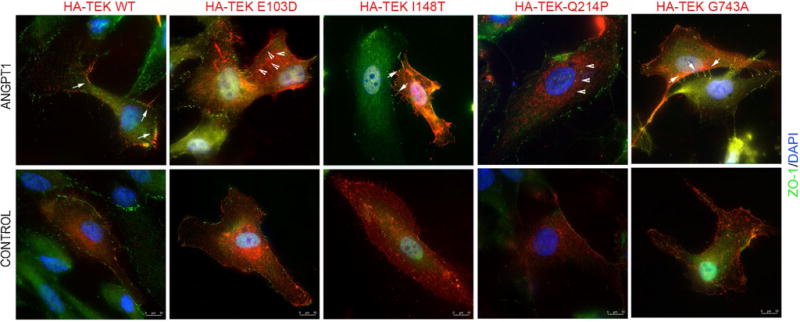
Mutant TEK localization in HUVECs. HUVECs transiently transfected with WT (wild-type) HA-TEK or indicated mutants were treated with Angiopoietin-1 (ANGPT-1) for 30 min followed by staining with HA (red) and ZO-1 (green; cell–cell junction marker). Control cells were treated with the vehicle. Nuclei are stained with Hoechst (blue). Arrows indicate the punctate staining of TEK along cell–cell junctions in the ANGPT-1-treated cells. Arrowheads indicate majority cytosolic localization of TEK E103D mutant after ANGPT-1 stimulation (color figure online)
Discussion
Despite remarkable investigations on identifying genes associated with PCG (Souma et al. 2016; Ali et al. 2009; Vasiliou and Gonzalez 2008), our efforts to delineate the genetic defects have been hampered by the variable penetrance and expression of the effect of the genetic mutations. Such effects were further compounded by genetic interactions between genes that encode for proteins involved in common intracellular pathways. Here, we have provided evidence of genetic and physical interaction between two known PCG genes TEK and CYP1B1. This interaction is associated with relatively severe pathogenesis. Furthermore, the mutations not only altered the ability of TEK to interact with CYP1B1 but also perturbed its ligand responsiveness.
Consistent with a previous study reporting heterozygous TEK mutations in PCG (autosomal dominant mode of inheritance), we identified novel heterozygous TEK mutations in our PCG cohort. However, identification of heterozygous CYP1B1 mutations in the TEK-positive PCG cases provided evidence for genetic interaction between these two genes. For these genes to be involved in the same disease pathway, the corresponding proteins should interact with each other. Our studies not only showed that TEK and CYP1B1 could genetically and physically associate with each other; they also provided evidence that the mutant combinations perturbed this interaction. Similar studies have been reported in some cases of glaucoma as well as retinal degenerative diseases, indicating that digenic inheritance underlies the complex etiology of genetic diseases (Kajiwara et al. 1994; Khanna et al. 2009; Schaffer 2013; Vincent et al. 2002).
While the molecular mechanisms underlying glaucoma remain obscure, increased IOP is a major risk factor (Tamm 2009; Wang and Wiggs 2014). Recent remarkable studies have identified a link between TEK and IOP (Thomson et al. 2014). TEK is highly expressed in the Schlemm’s canal endothelium and also regulates its development (Karpinich and Caron 2014). Our data indicated a role of TEK-CYP1B1 interaction in regulating this function. The TEK mutations that exhibited perturbed interaction with CYP1B1 or localization were associated with disease in only cases carrying the heterozygous CYP1B1 mutations. It is possible that the other heterozygous TEK alleles might show a similar effect on the interaction or localization of TEK; however, the absence of a second mutant allele in CYP1B1 or in another glaucoma gene (Supplementary Table 1) necessitates further screening of larger cohorts across different ethnicities.
CYP1B1 mutations are widely associated with PCG (Vasiliou and Gonzalez 2008; Wang and Wiggs 2014). Although the molecular mechanism of CYP1B1-mediated pathology is not clear, it is implicated in regulating intracellular metabolism and oxidative stress during anterior segment development (Vasiliou and Gonzalez 2008). Two out of the three CYP1B1 variants (p.E229K and p.R368H) that co-occurred with TEK were reported to exhibit altered estradiol and retinoid metabolism activity (Banerjee et al. 2016). While the activity of CYP1B1 A115P is yet to be determined, a disease-associated mutation in the adjacent residue R117P resulted in severely compromised estradiol and retinoid metabolizing activities (Banerjee et al. 2016). We, therefore, predict that the p.A115P mutation would have similar effects on CYP1B1 activity. However, further studies are needed to test this hypothesis.
