

Abstract
Paclitaxel (PTX) has gained widespread clinical use yet its administration is associated with significant toxicity. In the present study, the toxicity and anti-tumor efficacy of tyrosine-derived nanospheres (NSP) for the delivery of PTX was compared to a clinical formulation of PTX in PBS-diluted Cremophor® EL (PTX–CrEL-D). Maximum tolerated dose was determined using a concentration series of PTX in NSP and CrEL-D, with toxicity assessed by measuring changes in body weight. Healthy mice administered PTX–NSP continued to gain weight normally while treatment with PTX–CrEL-D resulted in significant weight loss that failed to recover following treatment. Even at the dose of 50 mg/kg, PTX–NSP showed better tolerance than 25 mg/kg of PTX–CrEL-D. Xenograft studies of breast cancer revealed that the anti-tumor efficacy of PTX–NSP was equal to that of PTX–CrEL-D in tumors originating from both MDA-MB-435 and ZR-75-1 cancer lines. Larger volume of distribution and longer half-life were measured for PTX–NSP administration compared to those reported in the literature for a CrEL formulation. This trend suggests the potential for improved therapeutic index of PTX when administered via NSP. The findings reported here confirm that the NSP formulation is an efficient method for PTX administration with significant increase in maximum tolerated dose, offering possible clinical implications in the treatment of breast tumors.
Keywords: Tyrosine-derived nanospheres, Drug delivery, Paclitaxel, Chremophor, Toxicity, Anti-tumor efficacy
1. Introduction
Paclitaxel (PTX) is one of the most powerful chemotherapeutic drugs for the treatment of breast, ovarian, non-small cell lung carcinoma, bladder, prostate, melanoma, esophageal, as well as other types of solid tumor cancers (Gligorov and Lotz, 2004). However, adverse effects of standard paclitaxel treatment (Taxol®) create constraints on therapy. Severe side effects of PTX, such as neutropenia, neuralgia, and gastrointestinal toxicity (Gligorov and Lotz, 2004; Guastalla et al., 1994; Ohtsu et al., 1995), impact dose and schedule of administration. Additionally, PTX is poorly soluble in an aqueous medium: only 10.6 mM in water (Goldspiel, 1997; Mitra and Lin, 2003). To obtain clinical dosage, the pharmaceutical formulation of PTX is dissolved in a mixture (50:50, v/v) of the non-ionic surfactant Cremophor® EL (polyethoxylated castor oil) and absolute alcohol, which is further diluted in isotonic saline solution before intravenous (IV) administration (Adams et al., 1993). Unfortunately, Cremophor® EL (CrEL) has its own dose-limiting side effects increasing patient toxicity, including nephrotoxicity and neurotoxicity (Gelderblom et al., 2001; ten Tije et al., 2003). At nearly every step in both preclinical and clinical testing, CrEL has been shown to cause fatal hypersensitivity reactions, especially in bolus administration and more frequent infusion schedules (Weiss et al., 1990).
In order to reduce the adverse effects caused by CrEL in PTX treatments, alternative solubilization and delivery methods of PTX have been studied intensively in the past decade. Nanotechnology, one of the fastest growing fields in medical science, holds the potential to overcome the problem of PTX bioavailability and perhaps improve its therapeutic index (Farokhzad and Langer, 2009).
ABI-007 (Abraxane®) is the first FDA-approved albumin-bound, 130 nm particle formulation of PTX for the treatment of metastatic breast cancer (Desai et al., 2006). Although Abraxane® notably improved both toxicity profile and maximum tolerated dose compared to standard Taxol® (PTX–CrEL), only a fractional improvement in breast cancer progression-free survival was achieved (Gradishar et al., 2005). Hence, the search for the ideal PTX nanocarrier system continues.
Development of novel PTX delivery systems include, but is not limited to, nanoparticles (Cheng et al., 2011; Damascelli et al., 2003; Desai et al., 2006; Liang et al., 2011; Milane et al., 2011; Potineni et al., 2003; Reul et al., 2011; Shenoy et al., 2005), micelles (Liggins and Burt, 2002; Tsallas et al., 2010), liposomes (Heney et al., 2010; Kunstfeld et al., 2003; Rivera, 2003), dendrimers (Lim and Simanek, 2008; Minko et al., 2010; Ooya et al., 2004), superparamagnetic nanoparticles (Dilnawaz et al., 2010; Johnson et al., 2010; Zhao et al., 2010), polymer-based nanoshells (Zahr and Pishko, 2007), silica-based nanoparticles (Lu et al., 2007), and carbon nanotubes (Liu et al., 2008). Among these, nanoparticles based on biodegradable synthetic amphiphilic polymers have received the majority of attention due to ease of fabrication, stability (compared to liposomes), specificity, significant drug encapsulation/solubilization, and biocompatibility as a result of their material composition (Grodzinski et al., 2006). Moreover, such nanoparticles can be “tailor made” to achieve both controlled drug release and targeting by adjusting the polymer characteristics and surface chemistries. The choice of amphiphilic block copolymers is based on their ability to self-assemble in aqueous medium to form a core–shell architecture. The hydrophobic core serves as the depot for loading PTX while the hydrophilic shell enables stable dispersion in an aqueous environment (Allen et al., 1999; Haag, 2004; Torchilin, 2001; Wang et al., 2008). A variety of cores forming biodegradable polymers have been evaluated for the delivery of PTX with polyester being the most common choice (Chan et al., 2009; Forrest et al., 2008; Gaucher et al., 2010; Letchford et al., 2009; Milane et al., 2011; Reul et al., 2011; Sheihet et al., 2007). By the adjustment of the chemical composition and molecular weight (Mw) of the polymer, the degradation time of the core and/or release kinetics of the encapsulated agent can be controlled (Holland et al., 1986). Poly(ethylene glycol), PEG, and methoxy poly(ethylene glycol), mPEG, are typically chosen as hydrophilic shell of the nanoparticle that extends into the aqueous physiological environment, repelling proteins and decreasing antibody formation by steric repulsion mechanism, resulting in a prolonged circulation time in blood (Greenwald et al., 2003; Stolnik et al., 1994; Xu et al., 2005). Further, nanoparticle sizes of <200 nm tend to spontaneously accumulate via the enhanced permeability and retention (EPR) effect in pathological tissues through “leaky” vasculature, characteristic of solid tumors and inflamed tissue (Dreher et al., 2006; Maeda, 1992, 2001). Thus, the advantages of using synthetic biodegradable polymer-based nanoparticles for PTX delivery are the result of three properties: size, capability of PTX solubilization and controlled delivery through adjustable chemical composition of the nanoparticles.
We have previously reported on the design and synthesis of unique ABA-type amphiphilic tyrosine-derived triblock copolymers where the hydrophilic A-blocks consist of mPEG and the hydrophobic B-block consists of oligomers of desaminotyrosyl-tyrosine octyl esters (DTO) and non-toxic diacids (Scheme 1) (Nardin et al., 2004; Sheihet et al., 2005, 2007). These copolymers self-assemble into spherical structures (nanospheres, NSP) with hydrodynamic diameters of 60–70 nm and critical aggregation concentration that is significantly lower than values reported for other self-assembling oligomer systems (Nardin et al., 2004). Molecular dynamics simulations and docking calculations studies in water environment suggest that the self-assembly of tyrosine-derived triblocks results in core–shell architecture where PEG corona is not involved in the core assembly and the drug binding within core is governed only by the oligo (DTO–SA) hydrophobic core and solute interactions (Costache et al., 2009). The hydrophobic B-block of this oligomer is composed of naturally occurring metabolites, and the unique design of this polyarylate oligomer allows for systematic variations in the polymer backbone structure. Copolymerization of different desamintyrosyl-tyrosine esters with a variety of diacids gives rise to an array of structurally related copolymers. Hence, the desired binding efficiency of the drug (in this case PTX) can be achieved through the adjustment of polymer composition (Sheihet et al., 2007).
Scheme 1.
