

SUMMARY
Progress toward understanding Zika virus (ZIKV) pathogenesis is hindered by lack of immunocompetent small animal models, in part because ZIKV fails to effectively antagonize Stat2-dependent interferon (IFN) responses in mice. To address this limitation, we first passaged an African ZIKV strain (ZIKV-Dak-41525) through Rag1−/− mice to obtain a mouse-adapted virus (ZIKV-Dak-MA) that was more virulent than ZIKV-Dak-41525 in mice treated with an anti-Ifnar1 antibody. A G18R substitution in NS4B was the genetic basis for the increased replication, and resulted in decreased IFN-β production, diminished IFN-stimulated gene expression, and the greater brain infection observed with ZIKV-Dak-MA. To generate a fully immunocompetent mouse model of ZIKV infection, human STAT2 was introduced into the mouse Stat2 locus (hSTAT2 KI). Subcutaneous inoculation of pregnant hSTAT2 KI mice with ZIKV-Dak-MA resulted in spread to the placenta and fetal brain. An immunocompetent mouse model of ZIKV infection may prove valuable for evaluating countermeasures to limit disease.
eTOC Blurb
An immunocompetent mouse model of ZIKV infection is needed. Gorman et al generated ZIKV-Dak-MA, a strain with a mutation in NS4B that causes greater disease in mice than the parental virus. HumanSTAT2 knock-in mice were generated and challenged with ZIKV-Dak-MA to establish a fully immunocompetent ZIKV mouse model.
INTRODUCTION
Zika virus (ZIKV) is a mosquito-, sexually-, and vertically-transmitted Flavivirus in the Flaviviridae family and was isolated originally in 1947 from a febrile sentinel rhesus monkey in Uganda. Historically, ZIKV caused a self-limiting febrile illness in about 20% of adults, with the remainder of infections being subclinical (Weaver et al., 2016), and was understudied until an outbreak in 2013–2014 in French Polynesia, and an epidemic in Brazil in 2015–2016 (Cao-Lormeau et al., 2014; Heymann et al., 2016; Marrs et al., 2016). The recent epidemics have been associated with ZIKV congenital syndrome and microcephaly in fetuses of infected mothers and Guillain-Barré syndrome in adults.
In the developing fetal brain, ZIKV infects neocortical neuroepithelial cells, and neuronal stem cells (NSCs). ZIKV infection leads to apoptosis, reduced proliferation, and transcriptional alterations (Li et al., 2016a; Onorati et al., 2016; Wu et al., 2017), all of which result in cortical thinning and neurodevelopmental anomalies. ZIKV also has been detected in tissues outside of the central nervous system (CNS), including the testis, epididymis, eyes, and female reproductive tract organs (Govero et al., 2016; Joguet et al., 2017; Ma et al.; Miner et al., 2016). The basis of ZIKV cellular tropism remains unknown although expression of phosphatidylserine receptors (Meertens et al., 2017; Richard et al., 2017), Musashi proteins in neurons (Chavali et al., 2017), and the ability to evade innate immune host defense pathways correlate with infectivity.
The type I IFN signaling pathway is a primary response to RNA virus infections in mammals, and is triggered by RIG-I-like and Toll-like recognition receptors. The resultant induction of type I (e.g., IFNα and IFNβ) and type III (e.g., IFN-λ) IFNs and engagement of their respective heterodimeric receptors (IFNAR1/IFNAR2 and IFNLR1/IL10Rβ) results in Janus (Jak) and tyrosine (Tyk) kinase phosphorylation, dimerization and nuclear translocation of STAT1 and STAT2, and the production of hundreds of antiviral and immunomodulatory IFN stimulated genes (ISGs). Although the ZIKV NS5 protein evades the human IFN signaling response by binding to and degrading STAT2, it is unable to bind efficiently to mouse Stat2 and thus lacks the ability to antagonize the mouse IFN response (Bowen et al., 2017; Grant et al., 2016; Kumar et al., 2016).
While existing mouse models of ZIKV infection in adult animals have provided insight into pathogenesis and disease outcome, they have been performed principally in mice with genetic or acquired deficiencies of IFN signaling (reviewed in (Morrison and Diamond, 2017)). It has been challenging to infect adult immunocompetent mice productively through a natural peripheral route; viral replication is readily aborted by the innate immune response before systemic dissemination (Aliota et al., 2016; Lazear et al., 2016). An immunocompetent mouse model of ZIKV infection that more closely reflects the human disease could improve our understanding of the pathogenesis of ZIKV and the ensuing host immune response, and have greater utility for evaluating vaccines and therapeutics. Here, we first generated a mouse-adapted strain of ZIKV with a key mutation in the viral NS4B gene, which facilitated less induction of IFN-β and greater replication in NSCs and the brains of mice. By combining this adapted-virus with a transgenic mouse that replaces mouse Stat2 with human STAT2 and thus allows evasion of IFN signaling cascades in infected cells, we created an immunocompetent mouse model of ZIKV infection that allowed for trans-placental transmission and fetal infection.
RESULTS
A mouse adapted ZIKV strain
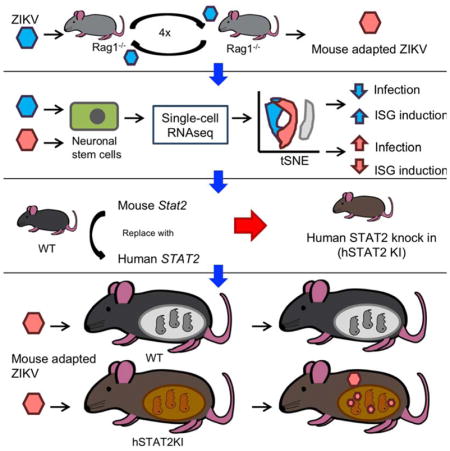
Adult immunocompetent wild-type (WT) mice are resistant to ZIKV infection (Lazear et al., 2016) in part because ZIKV cannot efficiently antagonize mouse Stat2 and type I IFN signaling (Grant et al., 2016; Kumar et al., 2016). In an attempt to overcome this restriction, we passaged an African strain of ZIKV (Dakar 41525, Senegal, 1984, GenBank: KU955591.1) in C57BL/6 Rag1−/− mice with an intact type I IFN response. As these mice lack mature B and T cell responses, we hypothesized that the sustained pressure of an innate immune response might facilitate evolution of viral escape mutants against IFN. We selected an African strain of ZIKV (ZIKV-Dak) because of its greater pathogenicity in WT neonatal mice than Asian strains (Lazear et al., 2016). We initially tested 4 to 6 week-old Rag1−/− mice for susceptibility to ZIKV-Dak, and all mice succumbed within 31 days of infection (Fig 1A). To adapt ZIKV-Dak, virus-containing brain homogenates isolated from moribund Rag1−/− mice were used for subcutaneous infection of naive Rag1−/− mice (Fig 1B). After four passages in Rag1−/− mice, we inoculated this mouse-adapted virus (ZIKV-Dak-MA) into four to five week-old WT mice after treatment with 2 mg of anti-Ifnar1 mAb and compared it to the parental virus. Anti-Ifnar1-treated WT mice were more vulnerable to infection with ZIKV-Dak-MA than ZIKV-Dak with a shorter time to death (Fig 1C). We investigated the basis for increased lethality by comparing viral RNA levels in serum, spleen, and the brain at 3 and 6 days after subcutaneous inoculation with 105 FFU of ZIKV-Dak or ZIKV-Dak-MA. In serum, no difference was observed at day 3 between the strains whereas a slightly higher level was measured at day 6 in ZIKV-Dak-MA-infected animals (Fig 1D). No difference in ZIKV-Dak and ZIKV-Dak-MA levels was detected in the spleen at days 3 or 6 after infection (Fig 1E). However, we observed higher levels of infection in the brains of ZIKV-Dak-MA than ZIKV-Dak infected mice on day 6 (Fig 1F). Thus, ZIKV-Dak-MA replicated to moderately higher levels in the brain of anti-Ifnar1-treated mice, and this phenotype was associated with an increase in lethality.
Figure 1. Generation of an adapted ZIKV with increased lethality and CNS viral burden in mice.

A. Four to six week-old Rag1−/− mice were inoculated subcutaneously with ZIKV-Dakar 41525 (ZIKV-Dak) (3 experiments, n = 8). B. Scheme of in vivo passaging of ZIKV-Dak. Brain homogenates (106 PFU) from Rag1−/− mice that succumbed to infection were inoculated subcutaneously into naive Rag1−/− mice. C. Four to five week-old WT C57BL/6 mice were treated with anti-Ifnar1 mAb one day prior to subcutaneous inoculation with 105 FFU of ZIKV-Dak or ZIKV-Dak-MA (3 experiments, n = 15, log rank test; ***, P < 0.001). D–F. ZIKV RNA levels in serum (D), spleen (E), and brain (F) of WT mice after treatment with anti-Ifnar1 mAb and subcutaneous inoculation of ZIKV-Dak or ZIKV-Dak-MA (3 experiments, n = 8–12, Mann-Whitney test; *, P < 0.05). Solid lines are median values, and dotted lines denote the limit of detection of the assay. G. RNA was isolated from ZIKV-Dak-MA and subjected to next generation sequencing. Mutations present in over 95% of genomes are shown. See also Table S1 and Fig S1.