Although CYP1B1 possesses metabolic activity towards intracellular substrates (Nishida et al. 2013), a yet unidentified function of CYP1B1 involving TEK-mediated signaling in the Schlemm’s canal cannot be ruled out. In fact, CYP1B1 was shown to regulate vascular homeostasis in endothelial cells (Palenski et al. 2013). Given an association between TEK and CYP1B1, it is plausible that mutant TEK and CYP1B1 may compromise their function. We also acknowledge that the perturbation of TEK-CYP1B1 interaction may not be completely causative. TEK function is modulated by its ligand ANGPT1, which is also involved in Schlemm’s canal development (Thomson et al. 2014). Although screening of the entire promoter and coding regions of the ANGPT1 gene did not reveal any mutations in our PCG cases, we cannot rule out the possibility of the other TEK ligand ANGPT2 or additional TEK-interacting proteins in mediating disease manifestation. In addition, the mutant TEK or CYP1B1 proteins that exhibit perturbed interactions with mutant CYP1B1 or mutant TEK, respectively, also showed altered interactions with their wild-type counterparts. Nonetheless, the TEK mutations identified in our study also altered its localization. These results suggested that TEK and CYP1B1 may function in overlapping as well as distinct mechanisms resulting in PCG.
Conclusions
Overall, our studies provided evidence for (1) a genetic interaction of TEK and CYP1B1 in PCG and; (2) a likely involvement of CYP1B1 in TEK function in the manifestation of PCG. Given the considerable genetic and phenotypic heterogeneity in PCG, our study provide clues to the involvement of multiple alleles as part of protein complexes and enable deciphering the molecular architecture of the associated variable expressivity.
Supplementary Material
Acknowledgments
We thank the PCG, POAG, and anterior segment dysgenesis patients and their families, and the normal volunteers for their participation in this study. The authors acknowledge the help of Drs. Ravi Thomas, Rajul Parikh, Harsha L Rao, and Garudadri C Sekhar for the diagnosis of some patients and controls; Drs. Aramati BM Reddy, Kiranpreet Kaur, Ratnakar Tripathi, and Mr. Chandan S Appikonda for sample collection; and Dr. Prathap Naidu and Mr. Hersh Parikh for the technical help. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about).
Funding: This work was supported by grants from the Department of Biotechnology, Government of India (BT/01/COE/06/02/10) to SC, and the National Eye Institute, NIH (EY022372) to HK. GP was supported by a fellowship from the Council of Scientific and Industrial Research, Government of India.
Footnotes
Electronic supplementary material: The online version of this article (doi:10.1007/s00439-017-1823-6) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest: The authors declare that they have no conflict of interest.
Data availability: All the molecular genetic, biochemical, and cellular data generated during this study are included in this manuscript and its supplementary files. However, the patients’ clinical data are confidential and will be available from the corresponding author on request.