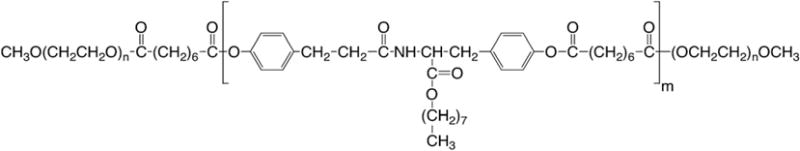
Chemical structure of PEG5K-b-oligo(desaminotyrosyl-tyrosine octyl ester suberate)-b-PEG5K triblock copolymer, DTO–SA/5K. The oligo B-block is distinguished by both their alkyl pendent chain “octyl” linked to the (desaminotyrosyl-tyrosine alkyl ester) unit and the SA “suberic acid” to form the oligomer of (DTO–SA). mPEG A-blocks are abbreviated as 5K, indicating the molecular weight of the PEG components.
These NSP, which are themselves non-cytotoxic, deliver PTX very effectively to tumor cells as measured by cell killing activity in vitro (Sheihet et al., 2007). Slight modification to the synthetic procedure of this triblock copolymer could enable covalent attachment of a wide variety of targeting ligands to selectively deliver PTX, thus significantly improving the therapeutic potential of PTX and alleviating some of the adverse side effects by reducing PTX accumulation in non-targeted tissue (manuscript in preparation, 2011). Moreover, it is important to note that the B-block of the triblock copolymer is closely related to the monomer repeat units of the tyrosine-derived polyarylate polymer used in two FDA-cleared medical devices: an anti-bacterial envelope for cardiac rhythm medical device (AIGISRX™, 2008) and a surgical hernia mesh (PIVI-TAB™, 2006). Thus, similar biocompatibility properties of the B-block copolymer are expected for the investigated herein nanospheres.
To further evaluate the clinical potential of these NSP, we assessed the in vivo comparison of toxicity and anti-tumor efficacy of PTX administered via tyrosine-derived NSP or in CrEL in two clinically relevant breast cancer estrogen receptor negative (ER−) and estrogen receptor positive (ER+) xenograft models.
2. Materials and methods
2.1. Materials
Methylene chloride (HPLC grade), methanol (HPLC grade) and 2-propanol were purchased from Fisher Scientific (Pittsburgh, PA). Suberic acid, 4-dimethylaminopyridinium-p-toluene sulfate (DMPTS), and poly(ethylene glycol) monomethyl ether (mPEG, Mw 5000), dimethyl sulfoxide (DMSO), and Dulbecco’s phosphate buffered saline (pH 7.4) were purchased from Aldrich Chemical Co. (Milwaukee, WI). Diisopropylcarbodiimide (DIPC) was purchased from Tanabe Chemicals (San Diego, CA). Paclitaxel was purchased from LC Laboratories (Woburn, MA). Cremophor EL was purchased from Sigma Chemical Co. (St. Louis, MO). N,N-dimethylformamide (DMF) and tetrahydrofuran (THF) were obtained from Merck (EM Science, Darmstadt, Germany). An internal standard (IS) for pharmacokinetics studies Baccatin III was purchased from Hande Tech (Houston, TX). All reagents were used as received.
2.2. Preparation of paclitaxel formulations
2.2.1. Preparation and characterization of the PTX–NSP and NSP alone formulations
ABA triblock copolymer (PEG5K-b-oligo(desaminotyrosyl-tyrosine octyl ester suberate)-b-PEG5K triblock copolymer) was synthesized and characterized as previously published (Sheihet et al., 2005, 2007). Paclitaxel-loaded nanospheres (PTX–NSP) were prepared according to previously established procedure (Sheihet et al., 2007) with the following modifications: (a) the feed ratio between the PTX and triblock copolymer changed and (b) to further increase the PTX in final formulation, re-suspended samples were combined, re-centrifuged, re-suspended, and finally, the solution was filter-sterilized (0.22 μm). This standard fabrication procedure produced 2.5 mg of PTX and 65 mg of NSP in 1 mL of PTX–NSP formulation. For determination of toxicity/maximum tolerated dose (MTD), different PTX doses in NSP (5, 10, 20, 25, 30, 40, and 50 mg/kg in 0.2 mL injection per 20 g animal) were obtained by keeping the polymer amount constant and increasing the drug to polymer feed ratios to 0.8, 1.6, 3.2, 4, 4.8, 6.4, and 8 ± 0.2% w/w, respectively.
PTX concentration in final purified PTX–NSP (loading efficiency, LE) was measured using previously established extraction and reverse phase high-performance liquid chromatography (HPLC) techniques (Sheihet et al., 2007) with modifications: (a) the mobile phase was a mixture of water (0.1% trifluoroacetic acid, TFA)/acetonitrile (0.1% TFA) with the ratio 48/52 (v/v) and (b) standard calibration samples were prepared at concentrations ranging from 1 to 20 μg/mL. The detection limits were evaluated on the basis of a signal to noise ratio of 3 and 0.01 μg/mL. Within- and between-day precision and accuracy determination of quality control samples were better than 10% across the range of the calibration curve.
The hydrodynamic diameters of the NSP or PTX–NSP were obtained by dynamic light scattering, DLS (DelsaNano S, Beckman Coulter Inc., NJ) (Sheihet et al., 2007) and transmission electron microscopy, TEM (JEM 100CX Transmission Electron Microscope, JEOL LTD, Peabody, MA) (Sheihet et al., 2008).
2.2.2. Preparation of the PTX–CrEL-D formulation
The paclitaxel–Cremophor (PTX–CrEL) formulation was prepared by first dissolving the desired amount of PTX in ethanol (PHARMCO-AAPER, Brookfield, CT) and then adding an equal volume of CrEL. It is important to note that following the first intravenous (iv) administration of CrEL alone (200 μL) clinical signs of toxicity were observed visually for appetite loss and tremors (more food left each day and obvious paw tremors and head shakes, respectively) whereas rapid weight loss was quantified. Therefore, to minimize toxicity and avoid animal mortality from vehicle-related side effects, all PTX–CrEL formulations were further diluted with sterile PBS (pH 7.4) to achieve a 1:1 (v:v) of organic to aqueous solution. In this study, PBS diluted solutions of CrEL will be referred to as CrEL-D. Using this standard fabrication procedure each mL of CrEL-D formulation (without or with the desired amount of PTX) contained 260 mg of CrEL, 25% v/v ethanol, and 50% v/v PBS. PTX concentration in CrEL-D formulations was confirmed using extraction and reverse phase HPLC technique described above. Prior to administration, all PTX–CrEL-D formulations were filter sterilized using 0.22 μm low protein binding durapore syringe filters (PVDF, Millipore, Bedford, MA).
For determination of maximum tolerated dose of PTX–CrEL-D, different PTX doses in CrEL-D (5, 10, 20, 25, and 30 mg/kg in 0.2 mL injection per animal) were obtained by subsequent dilution of 5 mg/mL PTX in CrEL first with CrEL and then PBS.
2.3. In vitro cytotoxicity
Breast cancer cell lines MDA-MB-435 and/or ZR-75-1 were obtained from ATCC (Manassas, VA) and cultured in Dulbecco’s modified Eagle medium (DMEM, Invitrogen Corp., Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Inc., Lawrenceville, GA), 2 mM glutamine (MP Biomedical LLC, Fisher Scientific, Suwanee, GA), and 0.025 mg/mL gentamycin (Gibco®, Carlsbad, CA). Toxicity of studied substances to these cell lines was assessed by the Alamar Blue® viability assay (AbD Serotec, Fisher Scientific, Suwanee, GA). Cells were seeded at 2000 cells per well in a 96-well plate and allowed to adhere and equilibrate to the culture conditions overnight. The following day the media was changed in all wells to the experimental conditions (NSP or CrEL-D or PTX–NSP or PTX–CrEL-D). Formulations of NSP or CrEL with PTX were diluted in media to obtain the indicated PTX doses, while the NSP alone or CrEL-D formulation were diluted in media to match the NSP and CrEL-D amounts used in PTX-containing formulations. Cells were allowed to culture for 72 h prior to washout of the test formulations and the cell viability assay. Metabolization of resazurin in the Alamar Blue to resorufin was quantified by the absorbance of the resorufin on a plate reader (TECAN-M200). Experiments were also performed to verify that the NSP alone do not affect the viability assay reagents. Cells incubated with PBS and media were used as negative control. Three independent experiments were conducted, which included all test formulations (NSP or CrEL-D or PTX–NSP or PTX–CrEL-D) for each respective experiment, to obtain statistical power.