To determine the genetic basis for the increased pathogenicity of ZIKV-Dak-MA in mice, we performed next-generation sequencing of the parental and adapted viruses. This analysis revealed three mutations in greater than 95% of the ZIKV-Dak-MA population (Fig 1G): a synonymous C to T change at nucleotide 1006, a non-synonymous A to G change at nucleotide 5808 resulting in a conservative lysine to arginine substitution at position 399 in NS3, and a non-synonymous G to A change at nucleotide 6965 resulting in a non-conservative glycine to arginine substitution at position 18 in NS4B. Analysis of the viral sequences obtained from the earlier sequential passages (P2 and P3) in Rag1−/− mice revealed that the mutations in NS4B arose during the second virus passage, with the NS3 mutation arising during the fourth passage (Table S1). These mutations in NS3 and NS4B in ZIKV-Dak-MA were not present in any ZIKV sequences available in the VIPR databases, but were present in some related mosquito and tick transmitted flaviviruses (Fig S1A and B).
ZIKV-Dak-MA replicates more efficiently in the brain and in NSCs
The increased infectivity of ZIKV-Dak-MA in brain tissue could reflect an enhanced ability to cross the blood-brain-barrier or an intrinsic ability to replicate more efficiently in neuronal cell targets. To evaluate these possibilities, we inoculated 4 to 5 week-old WT C57BL/6 mice with 104 FFU of ZIKV-Dak or ZIKV-Dak-MA via an intracranial route (no anti-Ifnar1 mAb treatment) and compared lethality and viral burden. Remarkably, the animals did not succumb to infection with either virus, suggesting that the changes in ZIKV-Dak-MA were not sufficient to result in lethal infection in immunocompetetent mice (Fig 2A). Although both strains replicated in different regions of the brains of WT mice, ZIKV-Dak-MA accumulated to greater levels (Fig 2B–E). At day 4, the midbrain of ZIKV-Dak-MA infected mice had higher viral RNA burden (Fig 2D) than ZIKV-Dak-challenged mice. By day 6, multiple regions of the brain had higher levels of ZIKV-Dak-MA than ZIKV-Dak RNA (brain stem, cerebellum, midbrain, and cortex) (Fig 2B–E). In contrast, the olfactory bulb showed no differences (Fig 2F).
Figure 2. Enhanced infectivity in the CNS and NSCs of ZIKV-Dak-Ma.

A. Four to five week-old WT C57BL/6 mice (without Ifnar1 blockade) were inoculated via intracranial route with ZIKV-Dak or ZIKV-Dak-MA and monitored for survival (3 experiments, n = 7). B–F. ZIKV RNA levels in (B) brainstem, (C) cerebellum, (D) midbrain, (E) cortex, and (F) olfactory bulb in WT mice at days 4 or 6 after intracranial challenge with ZIKV-Dak or ZIKV-Dak-MA (3 experiments, n = 6–9, Mann-Whitney test; *, P < 0.05; **, P < 0.01; ***, P < 0.001). Solid lines are median values, and dotted lines denote the limit of detection of the assay. G–I. Adult NSCs were inoculated at an MOI of 1 (H, I) or 0.01 (G) with ZIKV-Dak or ZIKV-Dak-MA. Infectious titers were determined by focus-forming assay (FFA). Viability of NSCs (I) was normalized to uninfected control cells (3 experiments performed in duplicate). Mean values ± standard error of the mean (SEM). A two-way and one-way ANOVA with multiple comparison correction were for viral titers and cell viability, respectively (*, P < 0.05; **, P < 0.01; P < 0.001). J–L. Viral RNA levels in the (J) brainstem, (J) cerebellum, (L) midbrain, (M) cortex, and (N) olfactory bulb in congenic Ifnar1−/− mice at day 2 and day 4 after intracranial challenge with ZIKV-Dak or ZIKV-Dak-MA (3 experiments, n = 8–13, Mann-Whitney test; *, P < 0.05; **, P < 0.01; ***, P < 0.001). Solid lines are median values, and dotted lines denote the limit of detection of the assay.
To corroborate the differential infectivity of ZIKV-Dak and ZIKV-Dak-MA, we performed infection studies in cell culture. As ZIKV replicates efficiently in NSCs (Li et al., 2016b; McGrath et al., 2017), we isolated these cells from the subventricular zone of 4 to 6 week-old WT C57BL/6 mice, and analyzed multi-step (multiplicity of infection (MOI) of 0.01) and single-step (MOI of 1) viral growth kinetics. ZIKV-Dak-MA replicated to higher levels than ZIKV-Dak at multiple time points (Fig 2G–H). The increased yield of ZIKV-Dak-MA was not due to greater cell survival, as ZIKV-Dak-MA resulted in more, not less, cell death than ZIKV-Dak (Fig 2I).
We hypothesized that compared to the parental virus, ZIKV-Dak-MA may have acquired an ability to evade a type I IFN response in the brain. To test this hypothesis, we compared infectivity of ZIKV-Dak and ZIKV-Dak-MA in different brain regions after intracranial inoculation of Ifnar1−/− mice. No differences in infection were observed between ZIKV-Dak and ZIKV-Dak-MA in different brain regions at day 2 after infection (Fig 2J–N). By day 4 after infection, minimal differences in viral RNA levels (< 2-fold) were observed between ZIKV-Dak and ZIKV-Dak-MA in different brain regions. Because ZIKV-Dak-MA infection resulted in up to 9-fold greater viral titers than ZIKV-Dak in different brain regions of WT mice (Fig 2B–E), in aggregate, these data suggest that majority of the replication disparity between ZIKV-Dak and ZIKV-Dak-MA in the brain is likely due to a differential ability to evade type I IFN responses. However, there is a small difference in relative infectivity that could reflect effects on type I IFN-independent host defenses.
The NS4B G18R mutation determines the increased pathogenicity of ZIKV-Dak-MA
ZIKV-Dak-MA had three mutations present in over 95% of the viral population, two of which (NS3 K399R and NS4B G18R) were non-synonymous changes. To evaluate which mutations were responsible for enhanced infection and disease, we engineered isogenic viruses from a quadripartite infectious clone of ZIKV-Dakar 41525 (Fig 3A, and see STAR Methods). We produced WT and isogenic variants with mutations in NS3 K399R, NS4B G18R, or NS3 K399R and NS4B G18R (DM). Four to five week-old WT mice were pretreated with anti-Ifnar1 and challenged with 105 FFU of the isogenic viruses (Fig 3B). Approximately 95% of mice succumbed to ZIKV-Dak NS4B G18R or ZIKV-Dak DM whereas only 5% of animals died after inoculation with ZIKV-Dak WT or ZIKV-Dak NS3 K399R (Fig 3B). Thus, the NS4B G18R mutation was necessary and sufficient for enhanced virulence. Viral burden analysis at day 6 after inoculation corroborated these findings. Although no differences were observed in serum (Fig 3C), ZIKV-Dak NS4B G18R and ZIKV-Dak DM replicated to slightly higher titers in the spleen than ZIKV-Dak WT (Fig 3D). Moreover, in the brain, at day 6, ZIKV-Dak NS4B G18R and ZIKV-Dak DM accumulated to higher levels than ZIKV-Dak WT (Fig 3E). Consistent with these results, ZIKV-Dak NS4B G18R and ZIKV-Dak DM produced higher viral yields in the supernatants of mouse and human NSCs (Fig 3F–H) and induced greater cell death than ZIKV-Dak-WT at 96 h post-infection (Fig 3I).
Figure 3. The NS4B G18R mutation is required for enhanced mouse lethality and increased NSC infection by ZIKV.

A. ZIKV Dakar 41525 infectious cDNA clone. B. Four to five week-old WT C57BL/6 mice were treated with anti-Ifnar1 mAb one day prior to subcutaneous inoculation with infectious clone derived WT ZIKV-Dakar 41525 (ZIKV-Dak-WT), ZIKV-Dak NS3 K399R, ZIKV-Dak NS4B G18R, or ZIKV-Dak NS3 K399R + NS4B G18R (ZIKV-Dak DM) (3–4 experiments, n=15–16, log rank test compared to ZIKV-Dak-WT; ZIKV-Dak DM and ZIKV-Dak NS4B G18R, P < 0.001; ZIKV-Dak NS3 K399R, P > 0.9). C–E. Viral burden in serum (C), spleen (D), and brain (E) at day 6 post subcutaneous inoculation with ZIKV-Dak-WT, ZIKV-Dak-DM, ZIKV-Dak NS3 K399R, or ZIKV-Dak NS4B G18R of four to five week-old WT mice after treatment with anti-Ifnar1 mAb (3 experiments, n = 12–14, one-way ANOVA with multiple comparisons correction; *, P < 0.05; **, P < 0.01; ***, P < 0.001). F–H. Mouse NSCs were inoculated at an MOI of 0.01 (F) or 1 (G, I) with ZIKV-Dak WT, ZIKV-Dak DM, or ZIKV-Dak NS4B G18R and analyzed by FFA (F–G) or cell viability assay (I). Human NSCs (hNSCs) were inoculated with ZIKV-Dak WT or ZIKV-Dak NS4B G18R (MOI of 0.1) and analyzed by plaque assay (H) (3 experiments performed in duplicate). For F–H mean ± SEM are shown. Viral titer and cell viability were analyzed (ANOVA with multiple comparisons corrections; *, P < 0.05; **, P < 0.01; ***, P < 0.001). J–K. Three week-old WT mice were treated with anti-Ifnar1 one day prior to subcutaneous inoculation with ZIKV-Brazil BeH819015 (ZIKV-Bzl-WT), ZIKV-Bzl NS3 K399R, or ZIKV-Bzl NS4B G18R and monitored for survival (J) and weight (K). The single animal that survived infection with ZIKV-Bzl NS4B G18R was omitted from the weight change data (K) (5 experiments, n=8–17). Survival and weight loss data were analyzed by the log-rank test and two-way ANOVA with multiple comparison correction, respectively (*, P < 0.05; **, P < 0.01; ***, P < 0.001; **** P < 0.0001).