References
- Ali M, McKibbin M, Booth A, Parry DA, Jain P, Riazuddin SA, Hejtmancik JF, Khan SN, Firasat S, Shires M, Gilmour DF, Towns K, Murphy AL, Azmanov D, Tournev I, Cherninkova S, Jafri H, Raashid Y, Toomes C, Craig J, Mackey DA, Kalaydjieva L, Riazuddin S, Inglehearn CF. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet. 2009;84:664–671. doi: 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DR. The development of the trabecular meshwork and its abnormality in primary infantile glaucoma. Trans Am Ophthalmol Soc. 1981;79:458–485. [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Chakraborty S, Chakraborty A, Chakrabarti S, Ray K. Functional and structural analyses of CYP1B1 variants linked to congenital and adult-onset glaucoma to investigate the molecular basis of these diseases. PLoS One. 2016;11:e0156252. doi: 10.1371/journal.pone.0156252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Barton WA, Tzvetkova-Robev D, Miranda EP, Kolev MV, Rajashankar KR, Himanen JP, Nikolov DB. Crystal structures of the Tie2 receptor ectodomain and the angiopoietin-2-Tie2 complex. Nat Struct Mol Biol. 2006;13:524–532. doi: 10.1038/nsmb1101. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Kaur K, Kaur I, Mandal AK, Parikh RS, Thomas R, Majumder PP. Globally, CYP1B1 mutations in primary congenital glaucoma are strongly structured by geographic and haplotype backgrounds. Invest Ophthalmol Vis Sci. 2006;47:43–47. doi: 10.1167/iovs.05-0912. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Devi KR, Komatireddy S, Kaur K, Parikh RS, Mandal AK, Chandrasekhar G, Thomas R. Glaucoma-associated CYP1B1 mutations share similar haplotype backgrounds in POAG and PACG phenotypes. Invest Ophthalmol Vis Sci. 2007;48:5439–5444. doi: 10.1167/iovs.07-0629. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Kaur K, Rao KN, Mandal AK, Kaur I, Parikh RS, Thomas R. The transcription factor gene FOXC1 exhibits a limited role in primary congenital glaucoma. Invest Ophthalmol Vis Sci. 2009;50:75–83. doi: 10.1167/iovs.08-2253. [DOI] [PubMed] [Google Scholar]
- Dandona L, Williams JD, Williams BC, Rao GN. Population-based assessment of childhood blindness in southern India. Arch Ophthalmol. 1998;116:545–546. [PubMed] [Google Scholar]
- deLuise VP, Anderson DR. Primary infantile glaucoma (congenital glaucoma) Surv Ophthalmol. 1983;28:1–19. doi: 10.1016/0039-6257(83)90174-1. [DOI] [PubMed] [Google Scholar]
- Dharmaraj N, Reddy A, Kiran V, Mandal A, Panicker S, Chakrabarti S. PAX6 gene mutations and genotype-phenotype correlations in sporadic cases of aniridia from India. Ophthalmic Genet. 2003;24:161–165. doi: 10.1076/opge.24.3.161.15607. [DOI] [PubMed] [Google Scholar]
- Dimasi DP, Hewitt AW, Straga T, Pater J, MacKinnon JR, Elder JE, Casey T, Mackey DA, Craig JE. Prevalence of CYP1B1 mutations in Australian patients with primary congenital glaucoma. Clin Genet. 2007;72:255–260. doi: 10.1111/j.1399-0004.2007.00864.x. [DOI] [PubMed] [Google Scholar]
- Gould DB, John SW. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum Mol Genet. 2002;11:1185–1193. doi: 10.1093/hmg/11.10.1185. [DOI] [PubMed] [Google Scholar]
- Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- Karpinich NO, Caron KM. Schlemm’s canal: more than meets the eye, lymphatics in disguise. J Clin Invest. 2014;124:3701–3703. doi: 10.1172/JCI77507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur K, Reddy AB, Mukhopadhyay A, Mandal AK, Hasnain SE, Ray K, Thomas R, Balasubramanian D, Chakrabarti S. Myocilin gene implicated in primary congenital glaucoma. Clin Genet. 2005;67:335–340. doi: 10.1111/j.1399-0004.2005.00411.x. [DOI] [PubMed] [Google Scholar]
- Khan AO. Genetics of primary glaucoma. Curr Opin Ophthalmol. 2011;22:347–355. doi: 10.1097/ICU.0b013e32834922d2. [DOI] [PubMed] [Google Scholar]
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, MacDon-ald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, Inglehearn CF, Lewis RA, Jacobson SG, Bergmann C, Beales PL, Attie-Bitach T, Johnson CA, Otto EA, Bhattacharya SS, Hildebrandt F, Gibbs RA, Koenekoop RK, Swaroop A, Katsanis N. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna RC, Murthy GV, Marmamula S, Mettla AL, Giridhar P, Banerjee S, Shekhar K, Chakrabarti S, Gilbert C, Rao GN, Andhra Pradesh Eye Disease Study G Longitudinal Andhra Pradesh Eye Disease Study: rationale, study design and research methodology. Clin Exp Ophthalmol. 2016;44:95–105. doi: 10.1111/ceo.12633. [DOI] [PubMed] [Google Scholar]