2.4. In vivo studies
All procedures performed in the in vivo studies were designed in accordance with the guidelines of the Care and Use of Laboratory Animals and were approved by the Animal Care Committee of the Rutgers University and University of Medicine and Dentistry of New Jersey and USAMRMC Animal Care and Use Review Office (ACURO). Female NCr (nu/nu strain), 6–8 weeks old mice, weighing about 20 g were purchased from Taconic Farms, Inc. (Germantown, NY) and were housed in cages under controlled laboratory conditions (temperature 22–24 °C, relative humidity 50 ± 10% and 12 h/12 h light/dark cycle) and allowed free access to sterilized rodent pellet diet and acidified drinking water. The animals were acclimatized for at least 72 h prior to any experiments, and “pre-numbered” ear tags were used to identify each mouse.
2.4.1. In vivo toxicity and maximum tolerated dose (MTD) determination
For MTD studies a series of PTX doses (N = 6 per dose) in NSP and CrEL-D were tested (Table 2). The same concentrations of drug free NSP and CrEL-D were administered as vehicle controls (Table 2, N = 6). Healthy female NCr (nu/nu) mice were weighed and then began receiving IV tail injections (~0.2 mL per mouse) of vehicle controls and different concentrations of PTX in NSP and CrEL-D using a q5dx4 (every 5 days four times) schedule. Mice were monitored and weighed on the day of the treatment, the day after, and every other day during the course of the entire trial. Toxicity was assessed as a percent of weight loss. The MTD was defined as the highest dose resulting in less than 15% body weight loss and not causing significant lethality or any prominent observable changes during the course of the trial (Ahmed et al., 2006; Chandna et al., 2010; Dharap et al., 2005). The difference in mean body weight was calculated with respect to the beginning of treatment (day 1) as: (mean body weight on day × − mean body weight on day 1)/(mean body weight on day 1) × 100.
Table 2.
Test formulations and experimental conditions in in vivo studies.
| Test formulations | Study and tested PTX dose (mg/kg)
|
||
|---|---|---|---|
| Toxicity/MTD | Anti-tumor activity
|
||
| ER(−) | ER(+) | ||
| PBS | 0 | 0 | Not tested |
| NSP | 0 | Not tested | 0 |
| CrEL-D | 0 | Not tested | 0 |
| PTX–NSP | 5, 10, 20, 25, 30, 40*, 50* | 20 | 20 |
| PTX–CrEL-D | 5, 10, 20, 25, 30 | 20 | 20 |
Healthy (toxicity/MTD study) or tumor-bearing (anti-tumor activity) female NCr (nu/nu) mice (N = 6) were treated on q5dx4 (every 5 days four times) schedule with ~0.2 mL of respective formulation.
“0” denotes carrier administered w/o PTX.
N = 4.
2.4.2. In vivo anti-tumor activity
Female NCr nu/nu mice were used as a human tumor xenograft model in the study. Tumors were established by inoculating ER(−) MDA-MB-435 (5 × 106/0.2 mL) or ER(+) ZR-75-1 cells (8 × 106/0.2 mL) into a flank of each mouse. Tumors were monitored visually twice weekly and then daily as their volumes approached 0.08–0.1 cm3. When tumors size reached 0.2–0.3 cm3 (approximately 6 weeks after inoculation), the animals were selected for experimental treatment. The tumor-bearing mice were randomly allocated to 3 or 4 different treatment groups (Table 2), depending on the xenograft model, with similar mean tumor volumes of each group (N = 6). PBS, CrEL-D alone (vehicle control), NSP alone (vehicle control), PTX–CrEL-D (20 mg/kg PTX) and PTX–NSP (20 mg/kg PTX) were IV administered (0.2 mL per animal) using a q5dx4 schedule. The mortality was monitored daily and the tumor growth was measured by a caliper on the day of the treatment, day after, and every other day during the course of the entire trial. Tumor volume was calculated as d2 × D/2 where d and D are the narrowest and widest diameter of the tumor in mm, respectively. According to the approved institutional animal use protocol when the tumor reached around 1.2–1.3 cm3, the mice were sacrificed.
2.4.3. Pharmacokinetics study
To study the pharmacokinetics of PTX–NSP, a dose equivalent of 15 mg PTX/kg was administered as single tail vein injection (~30 s, 0.2 mL) to the groups of 5 female NCr nu/nu mice. At the specified times after the injection, 20, 30, 40 min, and 1, 2, 3, 4, 6, 17, 24, and 48 h, mice were euthanized using carbon dioxide gas and blood was collected by cardiac puncture, and transferred to heparinized tubes. Plasma was obtained by centrifugation and stored at −80 °C until analysis. PTX content was determined using the validated HPLC method described below.
The HPLC system consisted of (Hitachi International 7000 series) autosampler (7200), quaternary pump (7100), UV detector 7400), and the Hitachi chromatography data station software (D7000) interfaced to computer. The samples were analyzed using 250 × 4.6 mm Synergi 4 Fusion-RP 80A column (Phenomenex, Torrance, CA USA), with a mobile phase containing water: acetonitrile: methanol (47:50:3) at a flow rate (0.8 ml/min), using UV detector set at 229 nm. The plasma samples were processed as previously described with some modifications (Toppmeyer et al., 2003). Briefly, plasma PTX calibration standards were prepared using drug-free control human plasma (NBAH Blood Center, New Brunswick, NJ). For the assessment of chromatographic linearity and limit of quantitation of PTX, calibration standards were spiked in plasma and further diluted serially to obtain concentrations ranging from 20 μg/mL to 0.025 μg/mL. Murine plasma samples (200 μL) and calibration standards (200 μL) were spiked with internal standard (baccatin III at 5 μg), and vortex-mixed for about 60 s. Where sample volume was less than 200 μL, human plasma was added to adjust the volume to 200 μL. The samples were extracted twice with three times volume of methanol. The samples were then centrifuged at 20,000g for 10 min at 4 °C. The supernatants were transferred to 1.5 mL polypropylene tubes and evaporated to dryness under stream of nitrogen at 45 °C. The residues were dissolved in water and analyzed by HPLC. The resulting PTX concentrations were adjusted to account for addition of human plasma. Several calibration standard curves were prepared and the correlation coefficient (mean ± SD, N = 3) for paclitaxel was 0.9971 ± 0.0025. The assay was linear from 0.05 to 20 μg/mL. The lowest limit of quantitation was 0.05 μg/mL and the lowest limit of detection was 0.025 μg/mL. Pharmacokinetic parameters for paclitaxel were calculated using non-compartmental methods and WinNonlin (version 5.2). Area under the concentration-time curve (AUC) was estimated by the trapezoidal rule up to the last measurable concentration (Clast). Total body clearance (CL) was calculated by dividing dose by AUC.
2.5. Statistical analysis
Statistical differences were determined using a one-way ANOVA, followed by Tukey’s post hoc test for comparison of treatments (NSP vs CrEL-D and/or different doses of PTX in NSP or CrEL across time). All analyses were carried out with Prism 4 statistical software (GraphPad, CA, USA). All data are presented as a mean value with its standard error indicated (mean ± SE). p-values of less than 0.001 (***), 0.01 (**) and 0.05 (*) were considered significant.
3. Results and discussion
3.1. Formulation design and characterization
The general chemical structure of the tyrosine-based triblock copolymer used to fabricate NSP is depicted in Scheme 1. The hydrophobic B-block was prepared by polymerization of tyrosine-derived monomer DTO with suberic acid (SA) resulting in oligomers with carboxylic acid end groups that were further reacted with mPEG using a previously established procedure (Nardin et al., 2004; Sheihet et al., 2007). The design of this copolymer system was based on the following rationale: the choice of the oligo (DTO suberate) for the middle block was based on its low glass transition temperature Tg (294 K), which ensures that each triblock copolymer molecule will be sufficiently flexible to self-assemble into a dynamic and non-frozen structure (Nardin et al., 2004). In addition, this middle block is degradable under physiological conditions to produce naturally occurring metabolites (Bourke and Kohn, 2003). mPEG as end blocks provides the modulation of cell behavior and non-fouling characteristics, prolonging the circulation time of the NSP in blood (Xu et al., 2005).