To assess whether the adaptive mutations could enhance the pathogenicity of other ZIKV strains, we generated isogenic viruses using an infectious clone of a Brazilian ZIKV strain (BeH819015) (Widman et al., 2017). Three week-old WT C57BL/6 mice were pre-treated with 2 mg of anti-Ifnar1 and inoculated subcutaneously with 105 FFU of the WT clone-derived ZIKV from Brazil (ZIKV-Bzl WT), a single NS3 K399R mutant (ZIKV-Bzl NS3 K399R), or a single NS4B G18R mutant (ZIKV-Bzl NS4B G18R). A majority of animals inoculated with ZIKV-Bzl NS4B G18R succumbed to infection whereas those infected with ZIKV-Bzl-WT or ZIKV-Bzl NS3 K399R did not (Fig 3J). Moreover, ZIKV-Bzl NS4B G18R infected animals lost weight whereas ZIKV-Bzl WT or ZIKV-Bzl NS3 K399R infected animals did not (Fig 3K).
Single-cell RNAseq of infected and uninfected murine NSCs
To begin to determine how the adaptive NS4B mutation in ZIKV impacted IFN responses, we first performed single-cell RNAseq analysis on uninfected and ZIKV-Dak WT-infected murine NSCs at 24 and 48 h time points (Fig 4A). We detected large transcriptional changes in the ZIKV-infected NSCs compared to the uninfected cells, particularly by 48 h (Fig 4B and Fig S2). As expected, many cells expressed neural stem cells markers including Id3, Rpl32, Cdk4, Nes, and Sox2, which confirmed their cellular identity (Fig 4C) (Dulken et al., 2017). Clustering analysis grouped transcriptionally similar cells from all uninfected and infected samples into six groups (Fig 4D). The clusters were then manually labeled based on which RNAs were highly differential between clusters (e.g. cell cycle, IFN-stimulated genes, and levels of ZIKV RNA) (Fig 4E). ZIKV-infected cells were defined as having greater than 0.03% of viral transcripts relative to total cellular transcripts (see STAR Methods), and increased from none (uninfected), to a minority of cells at 24 h (9%), to a majority of cells (67%) at 48 h after infection (Fig 4F). ISGs were increased in cells exposed to ZIKV (Fig S2), with some (e.g., Ifnb1) correlating with viral RNA and infection, and others (e.g., Isg15, Rsad2, and Cxcl10) expressed in both infected (ZIKV RNA+) and uninfected (ZIKV RNA−) NSCs (Fig 4E–G). By 48 h, most cells responded to type I IFN signals based on expression of Rsad2 and Isg15, but only a small fraction accumulated high level of Ifnb1 transcripts and these were among the most infected ones. The highly ZIKV-infected cells were the sole producers of pro-inflammatory chemokines including Ccl5 and Cxcl10. The overall IFN effect also manifested in the cell cycle arrest, as the majority of the cells at 48 h were in G0/G1 phase whereas at 0 and 2 4h cells were equally distributed across G0/G1, G2/M, and S cell cycle phases (Fig 4D). This analysis illustrates the utility of single cell RNAseq in determining the transcriptional changes in uninfected and virus-infected cell populations.
Figure 4. Single cell RNAseq of NSCs after infection with ZIKV-Dak.

A. Scheme for single cell RNAseq experiment. 100,000 NSCs (uninfected or 24 or 48 h after ZIKV-Dak WT infection) were collected from three technical replicates, pooled, and subsequently, 17,500 cells were subjected to microfluidic-based single cell RNA-seq library generation and sequencing. B. T-distributed stochastic neighbor embedding (tSNE) plots comparing uninfected and ZIKV-Dak WT-infected cells. C. Violin plots showing expression of NSC-specific markers and RLR-related genes in NSCs from all three conditions. D. tSNE plots of all conditions with cell type clusters identified with graph-based clustering. Clusters were manually named (e.g., cell cycle and IFN-responding) after their most highly differential genes. E. Expression of a subset of ISGs and cell cycle markers used to determine clusters of (D) overlaid on the tSNE analysis from (B). F. Expression of ZIKV RNA overlaid on the tSNE analysis from (B). G. Violin plots showing expression of selected ISGs in NSCs from all three conditions. See also Fig S2.
ZIKV-Dak NS4B G18R induces less of a type I IFN response
We next used our single-cell RNAseq analysis to define transcriptional differences between ZIKV-Dak WT and ZIKV-Dak NS4B G18R infected murine NSCs (Fig 5A). Large transcriptional differences were found in cells infected with either ZIKV strain at the 48 h time point compared to uninfected cells (Fig 5B and Fig S2). ZIKV RNA was present in NSCs inoculated with either ZIKV-Dak WT or ZIKV-Dak NS4B G18R, and the transcriptional signatures clustered similarly (Fig 5C). Although more ZIKV-Dak NS4B G18R RNA was detected at 24 and 48 h after infection (Fig 5D), many ISGs (e.g., Ifit1, Ifitm3, and Isg15) were induced at lower levels in ZIKV-Dak NS4B G18R than ZIKV-Dak WT infected NSCs (Fig 5E and Fig S3A). Single cell RNA-seq allowed separate transcriptional interrogation of the uninfected and ZIKV-infected NSC populations. At 24 and 48 h, ISGs were induced at a higher level in uninfected cells from the ZIKV-Dak WT group than the ZIKV-Dak NS4B G18R group (Fig 5E and Fig S3A); this suggested that the ZIKV-Dak NS4B G18R might antagonize induction of the type I IFN response more efficiently than the parental virus, which would have its greatest effect in cells responding to but not infected with ZIKV. At 24 and 48 h, ISG levels also were higher in the infected cell subset from the ZIKV-Dak WT compared to ZIKV-Dak NS4B G18R group, although the differences were less than in the uninfected cells (see Fig S3B).
Figure 5. ZIKV-Dak NS4B G18R induces less IFN-β and induction of ISGs in infected NSCs.

A. Scheme of experiment. 100,000 NSCs (uninfected, 24 or 48 h after ZIKV-Dak WT infection, or 24 or 48 h after ZIKV-Dak NS4B G18R) were collected from three technical replicates, pooled, and then 17,500 cells were subjected to microfluidic-based single cell RNA-seq library generation and sequencing. B. T-distributed stochastic neighbor embedding (tSNE) plots comparing uninfected and ZIKV-Dak WT and ZIKV-Dak NS4B G18R infected cells. C. Expression of ZIKV RNA overlaid on the tSNE plots from (B). D. Violin plots showing the expression of ZIKV viral RNA in all populations. Wilcoxon rank sum test with continuity correction was performed **, P < 10−10). E. Violin plots were generated showing expression of selected RLRs (Ddx58, Ifih1, and Mavs), type I IFNs (Ifna2 and Ifnb1), and ISGs (Eif2ak2, Ifit1, Ifit3, Ifitm2, Ifitm3, Isg15, Oasl1, Oasl2, and Rsad2) in uninfected (ZIKV RNA−) and infected (ZIKV-RNA+) cells in the culture at 24 h after inoculation with ZIKV-Dak WT or ZIKV-Dak NS4B G18R. MAST test was used to determine the statistical difference in gene expression between different conditions. (** P adjusted < 0.01). F. NSCs were inoculated with ZIKV-Dak WT and ZIKV-Dak NS4B G18R at an MOI of 1. Supernatant was collected and IFN-β was quantified by ELISA (3 experiments in duplicate, mean ± SEM, student’s t-test; **, P < 0.01) G. NSCs were inoculated with ZIKV-Dak WT and ZIKV-Dak NS4B G18R (MOI of 1). Cells were lysed, RNA was isolated, and 18S, Ifnb1, and Isg15 mRNA was quantified by qRT-PCR. Expression was compared to uninfected cells (3 experiments in duplicate, student’s t-test for ΔΔCt data; *, P < 0.05). H–J. NSCs were untreated (no mAb) (H–I) or pretreated with anti-Ifnar1 blocking mAb (J) or 3U/mL of IFN-β (K) for 16 h and then inoculated with ZIKV-Dak WT or ZIKV-Dak NS4B G18R (MOI of 0.01). Infectious titers were determined by FFA. In panel I, NSCs were inoculated with ZIKV-Dak or ZIKV-Dak NS4B G18R. At 96 h, cells were stained for ZIKV E protein and processed by flow cytometry. The data in G–K are from 3 experiments in duplicate (mean ± SEM, two-way ANOVA with multiple comparison corrections; *, P < 0.05). See also Fig S2 and S3.
To begin to address whether ZIKV-Dak NS4B G18R antagonized type I IFN responses and induction of ISGs, we first assessed the levels of IFN-β in the supernatant of ZIKV-Dak WT and ZIKV-Dak NS4B G18R-infected murine NSCs. Despite higher levels of infection (see Fig 3G), less IFN-β was detected in the supernatant of ZIKV-Dak NS4B G18R than ZIKV-Dak WT-infected cells at 48 h (Fig 5F). However, both RNAseq and qRT-PCR data failed to show differences in IFN-β mRNA induction after ZIKV-Dak WT and ZIKV-Dak NS4B G18R infection of NSCs (Fig 5E and G). The lack of significant changes in the IFN-β mRNA levels in the context of decreased IFN-β protein in the supernatant of ZIKV-Dak NS4B G18R infected cells suggests that the NS4B G18R mutation might contribute to an IFN-β translational block. Inhibition of IFN-β protein translation has been described as a ZIKV immune evasion mechanism in human dendritic cells (Bowen et al., 2017), although the mechanism currently remains unknown.