- Komatireddy S, Chakrabarti S, Mandal AK, Reddy AB, Sampath S, Panicker SG, Balasubramanian D. Mutation spectrum of FOXC1 and clinical genetic heterogeneity of Axenfeld-Rieger anomaly in India. Mol Vis. 2003;9:43–48. [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhou Y, Du L, Wei M, Chen X. Overview of Cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Exp Eye Res. 2011;93:572–579. doi: 10.1016/j.exer.2011.07.009. [DOI] [PubMed] [Google Scholar]
- Libby RT, Gould DB, Anderson MG, John SW. Complex genetics of glaucoma susceptibility. Annu Rev Genomics Hum Genet. 2005;6:15–44. doi: 10.1146/annurev.genom.6.080604.162209. [DOI] [PubMed] [Google Scholar]
- Liu Y, Allingham RR. Molecular genetics in glaucoma. Exp Eye Res. 2011;93:331–339. doi: 10.1016/j.exer.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida CR, Everett S, Ortiz de Montellano PR. Specificity determinants of CYP1B1 estradiol hydroxylation. Mol Pharmacol. 2013;84:451–458. doi: 10.1124/mol.113.087700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palenski TL, Gurel Z, Sorenson CM, Hankenson KD, Sheibani N. Cyp1B1 expression promotes angiogenesis by suppressing NF-kappaB activity. Am J Physiol Cell Physiol. 2013;305:C1170–C1184. doi: 10.1152/ajpcell.00139.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasilova M, Stoilov I, Sarfarazi M, Kadasi L, Ferakova E, Ferak V. Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary congenital glaucoma. J Med Genet. 1999;36:290–294. [PMC free article] [PubMed] [Google Scholar]
- Rao KN, Zhang W, Li L, Anand M, Khanna H. Prenylated retinal ciliopathy protein RPGR interacts with PDE6delta and regulates ciliary localization of Joubert syndrome-associated protein INPP5E. Hum Mol Genet. 2016;25:4533–4545. doi: 10.1093/hmg/ddw281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfarazi M, Stoilov I. Molecular genetics of primary congenital glaucoma. Eye (Lond) 2000;14(Pt 3B):422–428. doi: 10.1038/eye.2000.126. [DOI] [PubMed] [Google Scholar]
- Schaffer AA. Digenic inheritance in medical genetics. J Med Genet. 2013;50:641–652. doi: 10.1136/jmedgenet-2013-101713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, Feng L, Limviphuvadh V, Whisenhunt KN, Maurer-Stroh S, Yanovitch TL, Kalaydjieva L, Azmanov DN, Finzi S, Mauri L, Javadiyan S, Souzeau E, Zhou T, Hewitt AW, Kloss B, Burdon KP, Mackey DA, Allen KF, Ruddle JB, Lim SH, Rozen S, Tran-Viet KN, Liu X, John S, Wiggs JL, Pasutto F, Craig JE, Jin J, Quaggin SE, Young TL. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest. 2016;126:2575–2587. doi: 10.1172/JCI85830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet. 1997;6:641–647. doi: 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- Tamm ER. The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res. 2009;88:648–655. doi: 10.1016/j.exer.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Thomson BR, Heinen S, Jeansson M, Ghosh AK, Fatima A, Sung HK, Onay T, Chen H, Yamaguchi S, Economides AN, Flenniken A, Gale NW, Hong YK, Fawzi A, Liu X, Kume T, Quaggin SE. A lymphatic defect causes ocular hypertension and glaucoma in mice. J Clin Invest. 2014;124:4320–4324. doi: 10.1172/JCI77162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V, Gonzalez FJ. Role of CYP1B1 in glaucoma. Annu Rev Pharmacol Toxicol. 2008;48:333–358. doi: 10.1146/annurev.pharmtox.48.061807.154729. [DOI] [PubMed] [Google Scholar]
- Vincent AL, Billingsley G, Buys Y, Levin AV, Priston M, Trope G, Williams-Lyn D, Heon E. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet. 2002;70:448–460. doi: 10.1086/338709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Wiggs JL. Common and rare genetic risk factors for glaucoma. Cold Spring Harb Perspect Med. 2014;4:a017244. doi: 10.1101/cshperspect.a017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.