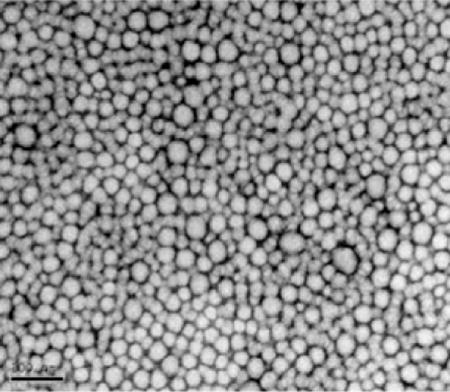
Under an established synthetic procedure (PEG5K-b-oligo(desaminotyrosyl-tyrosine octyl ester suberate)-b-PEG5K triblock copolymers (DTO–SA/5K, Scheme 1) are obtained with narrow molecular weight distributions centered around 25 kDa (Mw/Mn = 1.3) (Sheihet et al., 2007). The spherical morphology and nano-scale dimension of the NSP ± PTX were demonstrated by electron microscopy and dynamic light scattering measurements, revealing a narrow size distribution centered around 67 nm (poly-dispersity index, PDI, of 0.22), indicating that neither the presence of PTX nor its concentration and neither extensive purification nor “super-concentration” have a significant effect on NSP size (Table 1). Uniform dispersion and spherical morphology of PTX–NSP (2.5 mg PTX/1 mL of formulation) with sizes ranging between 40 and 70 nm were also confirmed by transmission electron microscopy (Table 1). The slight difference in the size distribution obtained by TEM and DLS can be attributed to the drying of the specimens prior to imaging that leads to dehydration of the nanospheres and outer PEG layer, some shrinkage and/or agglomeration. Zeta potential value of NSP is close to neutrality confirming the presence of PEG on the surface of the NSP (unpublished). Further, surface charge values remained in the same range regardless of tyrosine-derived oligomer chemistry, presence of the encapsulant or NSP concentration (unpublished). We have previously reported that the release kinetics of PTX from NSP are dependent upon the pH of the outer dialysis solutions: after 72 h the release of PTX is 72% at pH 5.5 and only 48% at pH 7.4 (Sheihet et al., 2007). The pH dependence of PTX release profiles is expected to have an important therapeutic advantage: at pH 7.4, which is typical of the blood stream, PTX will remain bound to the NSP, but enhanced release will occur at pH 5.5, which is typical of the intracellular environment. All reported characterizations of NSP confirm that this delivery system addresses the requirements of a proper carrier for PTX based on structural composition, polymer molecular weight distribution and particle size.
Table 1.
Morphology and size distribution of PTX–NSP formulation.
| PTX in NSP (mg/mL) | Hydrodynamic diameter (nm) | PDIa | Transmission electron micrograph of PTX–NSP (2.5 mg PTX/1 mL PTX–NSP, bar 100 nm)b |
|---|---|---|---|
| 0 | 66 | 0.21 |
|
| 0.5 | 65 | 0.21 | |
| 1 | 69 | 0.23 | |
| 2 | 71 | 0.21 | |
| 2.5 | 66 | 0.22 | |
| 3 | 65 | 0.22 | |
| 4 | 72 | 0.21 | |
| 5 | 68 | 0.22 |
Polydispersity index (PDI) value is for the PTX–NSP mean hydrodynamic diameter obtained for 3 independent preparations with five independent measurements of a single formulation.
Transmission electron microscopy (TEM) image was obtained using negative staining method (2% uranyl acetate) according to previously reported procedure (Sheihet et al., 2008).
3.2. In vitro toxicity of test formulations
The in vitro cytotoxicity of PTX in NSP against MDA-MB-435 breast cancer cells was compared with that of PTX in CrEL-D using Alamar Blue viability assay (Fig. 1). To achieve different doses of PTX and delivery vehicles, the standard formulations of PTX–NSP and PTX–CrEL-D and drug free formulations of NSP and CrEL-D (at equivalent amount of the vehicle mass) were diluted with the culture media, resulting in 2 and 6 ng/mL of NSP and 8 and 24 ng/mL of CrEL-D. NSP at these concentrations exhibited no toxicity compared to control cells. In contrast, CrEL-D treatment resulted in a concentration dependent toxicity with only 26% and 13% (p < 0.001) cell viability measured for 8 and 24 ng/mL of CrEL-D formulation respectively. Danhier et al. reported similar observations where CrEL concentration significantly affected the viability of HeLa cell line (Danhier et al., 2009). It is therefore likely that a significant portion of the cytotoxicity observed for the PTX–CrEL-D formulation (17% and 14% cell viability in 0.06 and 0.6 ng/mL of PTX in CrEL-D, respectively) can be attributed to the CrEL-D component. These results confirm the previous reports that dose-limiting side effects of CrEL-D significantly impact the efficiency Taxol® therapy (Gelderblom et al., 2001). On the other hand, the toxicity of PTX–NSP on MDA-MB-435 cells can only be attributed to the presence of PTX in the NSP formulation. Viability decreased to 60% and 31% for 0.06 and 0.6 ng/mL of PTX in NSP, respectively (p < 0.001, Fig. 1). These findings confirm that NSP exhibits no cellular toxicity in human breast cancer cells and maintain the pharmacological activity of PTX.
Fig. 1.
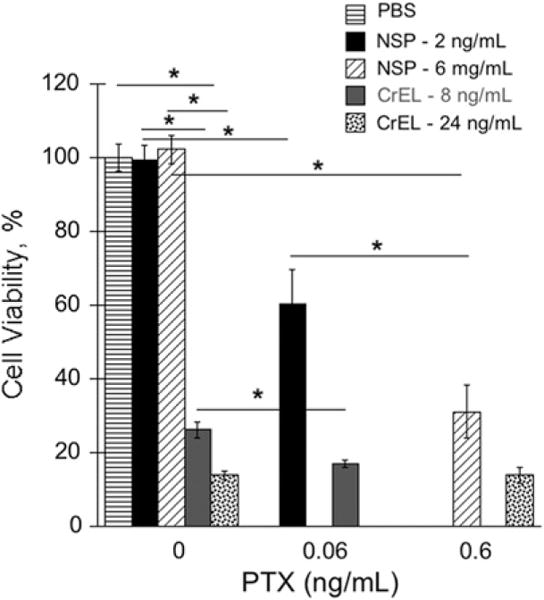
Viability of MDA-MB-435 breast cancer cells exposed to 0.06 and 0.6 ng/mL of PTX delivered via NSP (2 and 6 ng/mL), CrEL-D (8 and 24 ng/mL), and drug free formulations of NSP (2 and 6 ng/mL) and CrEL-D (8 and 24 ng/mL). Cell viability was measured after 72 h by AlamarBlue® cell proliferation indicator assay. Cells incubated with PBS and media were used as negative control. The results are expressed as mean value with its standard error indicated (mean ± SE) of 6 measurements of 3 independent experiments; p < 0.05 (*), 0.01 (**), and 0.001 (***) were considered to be statistically significant.
3.3. In vivo toxicity – maximum tolerated dose
Side effects of PTX treatment include specific toxicities such as neutropenia and more generalized toxicity such as body weight loss (Song et al., 2004). Thus, the toxicity/MTD of PTX administrated in the NSP and CrEL-D was evaluated by measuring body weight change of treated mice as a function of the fractional increase in PTX dose (Table 2) and time. The initial IV administration of drug free CrEL resulted in clear signs of toxicity (visually for appetite loss and tremors as more food left each day and obvious paw tremors and head shakes, respectively, whereas rapid weight loss was quantified) in 100% of the healthy mice with a significantly high number of deaths (62%). Vassileva et al. reported similar observations where 75% lethality was observed in nude mice bearing human ovarian xenografts (Vassileva et al., 2007). Therefore, to minimize toxicity and avoid mortality, all CrEL-based formulations tested here were further diluted with sterile PBS (pH 7.4) to achieve 1:1 (v:v) of organic to aqueous ratio and now will be referred to as CrEL-D. Further dilution of the CrEL with PBS (>50% v/v) was not possible due to insufficient solubility of tested PTX doses. In order to mimic clinically relevant dosing and schedule, IV bolus injections of PTX–NSP and PTX–CrEL-D with different PTX doses (Table 2) were performed on a q5dx4 schedule. Drug free NSP and CrEL-D were administered as vehicle controls.