To assess whether adapted ZIKV modulates type I IFN signaling responses, murine NSCs that were pre-treated with anti-Ifnar1 antibody or exogenous IFN-β were inoculated with ZIKV-Dak WT or ZIKV-Dak NS4B G18R. The greater infectivity phenotype seen with ZIKV-Dak NS4B G18R by viral yield assays or antigen staining (Fig 5H and I) was lost after anti-Ifnar1 mAb treatment, with both viruses replicating equivalently in NSCs lacking type I IFN signaling (Fig 5J); this data suggests that differences in replication in NSCs between ZIKV-Dak WT and ZIKV-Dak NS4B G18R are dependent on type I IFN signaling. However, pretreatment of NSCs with IFN-β resulted in equivalent restriction of ZIKV-Dak WT and ZIKV-Dak NS4B G18R (Fig 5K). As the WT and adapted ZIKV strains produced different amounts of IFN-β protein, showed infection differences that were abolished by anti-Ifnar1 mAb treatment, yet were equally sensitive to exogenous IFN-β pre-treatment, the NS4B G18R mutation in ZIKV likely acts through its ability to diminish IFN-β levels that initiate the antiviral IFN signaling pathway in infected NSCs.
An immunocompetent mouse model of ZIKV infection
Our experiments suggested that ZIKV-Dak-MA acquired an ability to minimize induction of type I IFN yet still remained sensitive to the antiviral effects of exogenous or paracrine-derived IFN, likely because of its inability to bind to and degrade mouse Stat2 (Bowen et al., 2017; Grant et al., 2016; Kumar et al., 2016; Tripathi et al., 2017). To overcome this limitation and create a fully immunocompetent mouse model of ZIKV pathogenesis, we inserted by homologous recombination a human STAT2 allele into the mouse Stat2 locus but retained the mouse Stat2 promoter: exon 1 and part of exon 2 were derived from the mouse gene (promoter sequences), and the remainder of exon 2 and all of exons 3 through 24 were from the human gene (coding sequences) (Fig 6A). Southern blotting and PCR corroborated the correct integration (Fig 6B–C), and Western blotting of splenocytes from mice treated with poly(I:C) confirmed the loss of mouse Stat2 and the expression of human STAT2 (Fig 6D). ISG induction (e.g., Oas1, Irf7, and Ifnb) in hSTAT2 KI mice downstream of IFN signaling was established by administration of poly I:C via intranasal route and interrogation of lungs 6 h later, and appeared equivalent to that observed in WT mice (Fig 6E).
Figure 6. hSTAT2 KI mice are susceptible to ZIKV-Dak-MA.

A. Diagram of targeting construct for hSTAT2 KI mice. B–C. Southern blot and PCR confirmation of integration of hSTAT2 allele. D. Western blot of WT, Stat2−/−, and hSTAT2 KI splenocytes from poly(I:C) treated mice. E. ISG (Oas1, Irf7, and, Ifnβ) induction in WT and hSTAT2 KI mice after administration of poly I:C via an intranasal route and interrogation of lungs 6 h later. F. Three week-old hSTAT2 KI mice were inoculated subcutaneously with ZIKV-Dak or ZIKV-Dak-MA (3 experiments, n = 12–26, log-rank test, P = 0.06). G–I. Viral burden in serum (G), spleen (H), and brain (I) of 3 week-old hSTAT2 KI at day 9 after inoculation with ZIKV-Dak or ZIKV-Dak-MA (2 experiments, n = 9, Mann-Whitney test; *, P < 0.05; **, P < 0.01; ***, P < 0.001). Solid lines are median values, and dotted lines denote the limit of detection of the assay. J–M. Eleven week-old hSTAT2 KI or WT dams mated to hSTAT2 KI and WT sires, respectively were infected with ZIKV-Dak-MA on E6.5 and tissues were harvested on E13.5. Viral burden in maternal serum (J), maternal spleen (H), placenta (L), or fetal heads (M) is shown (2 experiments, n = 9 WT and 10 hSTAT2 KI dams, n = 36 WT and 32 hSTAT2 KI placentas and fetal heads, Mann-Whitney test; **, P < 0.01; ****, P < 0.0001). Solid lines are median values, and dotted lines denote the limit of detection of the assay. See also Fig S4 and S5.
We investigated the pathogenic potential of ZIKV-Dak-MA in hSTAT2 KI mice without IFN blockade. Three week-old hSTAT2 KI mice were inoculated subcutaneously with ZIKV-Dak or ZIKV-Dak-MA and monitored. Thirty percent of hSTAT2 KI mice succumbed to lethal ZIKV-Dak-MA infection whereas none died after inoculation with parental ZIKV-Dak (Fig 6F). Viral burden analysis revealed higher levels of ZIKV-Dak-MA than ZIKV-Dak in the spleen and brain, but not in serum at day 9 (Fig 6G–I). We next tested if pregnant female hSTAT2 KI mice could transmit ZIKV efficiently to developing fetuses. Eleven week-old WT or hSTAT2 KI dams that had been mated to WT or hSTAT2 KI sires, respectively (fetuses are homozygous WT or STAT2 KI) were inoculated with ZIKV-Dak or ZIKV-Dak-MA via subcutaneous route on day E6.5, and maternal and fetal tissues were collected on E13.5. Higher levels of ZIKV-Dak-MA RNA were present in maternal serum and spleen in hSTAT2 KI compared to WT dams (Fig 6J–K). ZIKV-Dak-MA infected the placenta and fetal head to significantly higher levels in hSTAT2 KI compared to WT mice (Fig 6L–M). Indeed, ZIKV RNA was detected in the junctional layer of the placenta and uterine lining from hSTAT2-infected dams (Fig S4). In comparison, hSTAT2 KI dams infected with parental ZIKV-Dak had lower viral RNA levels in maternal and fetal tissues (Fig S5A–E) than ZIKV-Dak-MA infected hSTAT2 KI dams (Fig 6J–M). Expression of the human STAT2 allele for transmission was important for the dam, as placentas and fetuses from hSTAT2 KI female mice mated to WT males (fetuses are heterozygous for hSTAT2 allele) also accumulated high levels of ZIKV (Fig S5F–J); this data suggests that evasion of IFN signaling by ZIKV is necessary for efficient dissemination of ZIKV to the maternal-fetal interface but is largely dispensable for infection of the placenta and fetus. Thus, by combining the adapted ZIKV strain with hSTAT2 KI mice (Fig S5K), we generated lethality and placental transmission models in immunocompetent mice after subcutaneous inoculation
DISCUSSION
ZIKV has emerged as an important global health concern due to its teratogenic potential. With no currently approved vaccine or treatment, there is an urgent need for the development of animal models that recapitulate key features of human disease to understand the biology of this unique flavivirus and facilitate the development of countermeasures. Here, we generated an adapted ZIKV strain through passage in Rag1−/− mice. This strain had one critical mutation in the NS4B gene, which allowed it to replicate to higher titers in NSCs and in the brains of mice, and to some degree evade type I IFN responses. The NS4B G18R substitution was necessary and sufficient to enhance the pathogenicity of both African and Brazilian strains of ZIKV in mice. Single-cell RNAseq analysis of ZIKV-Dak and ZIKV-Dak-MA infected NSCs revealed transcriptional changes between infected and uninfected cells and confirmed a reduced type I IFN response in ZIKV-Dak-MA infected cells. We combined the pathogenic features of this adapted strain with hSTAT2 KI mice, which overcome the inherent inability of ZIKV strains to bind and degrade mouse Stat2 (Bowen et al., 2017; Grant et al., 2016; Kumar et al., 2016), to generate an immunocompetent small animal model of ZIKV pathogenesis.
RNA viruses rapidly mutate to overcome selective immune or drug pressures, gain host tropism, or acquire immune evasive functions. Several recent studies have identified mutations in ZIKV that correlate with its epidemic emergence. One report used phylogenetic analysis to predict positively-selected mutations in the N-terminus (residue 26) and second ER loop (residues 87 and 88) of NS4B (Sironi et al., 2016), regions that inhibit type I IFN responses and STING activity and also bind NS1 (Muñoz-Jordán et al., 2005; Youn et al., 2012). A second study used an evolutionary approach to identify two amino acid substitutions in NS4B (V180I and L182S) present in epidemic strains (Zhu et al., 2016). Neither of these studies identified the NS4B G18 residue as under strong positive selection, and indeed G18R substitution is absent from all historical and circulating ZIKV strains. Several explanations for this are possible, including that residue 18 of NS4B is under strong purifying selection due to compromising effects on virulence in the epidemic or enzootic human, non-human primate, or mosquito hosts. Alternatively, the substitution could be specific for certain hosts, which might explain why some mosquito and tick-borne flaviviruses with other natural reservoirs (e.g., rodent or marsupial) have an arginine at this position (Fig S1), and there is no selection pressure to retain it in human/non-human primate/mosquito transmission cycles.
The flavivirus NS4B protein contributes to remodeling of the endoplasmic reticulum, which facilitates viral replication (Kaufusi et al., 2014; Zmurko et al., 2015) and modulation of host defense responses. Dengue virus NS4B inhibits IFN-α/β induction by altering mitochondrial morphology and RIG-I activation or by inhibiting TBK1 and IRF3 activation (Chatel-Chaix et al., 2016; Dalrymple et al., 2015). ZIKV NS4B also has been reported to diminish innate immune responses by preventing or altering activation of TBK1 (Onorati et al., 2016; Wu et al., 2017). Our studies with ZIKV-Dak and ZIKV-Dak-NS4B G18R in Ifnar1−/− mice, anti-Ifnar1 mAb treated NSCs, and IFN-β-pretreated NSCs suggest that the adaptive substitution in NS4B does not affect sensitivity to type I IFN or ISGs but rather impacts IFN-β induction. Our single-cell RNA-seq data in murine NSCs showed decreased ISG induction in ZIKV-Dak-NS4B G18R-infected cells at 24 and 48 h despite higher levels of intracellular viral RNA and extracellular virus compared to ZIKV-Dak WT. Because we distinguished transcriptional signatures of uninfected and ZIKV-infected cells, we were able to observe that higher levels of ISGs were induced in uninfected NSCs from the ZIKV-Dak WT group compared to ZIKV-Dak NS4B G18R group, which likely rendered the uninfected cells in ZIKV-Dak WT group less permissive (Fig S3B). Our data is consistent with a recent study in human dendritic cells showing that ZIKV evades type I IFN immunity in part by diminishing translation of IFN-β protein (Bowen et al., 2017). Given that IFN-β mRNA levels were similar in ZIKV-Dak-MA and ZIKV-Dak-NS4B G18R-infected cells, we speculate that the NS4B G18R mutation may enhance the efficiency of a ZIKV-induced IFN-β translational block. The precise means by which changes in NS4B sequence differentially affect IFN-β translation remains undetermined, as the mechanism by which ZIKV diminishes translation of IFN-β protein (Bowen et al., 2017) is unknown.