Fig. 2 depicts the effect of the vehicle treatment on the body weight change of healthy mice. Although significant changes (loss in the case of CrEL-D) in body weight were measured throughout the treatment (Fig. 2), here we discuss the results for only the day and week after the last injection (days 14 and 22), which represent the tolerability/safety of the overall treatment. NSP did not exhibit any visual and measurable toxicities and an average of 7% (p < 0.001) weight gain was registered during the 2 weeks of treatment. In contrast, mice receiving CrEL-D encountered significant toxicity throughout the whole study with 6% average weight loss by the end of the treatment (day 14, p > 0.05). It was also noted that majority of the weight was lost immediately after the first treatment (9% by day 3, p < 0.01) suggesting that CrEL-D toxicity is acute. During the post-treatment observation (days 15–22), five out of six CrEL-D mice showed an increase in weight, averaging 8% (p < 0.05) compared to an average weight gain of 16% for NSP (p < 0.001). These results confirmed the clinical biocompatibility and superior safety of NSP as delivery vehicle compared to CrEL-D.
Fig. 2. In vivo.
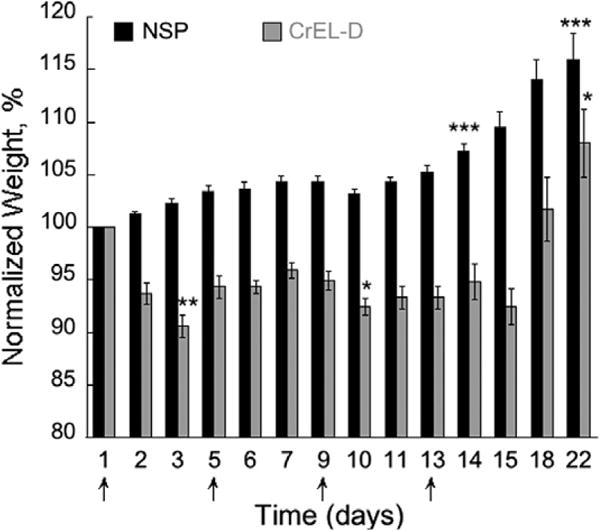
toxicity of NSP and CrEL-D as a function of treatment schedule in healthy NCr nu/nu mice (N = 6 for each treatment). Treatments (0.2 mL, IV) were administered on days 1, 5, 9, and 13 (arrows, q5dx4). Concentration of NSP and CrEL in each treatment was 13 and 52 mg/0.2 mL, respectively. Each data represents the mean value with standard error (mean ± SE); p < 0.05 (*), 0.01 (**), and 0.001 (***) were considered to be statistically significant in the body weight change on the selected times comparing each group to pre-treatment weight.
The change in body weight as an effect of PTX treatment is shown in Fig. 3. As mentioned above, while some significant changes in mice body weight compared to day 1 were measured throughout each treatment (significance of gain or loss are marked in Fig. 3), here we discuss the statistical analysis only for the day and week after the last injection (days 14 and 22) compared to the pre-treatment weight on day 1. Notably, all PTX doses when administered as PTX–NSP were well tolerated, during and post-treatment, with a direct correlation between the delay in the average weight gain and increase in PTX dose (Table 2). The significant weight gain of mice in all PTX–NSP treatments on the day after last injection (day 14) was: +17 (p < 0.001), +16 (p < 0.001), +12 (p < 0.05), +6, and +5% for 5, 10, 20, 25, and 30 mg/kg of PTX in NSP, respectively. Moreover, in the week of post-treatment mice treated with high PTX–NSP doses of 25 and 30 mg/kg gained an additional 10% body weight, respectively (Fig. 3D and E). In contrast, the mice throughout and after the treatment poorly tolerated PTX–CrEL-D (Fig. 3A–E and Table 2). The recorded weight change on the day 14 was: +2%, +1%, −5%, −6%, and −15% (p < 0.05) for 5, 10, 20, 25, and 30 mg/kg of PTX in CrEL-D, respectively. During the week of post treatment, the mice that received 25 or 30 mg/kg PTX–CrEL-D did not recover to their initial weight in addition to displaying the signs of behavioral distress (e.g. fatigue, allodynia). Lethal toxicity (5–10% mortality) was observed in all PTX–CrEL-D treatments, regardless of PTX dose, by the time of the third treatment. These toxicity observations are in agreement with previously reported tolerability of similar doses of PTX− CrEL-D in CD-1 mice (Vassileva et al., 2007). Previous findings have indicated that PTX has a toxic effect on gastrointestinal (GI) activity (Daniels et al., 2008), suggesting a likely explanation for the outcome of the experiments measuring mouse weight. Accordingly, CrEL is known to decrease the clearance of PTX from the gut, hence decreasing its inclusion to the systemic circulation and ultimate clearance. This phenomenon, observed in several animal models, is based on the tendency of CrEL to form micelles in aqueous solution entrapping PTX within their hydrophobic core upon the administration (Sparreboom et al., 1999). Bardelmeijer et al. reported significant concentration of such micelles being formed in gastrointestinal lumen, small intestinal contents, and caecum (Bardelmeijer et al., 2002). Our data suggests that the incorporation of PTX in NSP most likely reduced the drug exposure to the GI tract, hence alleviating some of the undesired toxicity observed in PTX–CrEL-D. To further evaluate the safety of PTX–NSP treatment, even higher doses of PTX (40 and 50 mg/kg) were tested. As shown in Fig. 3F, even at the dose of 50 mg/kg, PTX administrated via NSP showed better tolerance than half of this dose delivered via CrEL-D (Fig. 3D).
Fig. 3.
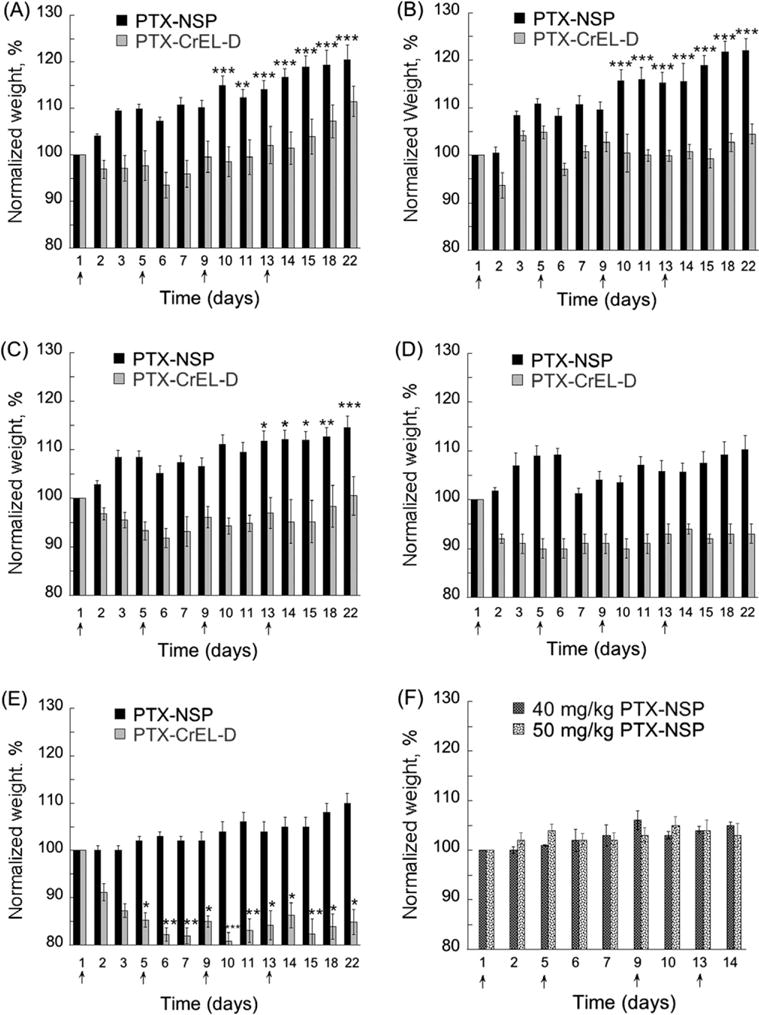
Determination of toxicity/MTD for PTX delivered via NSP and CrEL-D. Mice (N = 6 per group) were injected (0.2 mL) on days 1, 5, 9, and 13 with (A) 5, (B) 10, (C) 20, (D) 25, (E) 30 mg/kg of PTX in each vehicle: (
 ) PTX-NSP; (
) PTX-NSP; (
 ) PTX-CrEL-D. (F) 40 (
) PTX-CrEL-D. (F) 40 (
 ) and 50 (
) and 50 (
 ) mg/kg of PTX in NSP. Concentration of NSP and CrEL-D in each treatment was 13 and 52 mg/0.2 mL, respectively. Lethal toxicity (5-10% mortality) was observed in all PTX-CrEL-D treatments, regardless of PTX dose, by the time of the third treatment. The statistical data is expressed as the mean value ± S.E. (standard error); p < 0.05 (*), 0.01 (**), and 0.001 (***) were considered to be statistically significant in the body weight change compared to the initial pre-treatment weight.
) mg/kg of PTX in NSP. Concentration of NSP and CrEL-D in each treatment was 13 and 52 mg/0.2 mL, respectively. Lethal toxicity (5-10% mortality) was observed in all PTX-CrEL-D treatments, regardless of PTX dose, by the time of the third treatment. The statistical data is expressed as the mean value ± S.E. (standard error); p < 0.05 (*), 0.01 (**), and 0.001 (***) were considered to be statistically significant in the body weight change compared to the initial pre-treatment weight.
Based on the results of the body weight changes described above, 20 mg/kg was selected as an MTD for PTX–CrEL-D treatment. In contrast, MTD for PTX–NSP could not be established since all tested PTX doses in NSP were safe: no signs of behavioral distress were observed and no weight loss was measured. The above data suggests that MTD for PTX–NSP will be higher than 50 mg/kg. For the direct comparison of the effect of PTX delivery vehicles, the same PTX dose (20 mg/kg – highest PTX dose with minimum toxicity in CrEL-D) in CrEL-D or NSP was tested in the subsequent anti-tumor studies.
The results of the superior safety of NSP are in agreement with the results obtained for ABI-007 (Abraxane®), the first FDA-approved albumin-bound nanoparticle formulation of PTX for the treatment of metastatic breast cancer (Desai et al., 2006). Abraxane® notably improved both the toxicity profile and maximum tolerated dose when compared to standard Taxol® (PTX–CrEL) (Gradishar et al., 2005).
3.4. In vivo and in vitro anti-tumor efficacy
Anti-tumor efficacy studies were performed in NCr nu/nu mice bearing subcutaneous ER(−) MDA-MB-435 and ER(+) ZR-75-1 breast human cancer xenografts. When tumor size reached 0.2–0.3 cm3 (approximately 6 weeks after the instillation of the cells), the animals were randomly allotted to 3 and 4 different treatment groups (Table 2), depending on the xenograft model, with similar mean tumor sizes for each group (N = 6). In the case of the ER(−) xenograft study (Fig. 4A), the size of the tumor in the PBS treated group increased significantly by the end of the treatment schedule (2 weeks). An accelerated growth rate of tumors in the PBS group led to the early termination of the experiments at day 32 (with a 13-fold increase in tumor volume when compared with starting volume). In contrast, both PTX treatments significantly delayed tumor growth (p < 0.01), with average increases of only 4.4- and 4.3-fold tumor sizes in mice treated with PTX–NSP and PTX–CrEL-D, respectively, on day 46. No statistical difference between the anti-tumor efficiency of both PTX treatments was found (p > 0.05). Using the T–C method (Corbett et al., 1997), the growth delay (measured in days) between the median time required for tumors to reach four times the size at the initiation of therapy was calculated to be 37 ± 1.2, 41 ± 0.8, and 10 ± 1.4 days for PTX–NSP, PTX–CrEL-D, and PBS, respectively. These findings show that PTX delivered via NSP reached its intended destination in sufficient concentration to inhibit tumor growth. Given the significantly higher tolerability to PTX–NSP treatment (Fig. 3) and lack of toxicity of the NSP carrier (Fig. 1), we hypothesize that antitumor efficacy could be further improved by using a higher dose of PTX in NSP.
Fig. 4.
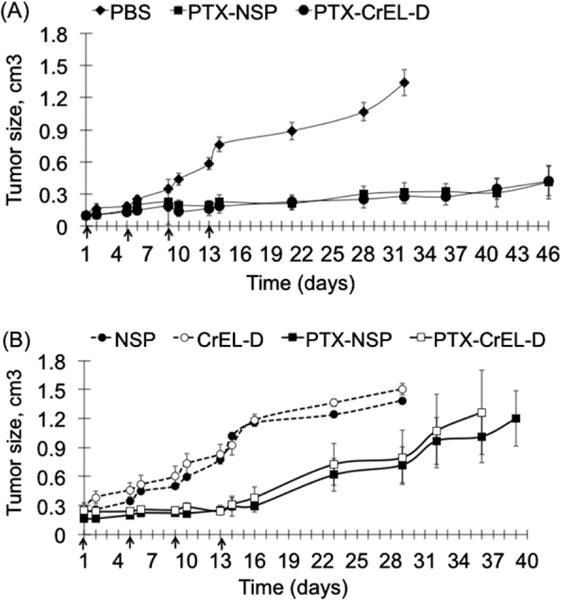
Anti-tumor activity in NCR nu/nu mice bearing subcutaneous (A) MDA-MB-435 estrogen-independent (ER−) and (B) ZR-75-1 estrogen dependent (ER+) breast cancer xenografts. Mice (N = 6 per group) were treated (0.2 mL) on days 1, 5, 9, and 13 with 20 mg/kg of PTX in NSP (13 mg) or CrEL-D (52 mg) in both studies. The control groups received PBS (MDA-MB-435 study, N = 6) and NSP and CrEL-D (ZR-75-1 study, N = 6) using the same administration schedule. Lethal toxicity (10–15% mortality) was observed in all PTX–CrEL-D treatments by the time of the third treatment. The data is presented as a mean value with standard error (mean ± SE) of tumor size in each tested group with no statistical difference seen between both treatments.
Lower anti-tumor activity of PTX–NSP and PTX–CrEL-D was observed in tumors derived from ER(+) ZR-75-1 breast cancer xeno-grafts compared to ER(−) MDA-MB-435 (Fig. 4B and Table 2). Using the same PTX dose and schedule, the inhibition of tumor growth lasted for a shorter period. No statistical difference in the efficiency of PTX–NSP and PTX–CrEL-D (p > 0.05) was observed and excessive tumor growth (⩾1 cm3) was measured on days 36–39 in both treatments. In agreement with the above-discussed toxicity studies, mice that received PTX–CrEL-D showed signs of physical wasting compared to mice that treated with PTX–NSP. Mortality was observed in both treatments with 5% occurring by the days 26 and 18 in PTX–NSP and PTX–CrEL-D groups, respectively. As expected, no anti-tumor activity was measured in both control groups that received drug free delivery vehicles, NSP and CrEL-D. Lethal toxicity evident by 15% and 29% mortality on days 29 and 20 was observed in NSP and CrEL-D treated groups, respectively. T–C growth delay was calculated to be 21 ± 2.3 and 23 ± 2.7 days for PTX–NSP and PTX–CrEL-D, respectively, and 14 ± 1.3 for both NSP and CrEL-D. The anti-tumor studies suggest that in tested xenograft models the PTX anti-tumor activity is, in part, dependent upon the inherent properties of the tumor originating cells. To test this hypothesis, in vitro cytotoxicity of PTX− NSP (2.5 mg/mL (3 mM of PTX) equivalent to the dose of 25 mg/kg in mice) and drug free NSP were diluted in media to the effective dose ranges for MDA-MB-435 and ZR-75-1 cells. As shown in Fig. 5, NSP alone did not show any toxicity toward MDA-MB-435 or ZR-75-1 cells. Incorporation of PTX into NSP confirmed that these two cell lines have very different susceptibilities to this drug. MDA-MB-435 cells showed an IC50 of approximately 0.1 nM (0.085 ng/mL) whereas ZR-75-1 cells were significantly more resistant with IC50 being in orders of magnitude higher 1 μM (0.85 μg/mL). While both of these cell lines originate from human breast cancers, there can be many distinguishing features to account for their respective chemotherapeutic profiles. Supportive data throughout the literature and clinical observations indicate that the relative abundance of the ER on the cancer cells is a key-mitigating factor to PTX susceptibility (Henderson et al., 2003; Sui et al., 2007). Al-Ghananeem et al., found that PTX-loaded hyaluronan nanospheres disproportionately affected ER(−) MDA-MB-231 with a significant decrease in cytotoxicity on the ER(+) ZR-75-1 (Al-Ghananeem et al., 2009). Enrichment of the ER, as in the ZR-75-1 cell line, is associated with elevated levels of the anti-apoptotic protein bcl-2 (Tabuchi et al., 2009) and activity of the NF-κB transcription pathway (Ciucci et al., 2006; Emi et al., 2005) that reduce PTX apoptotic activity. Thus, the differential susceptibilities of the MDA-MB-435 and ZR-71-1 to PTX are well documented and correspond to the differences in the efficacy of PTX treatments in our xenograft models. Furthermore, we note that in PTX–NSP formulations NSP is a benign carrier and that all toxicity associated with cells in vitro is due to PTX.