One caveat to our experiments with ZIKV-Dak and ZIKV-Dak-MA is the relative disparity of phenotype between peripheral organs and the brain after subcutaneous virus inoculation. As many of these studies were performed in anti-Ifnar1-mAb treated mice, the differences in viral titers in peripheral organs (serum and spleen) are small, as expected, between the WT and adapted ZIKV virus, analogous to our NSC experiments (see Fig 5J). In comparison, because, anti-Ifnar1 mAb does not appreciably cross the blood-brain-barrier, the phenotype of the mutant ZIKV-Dak-MA is more prominent in the brain. Notwithstanding these data, ZIKV-Dak-MA still replicated slightly more effectively than ZIKV-Dak in the CNS of Ifnar1−/− mice after direct intracranial inoculation, suggesting there may be an additional pathway, independent of type I IFN signaling, by which the adaptive mutation functions; NS4B G18R could antagonize or co-opt other host defense pathways including type II or III IFN signaling or autophagy in cells of the brain. Indeed, ZIKV NS4B reportedly modulates Akt-mTOR signaling and autophagy induction (Liang et al., 2016), and autophagy confers a proviral advantage for ZIKV (Cao et al., 2017).
Most adult mouse models of ZIKV infection require eliminating or inhibiting the mouse type I IFN response to achieve infection and pathogenesis (reviewed in Morrison and Diamond, 2017). Nonetheless, several immunocompetent mouse models of ZIKV infection have been described, although each has limitations. These include: (a) peripheral infection of neonatal (<1 week-old) wild-type mice (Lazear et al., 2016; Manangeeswaran et al., 2016), which have immature immune responses; (b) direct intrauterine injection of wild-type pregnant dams (Vermillion et al., 2017), which bypasses trans-placental transmission; (c) direct injection of fetal brains during pregnancy (Wu et al., 2016), which bypasses the placenta entirely; and (d) administration of high-dose ZIKV via an intravenous route to pregnant dams (Cugola et al., 2016), which bypasses peripheral immune responses. To overcome the lack of Stat2 antagonism, which limits ZIKV replication in the peripheral tissues likely due to sustained type I and III IFN responses, we engineered a transgenic mouse that replaced most of mouse Stat2 with human STAT2. This mouse showed normal immune responses to poly(I:C), a stimulus that activates IFN induction. ZIKV, especially the NS4B G18R adapted strain, was able to replicate, spread to the brain, and cause lethality in juvenile (3 week-old) hSTAT2 KI mice, and result in transplacental transmission and fetal infection in pregnant dams. Of note, the pregnant dams themselves, despite vertically transmitting ZIKV, did not sustain infection in the brain, which is characteristic of human infection. Thus, ZIKV infection in adult hSTAT2 KI mice also did not recapitulate the extreme vulnerability and lethality of Ifnar1−/− (Lazear et al., 2016) or Stat2−/− mice (Tripathi et al., 2017); this could reflect cell-extrinsic antiviral effects of IFN signaling in uninfected cells (as seen in our single-cell RNA-seq data) or the lack of additional species-specific evasion mechanisms that are not accounted for by the NS4B mutation (e.g., DENV antagonizes human but not mouse STING (Aguirre et al., 2012)). Alternatively, the absence of brain infection in adult hSTAT2 KI mice could reflect the age-dependent maturation of the blood-brain barrier and the tightening effects of type I and III IFNs, both of which signal through Stat2 (Daniels et al., 2014, 2017), which would be absent in Ifnar1−/− and Stat2−/− mice.
In summary, we identified an adaptive mutation in ZIKV NS4B that confers enhanced virulence in mice and NSCs for African and American ZIKV strains. We applied single-cell RNA-seq analysis in uninfected and ZIKV-infected NSCs to define unique cell-intrinsic transcriptional signatures, and used this information to establish mechanistically how the adaptive NS4B mutation enhanced infectivity and virulence. By combining the adapted virus with transgenic mice that exchanged human and mouse STAT2, which restricts ZIKV infection in a species-dependent manner, we developed an immunocompetent model of ZIKV infection and pathogenesis after peripheral inoculation that more closely recapitulates features of human infection. This immunocompetent small animal model of ZIKV infection may prove a resource for determining mechanisms of pathogenesis, defining correlates of innate and adaptive immune protection, and evaluating countermeasures to limit disease.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Michael S. Diamond (diamond@wusm.wustl.edu).
Experimental Model and Subject Details
Animals and ethics
Wild-type (WT) C57BL/6 (000664) mice were purchased from Jackson Laboratory. Congenic Rag1−/−, Ifnar1−/−, Stat2−/− (Park et al., 2000), and hSTAT2 KI C57BL/6 mice were bred in a specific-pathogen-free facility at Washington University or the Icahn School of Medicine at Mount Sinai. 4 to 6 week-old mice, or 11 week old pregnant female mice were used. Littermates of the same sex were randomly assigned to experimental groups. This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance number A3381-01). Animal studies also were approved by the Institutional Animal Care and Use Committee of Icahn School of Medicine at Mount Sinai (Assurance number A311-01). Virus inoculations were performed under anaesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering.
Cell lines
Vero cells were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 5% fetal bovine serum (FBS) and 25mM HEPES.
Primary cell cultures
NSCs were isolated from 4 to 6 week-old WT C57BL/6 mice. NSCs were cultured in Neurobasal media (Thermo Fisher) without phenol red, supplemented with B27 without vitamin A (Thermo Fisher) and N-2 supplement (Thermo Fisher) at 37°C in 5% CO2. NSCs were plated at 105 cells in 2 ml of Neurobasal media per well in 6 well plates treated with matrigel (Corning, 08-774-552).
Viruses
ZIKV strain Dakar 41525 (Senegal, 1984, GenBank: KU955591) was provided by the World Reference center for Emerging Viruses and Arboviruses (R. Tesh, University of Texas Medical Branch). ZIKV stocks were propagated in Vero cells, and cell supernatants were harvested 66–72 h post inoculation. Virus stocks were titrated by focus forming assay (FFA) on Vero cells as previously described (Lazear et al., 2016) and stored aliquoted at −80°C.
Method Details
Mouse experiments
In some experiments, mice were treated with 2 mg of an Ifnar1-blocking antibody (MAR1-5A3, Leinco Technologies) by intraperitoneal injection one-day before virus infection. Mice were inoculated with ZIKV by a subcutaneous (via footpad) route with 104 to 106 FFU PFU of ZIKV in a volume of 50 μl PBS or by an intracranial route with 104 FFU of ZIKV in a volume of 10 μl PBS. For survival studies, mice were monitored for 21–45 days. For enhanced safety, all mouse infection experiments with ZIKV were performed under A-BSL3 conditions.
hSTAT2 KI mouse generation
The hSTAT2 KI mouse line was generated commercially by TaconicArtemis Gmbh. The targeting vector was constructed using bacterial artificial chromosome clones C57BL/6J RPCIB-731 and human RPCIB-753, transfected into the TaconicArtemis C56BL/6N Tac ES cell line, and selected with neomycin and puromycin. The integration in ES cells was confirmed by Southern blotting and PCR. Germline pups were bred to C57BL/6-Tg(CAG-Flpe)2 Arte (Schaft et al., 2001) for removal of the neomycin and puromycin cassettes, and the Flpe transgene was bred away by backcrossing. Mice were genotyped with primers CTGAGGTAGAATCACTTTGACTTCC-5079-42 and aGATGGCTCAGAGGTTAAGAGC-5079-43, or GGCAAAGCCAAGACATAAACC-5080-46 and ACAGGTTCCAGGCCATCAAG-5080-47 and PCR conditions: 94°C 30 sec, 60°C 30 sec, 72°C 1 min. Western blotting for human STAT2 (Santa Cruz sc-476) or mouse Stat2 (Cell signaling 4597) was performed on splenocytes isolated 24 h after intravenous injection of 100 μg of poly(I:C).
IFN responses in mice
WT, hSTAT2 KI, and Stat2−/− mice were administered 50 μg of poly(I:C) (Invivogen, tlrl-pic) via an intranasal route in 50 μl of PBS, and lungs were harvested 6 h later. Lungs were homogenized in RLT buffer (Qiagen RNAeasy kit), and total RNA was isolated from 200 μl of tissue homogenate. RT-PCR was performed as described (Tripathi et al., 2017). ISGs were detected with the following primer sets: mouse Oas1a Forward 5′-ATGGAGCACGGACTCAGGA-3′ mouse Oas1a Rev 5′-TCACACACGACATTGACGGC-3′; mouse Ifnβ1 Forward 5′-CAGCTCCAAGAAAGGACGAAC-3′ mouse Ifnβ1 Reverse 5′-GGCAGTGTAACTCTTCTGCAT-3′; mouse Irf7 Forward 5′-GAGACTGGCTATTGGGGGAG-3′ mouse Irf7 Reverse 5′-GACCGAAATGCTTCCAGGG-3′; mouse 18s rRNA Forward 5′- GTAACCCGTTGAACCCCATT-3′ mouse 18s rRNA Reverse 5′-CCATCCAATCGGTAGTAGCG-3′
Viral burden measurements
ZIKV-infected mice were anesthetized with ketamine, blood was collected, and animals were perfused with at least 15 ml of PBS at specified time points after infection. The spleen and brain were harvested, weighed, and homogenized by bead dissociation using a MagNA Lyzer (Roche) in 1 ml of Dulbecco’s Modified Eagle’s Medium (DMEM) containing 2% fetal bovine serum (FBS). Blood was allowed to clot at room temperature and serum was separated by centrifugation. Viral RNA was isolated using the RNeasy 96 Kit (Qiagen) as described by the manufacturer. ZIKV RNA levels were determined by TaqMan one-step quantitative reverse transcriptase PCR (qRT-PCR) as described previously (Govero et al., 2016). A published primer set (Govero et al., 2016) was used to detect ZIKV RNA: Forward 5′-CCACCAATGTTCTCTTGCAGACATATTG-3′; Reverse 5′- TTCGGACAGCCGTTGTCCAACACAAG-3′; Probe 5′-/56-FAM/AGCCTACCT/ZEN/TGACAAGCAGTC/3IABkFQ/-3′ (Integrated DNA Technologies). For some samples, viral titer was determined by plaque assay as described (Lazear et al., 2016). Briefly, brains were harvested, weighed, and homogenized by bead dissociation using a MagNA Lyzer (Roche) in 1 ml of DMEM containing 2% FBS. Samples were clarified by centrifugation (2,000× g at 4°C for 10 min), diluted serially, and then add ed to Vero cell monolayers in 6 well plates. Cells were overlaid with 2% low melting point agarose (Lonza) and 4 days later were fixed with 10% formaldehyde and stained with crystal violet for manual counting on a lightbox.
NSC isolation and infection
NSCs were isolated from 4 to 6 week-old WT C57BL/6 mice. The subventricular zone of the brain was digested in 0.05% trypsin (Invitrogen) for 30 min at 37°C. Cells were resuspended after pipetting up and down twenty times. NSCs were cultured in Neurobasal media (Thermo Fisher) without phenol red, supplemented with B27 without vitamin A (Thermo Fisher) and N-2 supplement (Thermo Fisher) at 37°C in 5% CO2. NSCs were plated at 105 cells in 2 ml of Neurobasal media per well in 6 well plates treated with matrigel (Corning, 08-774-552) and inoculated with ZIKV at an MOI of 0.01 or 1. For some experiments, NSCs were pretreated with 25 μg/mL of Ifanr1 blocking antibody (MAR-1 5A3), 3 or 10 U/mL of IFN-β (PBL Assay science, 12400-1) for 16 h before infection. Supernatants were harvested and medium was replaced at indicated time points. Viral burden in the supernatant was determined by focus-forming assay (FFA). NSCs were harvested at 96 h after infection, fixed in Foxp3/transcription factor staining buffer (eBioscience), and stained with a combination of anti-ZIKV murine mAbs (ZIKV-2 and ZIKV-13) or only CHK-152 as an isotype control using 200 ng of mAb per 50 μl. Cells were then incubated with goat anti-mouse Alexa Fluor 647 antibody (ThermoFisher Scientific) and analyzed on a MACsQuant8 flow cytometer (Miltenyi Biotec).
IFN-β production in NSCs during ZIKV infection
NSCs were cultured in Neurobasal media without phenol red, supplemented with B27 without vitamin A and N-2 supplement at 37°C in 5% CO2. NSCs were plated at 105 cells in 2 mL of Neurobasal media per well in 6 well plates treated with matrigel and inoculated with ZIKV at an MOI of 1. After 4 h, cells were washed three times in PBS, and media was replaced with 2 mL of fresh Neurobasal media. At the indicated time points, supernatant was collected and Ifnβ in the supernatant was measured using VeriKine™ Mouse Ifnβ ELISA (PBL Assay Science, 42400-1) according to the manufacturer’s protocol. In brief, 100ul of supernatant was added to precoated wells and incubated for 1 h at room temperature. Detection, development, and washing steps were performed as described by the manufacturer’s protocol. OD450 was recorded using a Tristar LB 941 reader (Berthold Technologies). Ifnβ concentration was determined by comparing OD450 values to a standard curve. Limit of detection (LOD) was determined by calculating the mean and standard deviation (SD) of blank controls, and setting the LOD at three SDs above the mean of the blank controls.
Cell viability assay
Cell viability was measured using CellTiter-Glo reagents (Promega) according to the manufacturer’s protocol. Briefly, NSCs were plated at 105 cells in 2 mL of Neurobasal media in 6 well plates treated with matrigel. Cells were inoculated with ZIKV at an MOI of 1. Ninety-six hours after infection, 1.7 ml of media was removed and 300 μl of CellTiter-Glo reagent was added to each well. Plates were incubated for 15 min at room temperature and luminescence was recorded using a Synergy H1 Hybrid Reader (Bio-tek).
Next-generation sequencing of ZIKV
Preparation of sequencing libraries was performed as described (Miner et al., 2016). Briefly, RNA was extracted from the input or passaged virus, reverse-transcribed into cDNA, and then amplified in eight segments using Q5 DNA polymerase (New England Biolabs). DNA was fragmented using dsDNA fragmentase (New England Biolabs), and adapters and indexes were added to prepare Illumina compatible libraries using the NEBNext Ultra II DNA library prep kit (New England Biolabs). Libraries were sequenced on an Illumina Nextseq 500. The demultiplexed fastq files for the input were aligned to the ZIKV genome (Strain ArD_41519, Accession HQ234501) to create a consensus sequence. The input consensus sequence was used as a reference file to assess mutations present in the passaged virus. Variant calling was performed with LoFreq (Wilm et al., 2012) and consensus sequences were generated with Geneious software. All sequences have been uploaded and will be published upon publication of a finalized manuscript.
ZIKV-Dak and ZIKV-Bzl infectious clones
We developed an infectious clone utilizing a published reverse genetics platform (Widman et al., 2017). We applied a unidirectional quadripartite assembly method using asymmetric, nonpalindromic, naturally encoded restriction enzyme sites located near known centers of toxicity for bacterial expression (Messer et al., 2012; Widman et al., 2017). Cleavage sites were located at nucleotide positions 3064, 4451, and 7220 of the ZIKV-Dak (strain 41525) genome and resulted in four fragments for expression in bacterial plasmids. Non-native modifications added to the consensus sequence included hepatitis D virus ribozyme following the native 3′ untranslated region (UTR) as well as a T7 promotor upstream of the 5′-UTR for in vitro transcription. Site-directed mutagenesis overlap PCR was performed to generate mutations at positions NS3 K339R and/or NS4B G18R in ZIKV-Dak-WT and ZIKV-Bzl-WT clones (Widman et al., 2017). Screening was performed via Sanger sequencing to confirm mutagenesis as well as genetic integrity of the rest of the fragment.
Bacterial cultures of MC1061 or Top10 competent E. coli cells containing the quadripartite wild-type or mutant fragments were grown overnight at 27.6°C in appropriate selection growth media. Plasmids were isolated and digested according to molecular construction design, and ligated together overnight at 4°C. After ligation, DNA was precipitated via chloroform extraction and alcohol precipitation. The DNA pellet was resuspended in molecular grade water, and used as template for RNA transcription via T7 polymerase (Ambion). Reactions were incubated at 30°C or 37°C for 3–4 h and then electroporated into Vero cells. After 2–4 days, depending on cytotoxicity, supernatant containing virus was collected and filtered through 0.22 μM filters. After harvest and filtration, viral RNA was isolated (Qaigen Viral Mini RNA Kit), transcribed into cDNA via RT-PCR (Invitrogen), and sequenced via whole genomic Sanger Sequencing to ensure identity. Infectious clone generated viral titers were comparable to the passaged natural isolates.
Single cell RNA-seq
NSCs were resuspended at 103 cells/μL in PBS containing 0.04% (w/v) BSA to minimize cell loss and aggregation. ~17,500 cells were partitioned into nanoliter-scale Gel Bead-In-EMulsions (GEMs) to achieve single cell resolution for a maximum of 10,000 individual cells per sample. Utilizing the v2 Chromium™ Single Cell 3′ Library Kit and Chromium instrument (10x Genomics), poly-adenylated mRNA from an individual cell was tagged with a unique 16 basepair 10x barcode and 10 basepair Unique Molecular Identifier. Full length cDNA was amplified to generate sufficient mass for library construction. Enzymatic fragmentation and size selection were used to optimize the cDNA amplicon size (~400 basepair) for the library. The final library was sequence-ready and contained four unique sample indexes. The concentration of the 10x single cell library was determined through qPCR (Kapa Biosystems). The libraries were normalized, pooled, and sequenced with a custom recipe (26-8-98) on the HiSeq4000 platform (Illumina). Five single cell libraries were sequenced across an entire HiSeq4000 flow cell targeting ~45,000 reads per cell.
Alignment, barcode assignment and unique molecular identifier (UMI) counting
The Cell Ranger Single-Cell Software Suite (version 2.0.2) (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger) was used to perform sample demultiplexing, barcode processing, and single-cell 3′ counting. Cellranger mkfastq was used to demultiplex raw base call files from the HiSeq4000 sequencer into sample-specific fastq files. Files was demultiplexed with 98%+ perfect barcode match, and 74%+ q30 reads. Subsequently, fastq files for each sample were processed with cellranger counts to align reads to the mm10 mouse genome. The default estimated cell count value of 10,000 was used for this experiment. Samples were subsampled to have equal numbers of confidently mapped reads per cell. A separate run of cellranger count was performed to align reads to the ZIKV Dakar 41525 genome (GenBank: HQ234501.1) to obtain viral UMI counts.