Fig. 5.
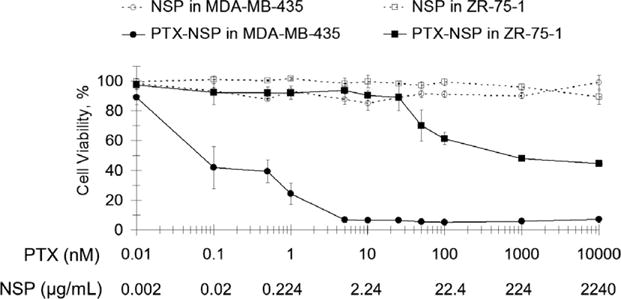
Viability of MDA-MB-435 and ZR-75-1 breast cancer cells exposed for 72 h to PTX–NSP and corresponding amounts of NSP alone. Cell viability was measured by AlamarBlue® cell proliferation indicator assay. Cells incubated with PBS and media were used as negative control and treatments data was normalized to these values. The results are expressed as mean value with its standard error (mean ± SD) indicated for 5 measurements of 3 independent experiments.
3.5. Pharmacokinetics study
The pharmacokinetics behavior of PTX–NSP at the dose of 15 mg/kg was assessed after a single treatment in healthy mice. The pharmacokinetic parameters (PK) of PTX were calculated using non-compartmental methods and WinNonlin (version 5.2). Plasma PK values for PTX–CrEL-D formulation, reported from a similar study of PTX–CrEL (11.25 mg/kg) (Eiseman et al., 1994), were used for comparison. As shown in Table 3, the pharmacokinetic properties of PTX are clearly affected by the composition of the delivery vehicle. A notably larger volume of distribution (Vd), higher clearance (CLt), and longer half-life (t½β) were measured in PTX–NSP treatment. The fact that the Vd and CL parameters are higher suggests that: (1) CrEL temporary inhibits PTX distribution early after administration, thereby potentially preventing the drug from reaching primary sites of action, and (2) non-lipophilic nanoparticle-based formulations are more suitable for the delivery of PTX. The decreased clearance of PTX–CrEL most likely caused by the drug entrapment by a highly hydrophobic interior of micellar-like structures of CrEL formed upon administration of PTX–CrEL (ten Tije et al., 2003). Further, longer t½β of PTX in PEGylated NSP could be attributed to the reduced uptake of formulation by the reticulo-endothelial system (RES) (Torchilin, 2001). It is important to note that a similar pharmacokinetic trend (faster CL and greater Vd) was reported for the clinically used an albumin-bound nanoparticle (ABI-007, Abraxane) and CrEL formulations (Sparreboom et al., 2005).
Table 3.
Pharmacokinetics parameters of PTX after single injection of PTX–NSP and PTX–CrEL.
| PTX formulation
|
||
|---|---|---|
| 15 mg/kg in NSP | 11 mg/kg CrEL* | |
| AUC (μg min/mL) | 1802 ± 16 | 1990 |
| Kel (min−1) | 0.008 ± 0.002 | 0.018 |
| t½β (min) | 67 ± 4 | 39 |
| Vd (mL/kg) | 10,200 ± 1295 | 320 |
| CLt (mL kg/min) | 84 ± 7 | 5 |
Historical PTX–CrEL data based on published by Eiseman et al. (1994).
4. Conclusion
The aim of this study was to evaluate the clinical potential of tyrosine-derived nanospheres (NSP) as delivery system for anticancer drug paclitaxel. PTX–NSP treatment showed an equivalent anti-tumor efficacy to the clinically used PTX formulation in Cremophor in two breast cancer xenograft models, while providing a superior safety and a significantly improved tolerability to higher PTX doses compared to clinically used Cremophor. Although additional studies are needed in order to fully characterize the nature of tumor uptake and biodistribution as well as assess the anti-tumor efficacy at higher PTX doses, current findings suggest that PTX–NSP could be a promising modality for solid mammary tumors.
Acknowledgments
The authors thank Drs. David Devore (US Army Institute for Surgical Research) and Eric H. Rubin (Clinical Oncology Research at Merck & Co., Inc.) for useful discussions and insights on efficacy and PK studies, Ms. Jin-Liern Hong (The Cancer Institute of New Jersey) for the assistance with PK studies, Dr. Hak-Joon Sung (former Rutgers University, NJCBM and current Vanderbilt University) and Mr. Jonathan Branch (Rutgers University, NJCBM) for the assistance with in vitro cytotoxicity studies. The work was supported by the Breast Cancer Research Program supported by the Office of the Congressionally Directed Medical Research Programs (Grant No. W81XWH-04-2-0031) and the New Jersey Center for Biomaterials.
Abbreviations
- DTO
desaminotyrosyl-tyrosine octyl ester
- SA
suberic acid
- PEG
poly(ethylene glycol)
- PTX
paclitaxel
- NSP
nanosphere(s)
- PTX-NSP
paclitaxel-nanospheres
- CrEL
chremophor
- CrEL-D
chremophor diluted with PBS
- ER(+) and ER(−)
estrogen receptor positive and estrogen receptor negative
- LE
loading efficiency
- MTD
maximum tolerated dose
- q5dx4
every 5 days four times treatment schedule
- IV
intravenous
- GI
gastrointestinal tract
- PK
pharmacokinetics
- SE
standard error
References
- Adams JD, Flora KP, Goldspiel BR, Wilson JW, Arbuck SG, Finley R. Taxol: a history of pharmaceutical development and current pharmaceutical concerns. J Natl Cancer Inst Monogr. 1993:141–147. [PubMed] [Google Scholar]
- Ahmed F, Pakunlu RI, Brannan A, Bates F, Minko T, Discher DE. Biodegradable polymersomes loaded with both paclitaxel and doxorubicin permeate and shrink tumors, inducing apoptosis in proportion to accumulated drug. J Controlled Release. 2006;116:150–158. doi: 10.1016/j.jconrel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Al-Ghananeem AM, Malkawi AH, Muammer YM, Balko JM, Black EP, Mourad W, Romond E. Intratumoral delivery of Paclitaxel in solid tumor from biodegradable hyaluronan nanoparticle formulations. AAPS PharmSciTech. 2009;10:410–417. doi: 10.1208/s12249-009-9222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf B: Biointerfaces. 1999;16:3–27. [Google Scholar]
- Bardelmeijer HA, Ouwehand M, Malingre MM, Schellens JH, Beijnen JH, van Tellingen O. Entrapment by Cremophor EL decreases the absorption of paclitaxel from the gut. Cancer Chemother Pharmacol. 2002;49:119–125. doi: 10.1007/s00280-001-0394-2. [DOI] [PubMed] [Google Scholar]
- Bourke SL, Kohn J. Polymers derived from the amino acid L-tyrosine: polycarbonates, polyarylates and copolymers with poly(ethylene glycol) Adv Drug Deliv Rev. 2003;55:447–466. doi: 10.1016/s0169-409x(03)00038-3. [DOI] [PubMed] [Google Scholar]
- Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC. PLGA-lecithin-PEG core–shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30:1627–1634. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Chandna P, Khandare JJ, Ber E, Rodriguez-Rodriguez L, Minko T. Multifunctional tumor-targeted polymer–peptide–drug delivery system for treatment of primary and metastatic cancers. Pharm Res. 2010;27:2296–2306. doi: 10.1007/s11095-010-0235-2. [DOI] [PubMed] [Google Scholar]
- Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Controlled Release, Epub ahead of print. 2011 doi: 10.1016/j.jconrel.2011.01.030. [DOI] [PubMed] [Google Scholar]
- Ciucci A, Gianferretti P, Piva R, Guyot T, Snape TJ, Roberts SM, Santoro MG. Induction of apoptosis in estrogen receptor-negative breast cancer cells by natural and synthetic cyclopentenones: role of the IkappaB kinase/nuclear factor-kappaB pathway. Mol Pharmacol. 2006;70:1812–1821. doi: 10.1124/mol.106.025759. [DOI] [PubMed] [Google Scholar]