Preprocessing analysis with Seurat package
For the analysis, the Seurat package (version 2.1) (Butler and Satija, 2017) was used. Cell Ranger filtered genes by barcode expression matrices were used as analysis inputs. Samples were pooled together using the AddSample function. The fraction of mitochondrial genes was calculated for every cell, and cells with a mitochondrial fraction > 5% were filtered out. Two sources of unwanted variation:UMI counts and fraction of mitochondrial reads -- were removed with RegressOut function. Expression measurements for each cell were normalized by total expression and then scaled to 10,000, after that log normalization was performed. Viral UMI counts were analyzed separately from reads mapped to the mm10 genome. Viral UMI counts were normalized to the total UMI count within the cell (both viral and non-viral) and then scaled to 10,000, and then log-normalization was performed. ZIKV-infected cells were defined arbitrarily as having greater than 0.03% of viral transcripts relative to total cellular transcripts since the background read level in naïve, uninfected cells was less than 0.02% of total cellular transcripts.
Dimensionality reduction and clustering
The most variable genes were detected using FindVariableGenes function. PCA was run only using these genes. Cells are represented with t-SNE (t-distributed Stochastic Neighbor Embedding) plots. We applied RunTSNE function to normalized data, using first 10 PCA components. For clustering, we used function FindClusters that implements SNN (shared nearest neighbor) modularity optimization based clustering algorithm on 10 PCA components with a resolution of 0.2. Seven clusters were detected, one of which contained poorly covered cells (lower number of UMIs and detected genes); this cluster was excluded from further analysis.
Identification of cluster-specific genes and marker-based classification
To identify marker genes, FindAllMarkers function was used with likelihood-ratio test for single cell gene expression. For each cluster only genes that were expressed in more than 25% of cells with at least 0.25-fold difference were considered. To characterize clusters, we used the following markers. NSCs: Id3, Clu, Rpl32, Egfr, Dlx2, Nes, Sox2, Gfap, and Rbfox3; Cell cycle: Cdk4, Cdk1, Tipin, Cenpa, Pcna, Mki67, Top2a, and Mcm5; ISGs: Ddx58, Ifih1, Rsad2, Isg15, Ccl5, and Cxcl10; and Viral infection: ZIKV RNA. NSC and cell cycle markers were selected based on published data (Dulken et al., 2017; Kowalczyk et al., 2015).
Single cell RNA-seq differential expression
To obtain differential expression between infected cells for 24 or 48 h with ZIKV-Dak-WT vs ZIKV-Dak-MA, a MAST test was performed and P value adjustment was performed using a Bonferroni correction (Finak et al., 2015).
qRT-PCR of IFN-β mRNA
NSCs were cultured in Neurobasal media without phenol red, supplemented with B27 without vitamin A and N-2 supplement at 37°C in 5% CO2. NSCs were plated at 105 cells in 2 ml of Neurobasal media per well in 6 well plates treated with matrigel and inoculated with ZIKV at an MOI of 1. After 4 h, cells were washed three times in PBS, and media was replaced with 2 ml of fresh Neurobasal media. At specified time points, supernatant was removed and cells were lysed in 500 μL of RLT. RNA was extracted using an RNeasy Mini Kit (Qiagen, 74104) and on-column DNase digestion was performed using RNase-Free DNase set (Qiagen, 79254) following the manufacturers protocol. 18S ribosomal and Ifnβ, were determined by Taqman one-step quantitative reverse transcriptase PCR (qRT-PCR). 18S ribosomal RNA was measured using an Eukaryotic 18S rRNA endogenous control (Thermo Fisher Scientific, 4319413E), and Ifnβ mRNA levels were determined using predesigned qPCR assay (IDT, Mm.PT.58.30132453.g). Gene expression was normalized to the 18S ribosomal housekeeping gene, and the 2−ΔΔCt method was used to calculate fold expression over mock.
Sequence alignment for ZIKV and related flaviviruses
Sequences for ZIKV strains and flavivirus strains were obtained from the NIAID Virus Pathogen Database and Analysis Resource (ViPR) (Pickett et al., 2012) through the website https://www.viprbrc.org/brc/vipr_genome_search.spg?method=ShowCleanSearch&decorator=flavi. Sequences were aligned using Geneius (http://www.geneious.com, version 11.0.4 (Kearse et al., 2012)) and MUSCLE sequence alignment algorithm. ZIKV-Dakar was used as the reference sequence to determine the position of amino acids.
Sanger sequencing of passaged ZIKV isolates
RNA from viral tissues was isolated using RNeasy Mini Kit (Qiagen, 74104) as described in the manufactures protocol. cDNA was generated using SuperScript III first-strand synthesis supermix (Thermo Fisher Scientific, 18080400), and cDNA was amplified using Phusion High Fidelity PCR Master Mix (New England Biolabs, M0531S). Primer set used to amplify cDNA ZIKV-Dakar 3500–7000 amplifying primer: 5′- AGA AAG GAA CCA GAG AGC AAC TTA-3′ ZIKV-Dakar 7100-3500 amplifying primer: 5′- GTGGTTACCGCATGTTGG-3′. Sanger sequencing was performed by Genewiz (https://www.genewiz.com/en/Public/Services/Sanger-Sequencing), and sequences were aligned by the ClustaW Method using DNAstar Megalign (https://www.dnastar.com/t-megalign.aspx). Primer set used to sequence regions of interest ZIKV-Dakar 5500 sequencing primer: 5′-TGCACGAGGATATATATCAACAAGGG-3′ ZIKV-Dakar 6500 sequencing primer: 5′- TTG CCA GGA CAC ATG ACA GAG-3′.
Viral RNA in situ hybridization
RNA ISH was performed using RNAscope 2.5 HD (Brown) (Advanced Cell Diagnostics) according to the manufacturer’s instructions. PFA-fixed, paraffin-embedded tissue sections were deparaffinized by incubating for 60 min at 60°C and endogenous peroxidases were quenched with H2O2 for 10 min at room temperature. Slides were then boiled for 15 min in RNAscope Target Retrieval Reagents and incubated for 30 min in RNAscope Protease Plus reagent prior to addition of a custom-designed ZIKV RNA probe (Advanced Cell Diagnostics, catalog #467771). Sections were counterstained with Gill’s hematoxylin and visualized by brightfield microscopy.
Quantification and Statistical Analysis
Statistical details of experiments are found in the corresponding Figure Legends. No statistical methods were used to predetermine sample size a priori and investigators were not blinded to allocation during experiments and outcome assessment. Survival experiments were analyzed by Kaplan-Meier survival analysis and the log-rank test. Viral burden in tissues was analyzed by the Mann-Whitney test. Viral growth kinetics was analyzed by two-way ANOVA with multiple comparison corrections as needed. The percentage of infected NSCs was analyzed by a student t-test. IFN-β production was analyzed by a student t-test. ISGs expression in infected NSCs was analyzed by a MAST test. Differential expression of viral RNA was analyzed by Mann-Whitney test. Cell viability data was analyzed by a one-way ANOVA. Statistical analyses were conducted with Prism 6 software (Graphpad). P values of < 0.05 were considered statistically significant.
Data and Software Availability
The single-cell RNAseq data can be accessed at Gene Expression Omnibus (accession number: GSE112711). The next-generation sequencing of ZIKV-Dakar MA has been deposited in the SRA database as BioProject ID: PRJNA413540. Consensus sequences for ZIKV-Dak (accession number: MG758785) and ZIKV-Dak MA (accession number: MG758786) have been uploaded in Genbank.
Supplementary Material
HIGHLIGHTS.
A mouse adapted ZIKV (Dak-MA) with enhanced brain and NSC infectivity generated
NS4B G18R mutation enhances pathogenicity of African and American ZIKV strains
Immunocompetent human STAT2 knock-in mice generated that are susceptible to ZIKV-Dak-MA
ZIKV-Dak-MA spreads to the placenta and fetus in immunocompetent hSTAT2 knock-in mice
Acknowledgments
We acknowledge the Genome Technology Access Center at Washington University for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 and by ICTS/CTSA Grant# UL1 TR000448. This work also was supported by grants from the NIH (R01 AI073755, R01 AI104972, and U19 AI083019 to M.S.D.; R01 HD091218 to I.U.M. and M.S.D.; U19 AI118610 and R21 AI129486 to A.G.-S; and R01 AI100625 and R01 AI107810 to R.S.B.) and by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH. K.Z. was supported by Government of Russian Federation grant 074-U01.
Footnotes
AUTHOR CONTRIBUTIONS
M.J.G., E.A.C, K.Z., J.W-L., M.B.U., S.T., G.E., S.M.B., M.A., and M.S.D designed the experiments. M.J.G, E.A.C., J.W-L., C.R., M.Y., B.C., S.J.R., K.L.M., M.U., and S.T. performed the experiments. J.M. contributed to the generation of the hSTAT2 KI mice. M.C.B., K.H.D.III, B.L.Y., Z.Z., J.Y., R.S.B, A.G-S. contributed key reagents including recombinant viruses. M.J.G., E.A.C, K.Z., M.B.U., B.C., I.U.M., S.T., J.W-L., C.R., G.E. analyzed the data. M.J.G. and E.A.C wrote the first draft, with M.S.D. providing major editorial comments. All authors participated in editing the final version of the manuscript.
DECLARATION OF INTERESTS
M.S.D. is a consultant for Inbios, Aviana, and Sanofi-Pasteur, and is on the Scientific Advisory Board of Moderna.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012;8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliota MT, Caine EA, Walker EC, Larkin KE, Camacho E, Osorio JE. Characterization of Lethal Zika Virus Infection in AG129 Mice. PLoS Negl Trop Dis. 2016;10:e0004682. doi: 10.1371/journal.pntd.0004682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017;13:e1006164. doi: 10.1371/journal.ppat.1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Satija R. Integrated analysis of single cell transcriptomic data across conditions, technologies, and species. bioRxiv. 2017;21:627–636. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Parnell LA, Diamond MS, Mysorekar IU. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J Exp Med. 2017;214:2303–2313. doi: 10.1084/jem.20170957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao-Lormeau V-M, Roche C, Teissier A, Robin E, Berry A-L, Mallet H-P, Sall AA, Musso D. Zika virus, French polynesia, South pacific, 2013. Emerg Infect Dis. 2014;20:1085–1086. doi: 10.3201/eid2006.140138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatel-Chaix L, Cortese M, Romero-Brey I, Bender S, Neufeldt CJ, Fischl W, Scaturro P, Schieber N, Schwab Y, Fischer B, et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe. 2016;20:342–356. doi: 10.1016/j.chom.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavali PL, Stojic L, Meredith LW, Joseph N, Nahorski MS, Sanford TJ, Sweeney TR, Krishna BA, Hosmillo M, Firth AE, et al. Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science. 2017;357:83–88. doi: 10.1126/science.aam9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JLM, Guimarães KP, Benazzato C, Almeida N, Pignatari GC, Romero S, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. 2016;534:267–271. doi: 10.1038/nature18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple NA, Cimica V, Mackow ER. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. MBio. 2015;6:e00553–15. doi: 10.1128/mBio.00553-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. MBio. 2014;5:e01476–14. doi: 10.1128/mBio.01476-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, White JP, Lazear HM, Gale M, Diamond MS, Klein RS. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J Clin Invest. 2017;127:843–856. doi: 10.1172/JCI88720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulken BW, Leeman DS, Boutet SC, Hebestreit K, Brunet A. Single-Cell Transcriptomic Analysis Defines Heterogeneity and Transcriptional Dynamics in the Adult Neural Stem Cell Lineage. Cell Rep. 2017;18:777–790. doi: 10.1016/j.celrep.2016.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govero J, Esakky P, Scheaffer SM, Fernandez E, Drury A, Platt DJ, Gorman MJ, Richner JM, Caine EA, Salazar V, et al. Zika virus infection damages the testes in mice. Nature. 2016;540:438–442. doi: 10.1038/nature20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe. 2016;19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann DL, Hodgson A, Sall AA, Freedman DO, Staples JE, Althabe F, Baruah K, Mahmud G, Kandun N, Vasconcelos PFCC, et al. Zika virus and microcephaly: why is this situation a PHEIC? Lancet (London, England) 2016;387:719–721. doi: 10.1016/S0140-6736(16)00320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joguet G, Mansuy J, Matusali G, Hamdi S, Walschaerts M, Pavili L, Guyomard S, Prisant N, Lamarre P, Dejucq-rainsford N, et al. Effect of acute Zika virus infection on sperm and virus clearance in body fluids: a prospective observational study. Lancet Infect Dis. 2017;3099:1–9. doi: 10.1016/S1473-3099(17)30444-9. [DOI] [PubMed] [Google Scholar]
- Kaufusi PH, Kelley JF, Yanagihara R, Nerurkar VR. Induction of endoplasmic reticulum-derived replication-competent membrane structures by West Nile virus non-structural protein 4B. PLoS One. 2014;9:e84040. doi: 10.1371/journal.pone.0084040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, Regev A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25:1860–1872. doi: 10.1101/gr.192237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016;17:1766–1775. doi: 10.15252/embr.201642627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS, Fernandez E, Miner JJ, Diamond MS. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe. 2016;19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin C, Xu Z. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell. 2016a;19:120–126. doi: 10.1016/j.stem.2016.04.017. [DOI] [PubMed] [Google Scholar]
- Li H, Saucedo-Cuevas L, Regla-Nava JA, Chai G, Sheets N, Tang W, Terskikh AV, Shresta S, Gleeson JG. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell. 2016b;19:593–598. doi: 10.1016/j.stem.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, Ge J, Wang S, Goldman SA, Zlokovic BV, et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell. 2016;19:663–671. doi: 10.1016/j.stem.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Li S, Ma S, Ma W, Li S, Ma S, Jia L, Zhang F, Zhang Y, Zhang J. Zika Virus Causes Testis Damage and Leads to Male Infertility in Mice Article Zika Virus Causes Testis Damage and Leads to Male Infertility in Mice. Cell. 167:1511–1518.e10. [Google Scholar]
- Manangeeswaran M, Ireland DDC, Verthelyi D. Zika (PRVABC59) Infection Is Associated with T cell Infiltration and Neurodegeneration in CNS of Immunocompetent Neonatal C57Bl/6 Mice. PLoS Pathog. 2016;12:e1006004. doi: 10.1371/journal.ppat.1006004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs C, Olson G, Saade G, Hankins G, Wen T, Patel J, Weaver S. Zika Virus and Pregnancy: A Review of the Literature and Clinical Considerations. Am J Perinatol. 2016;33:625–639. doi: 10.1055/s-0036-1580089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath EL, Rossi SL, Gao J, Widen SG, Grant AC, Dunn TJ, Azar SR, Roundy CM, Xiong Y, Prusak DJ, et al. Differential Responses of Human Fetal Brain Neural Stem Cells to Zika Virus Infection. Stem Cell Reports. 2017;8:715–727. doi: 10.1016/j.stemcr.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Labeau A, Dejarnac O, Gressens P, Schwartz O, Amara A, Meertens L, Labeau A, Dejarnac O, Cipriani S, et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses Report Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. 2017:324–333. [Google Scholar]
- Messer WB, Yount B, Hacker KE, Donaldson EF, Huynh JP, de Silva AM, Baric RS. Development and characterization of a reverse genetic system for studying dengue virus serotype 3 strain variation and neutralization. PLoS Negl Trop Dis. 2012;6 doi: 10.1371/journal.pntd.0001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JJ, Sene A, Richner JM, Ebel GD, Diamond MS, Apte RS, Miner JJ, Sene A, Richner JM, Smith AM, et al. Zika Virus Infection in Mice Causes Panuveitis with Shedding of Virus in Tears Article Zika Virus Infection in Mice Causes Panuveitis with Shedding of Virus in Tears. 2016:3208–3218. [Google Scholar]
- Morrison TE, Diamond MS. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. J Virol. 2017;91:e00009–17. doi: 10.1128/JVI.00009-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, Martínez-Sobrido L, Ashok M, Lipkin WI, García-Sastre A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol. 2005;79:8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorati M, Li Z, Liu F, Sousa AMM, Nakagawa N, Li M, Dell’Anno MT, Gulden FO, Pochareddy S, Tebbenkamp ATN, et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016;16:2576–2592. doi: 10.1016/j.celrep.2016.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/s1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, et al. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40:D593–8. doi: 10.1093/nar/gkr859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard AS, Shim B-S, Kwon Y-C, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, Choe H. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc Natl Acad Sci U S A. 2017;114:2024–2029. doi: 10.1073/pnas.1620558114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaft J, Ashery-Padan R, van der Hoeven F, Gruss P, Stewart AF. Efficient FLP recombination in mouse ES cells and oocytes. Genesis. 2001;31:6–10. doi: 10.1002/gene.1076. [DOI] [PubMed] [Google Scholar]
- Sironi M, Forni D, Clerici M, Cagliani R. Nonstructural Proteins Are Preferential Positive Selection Targets in Zika Virus and Related Flaviviruses. PLoS Negl Trop Dis. 2016;10:e0004978. doi: 10.1371/journal.pntd.0004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Balasubramaniam VRMTMT, Brown JA, Mena I, Grant A, Bardina SV, Maringer K, Schwarz MC, Maestre AM, Sourisseau M, et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. 2017;13:e1006258. doi: 10.1371/journal.ppat.1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermillion MS, Lei J, Shabi Y, Baxter VK, Crilly NP, McLane M, Griffin DE, Pekosz A, Klein SL, Burd I. Intrauterine Zika virus infection of pregnant immunocompetent mice models transplacental transmission and adverse perinatal outcomes. Nat Commun. 2017;8:14575. doi: 10.1038/ncomms14575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, Shi P-Y, Vasilakis N. Zika virus: History, emergence, biology, and prospects for control. Antiviral Res. 2016;130:69–80. doi: 10.1016/j.antiviral.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widman DG, Young E, Yount BL, Plante KS, Gallichotte EN, Carbaugh DL, Peck KM, Plante J, Swanstrom J, Heise MT, et al. A reverse genetics platform that spans the Zika virus family tree. MBio. 2017;8:1–15. doi: 10.1128/mBio.02014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K-Y, Zuo G-L, Li X, Ye Q, Deng Y-Q, Huang X-Y, Cao W-C, Qin C-F, Luo Z-G. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res. 2016;26:645–654. doi: 10.1038/cr.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, Yang H, Cui J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017;3:17006. doi: 10.1038/celldisc.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. Evidence for a Genetic and Physical Interaction between Nonstructural Proteins NS1 and NS4B That Modulates Replication of West Nile Virus. J Virol. 2012;86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chan JF-W, Tee K, Choi GK-Y, Lau SK-P, Woo PC, Tse H, Yuen K. Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg Microbes Infect. 2016;5:e22. doi: 10.1038/emi.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmurko J, Neyts J, Dallmeier K. Flaviviral NS4b, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention. Rev Med Virol. 2015;25:205–223. doi: 10.1002/rmv.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.