- Corbett TH, Valeriote FA, Demchik L, Lowichik N, Polin L, Panchapor C, Pugh S, White K, Kushner J, Rake J, Wentland M, Golakoti T, Hetzel C, Ogino J, Patterson G, Moore R. Discovery of cryptophycin-1 and BCN-183577: examples of strategies and problems in the detection of antitumor activity in mice. Invest New Drugs. 1997;15:207–218. doi: 10.1023/a:1005875015011. [DOI] [PubMed] [Google Scholar]
- Costache AD, Sheihet L, Zaveri K, Knight DD, Kohn J. Polymer–drug interactions in tyrosine-derived triblock copolymer nanospheres: a computational modeling approach. Mol Pharm. 2009;6(5):1620–1627. doi: 10.1021/mp900114w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damascelli B, Patelli GL, Lanocita R, Di Tolla G, Frigerio LF, Marchiano A, Garbagnati F, Spreafico C, Ticha V, Gladin CR, Palazzi M, Crippa F, Oldini C, Calo S, Bonaccorsi A, Mattavelli F, Costa L, Mariani L, Cantu G. A novel intraarterial chemotherapy using paclitaxel in albumin nanoparticles to treat advanced squamous cell carcinoma of the tongue: preliminary findings. AJR Am J Roentgenol. 2003;181:253–260. doi: 10.2214/ajr.181.1.1810253. [DOI] [PubMed] [Google Scholar]
- Danhier F, Lecouturier N, Vroman B, Jerome C, Marchand-Brynaert J, Feron O, Preat V. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Controlled Release. 2009;133:11–17. doi: 10.1016/j.jconrel.2008.09.086. [DOI] [PubMed] [Google Scholar]
- Daniels JA, Gibson MK, Xu L, Sun S, Canto MI, Heath E, Wang J, Brock M, Montgomery E. Gastrointestinal tract epithelial changes associated with taxanes: marker of drug toxicity versus effect. Am J Surg Pathol. 2008;32:473–477. doi: 10.1097/PAS.0b013e3181582331. [DOI] [PubMed] [Google Scholar]
- Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D, Noker P, Yao R, Labao E, Hawkins M, Soon-Shiong P. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of Cremophor-free, albumin-bound paclitaxel, ABI-007, compared with Cremophor-based paclitaxel. Clin Cancer Res. 2006;12:1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- Dharap SS, Wang Y, Chandna P, Khandare JJ, Qiu B, Gunaseelan S, Sinko PJ, Stein S, Farmanfarmaian A, Minko T. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci USA. 2005;102:12962–12967. doi: 10.1073/pnas.0504274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilnawaz F, Singh A, Mohanty C, Sahoo SK. Dual drug loaded superparamagnetic iron oxide nanoparticles for targeted cancer therapy. Biomaterials. 2010;31:3694–3706. doi: 10.1016/j.biomaterials.2010.01.057. [DOI] [PubMed] [Google Scholar]
- Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- Eiseman JL, Eddington ND, Leslie J, MacAuley C, Sentz DL, Zuhowski M, Kujawa JM, Young D, Egorin MJ. Plasma pharmacokinetics and tissue distribution of paclitaxel in CD2F1 mice. Cancer Chemother Pharmacol. 1994;34:465–471. doi: 10.1007/BF00685656. [DOI] [PubMed] [Google Scholar]
- Emi M, Kim R, Tanabe K, Uchida Y, Toge T. Targeted therapy against Bcl-2-related proteins in breast cancer cells. Breast Cancer Res. 2005;7:R940–R952. doi: 10.1186/bcr1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- Forrest ML, Yanez JA, Remsberg CM, Ohgami Y, Kwon GS, Davies NM. Paclitaxel prodrugs with sustained release and high solubility in poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelle nanocarriers: pharmacokinetic disposition, tolerability, and cytotoxicity. Pharm Res. 2008;25:194–206. doi: 10.1007/s11095-007-9451-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaucher G, Marchessault RH, Leroux JC. Polyester-based micelles and nanoparticles for the parenteral delivery of taxanes. J Controlled Release. 2010;143:2–12. doi: 10.1016/j.jconrel.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–1598. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- Gligorov J, Lotz JP. Preclinical pharmacology of the taxanes: implications of the differences. Oncologist. 2004;9:3–8. doi: 10.1634/theoncologist.9-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- Goldspiel BR. Clinical overview of the taxanes. Pharmacotherapy. 1997;17:110S–125S. [PubMed] [Google Scholar]
- Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliv Rev. 2003;55:217–250. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- Grodzinski P, Silver M, Molnar LK. Nanotechnology for cancer diagnostics: promises and challenges. Expert Rev Mol Diagn. 2006;6:307–318. doi: 10.1586/14737159.6.3.307. [DOI] [PubMed] [Google Scholar]
- Guastalla JP, Lhomme C, Dauplat J, Namer M, Bonneterre J, Oberling F, Pouillart P, Fumoleau P, Kerbrat P, Tubiana N, et al. Taxol (paclitaxel) safety in patients with platinum pretreated ovarian carcinoma: an interim analysis of a phase II multicenter study. Ann Oncol. 1994;5(Suppl. 6):S33–S38. [PubMed] [Google Scholar]
- Haag R. Supramolecular drug-delivery systems based on polymeric core–shell architectures. Angew Chem, Int Ed Engl. 2004;43:278–282. doi: 10.1002/anie.200301694. [DOI] [PubMed] [Google Scholar]
- Henderson IC, Berry DA, Demetri GD, Cirrincione CT, Goldstein LJ, Martino S, Ingle JN, Cooper MR, Hayes DF, Tkaczuk KH, Fleming G, Holland JF, Duggan, Carpenter JT, Frei E, 3rd, Schilsky RL, Wood WC, Muss HB, Norton L. Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol. 21:976–983. doi: 10.1200/JCO.2003.02.063. [DOI] [PubMed] [Google Scholar]
- Heney M, Alipour M, Vergidis D, Omri A, Mugabe C, Th’ng J, Suntres Z. Effectiveness of liposomal paclitaxel against MCF-7 breast cancer cells. Can J Physiol Pharmacol. 2010;88:1172–1180. doi: 10.1139/Y10-097. [DOI] [PubMed] [Google Scholar]
- Holland SJ, Tighe BJ, Gould PL. Polymers for biodegradable medical devices. 1. The potential of polyesters as controlled macromolecular release systems. J Controlled Release. 1986;4:155–180. [Google Scholar]
- Johnson B, Toland B, Chokshi R, Mochalin V, Koutzaki S, Polyak B. Magnetically responsive paclitaxel-loaded biodegradable nanoparticles for treatment of vascular disease: preparation, characterization and in vitro evaluation of anti-proliferative potential. Curr Drug Deliv. 2010;7:263–273. doi: 10.2174/156720110793360621. [DOI] [PubMed] [Google Scholar]
- Kunstfeld R, Wickenhauser G, Michaelis U, Teifel M, Umek W, Naujoks K, Wolff K, Petzelbauer P. Paclitaxel encapsulated in cationic liposomes diminishes tumor angiogenesis and melanoma growth in a “humanized” SCID mouse model. J Invest Dermatol. 2003;120:476–482. doi: 10.1046/j.1523-1747.2003.12057.x. [DOI] [PubMed] [Google Scholar]
- Letchford K, Liggins R, Wasan KM, Burt H. In vitro human plasma distribution of nanoparticulate paclitaxel is dependent on the physicochemical properties of poly(ethylene glycol)-block-poly(caprolactone) nanoparticles. Eur J Pharm Biopharm. 2009;71:196–206. doi: 10.1016/j.ejpb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Liang Y, Qiao Y, Guo S, Wang L, Zhai Y, Xie C, Hu R, Deng L, Dong A. Investigation on injectable, thermally and physically gelable poly(ethylene glycol)/poly(octadecanedioic anhydride) amphiphilic triblock co-polymer nanoparticles. J Biomater Sci Polym Ed. 2011 Jan 28; doi: 10.1163/092050610X552230. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Liggins RT, Burt HM. Polyether–polyester diblock copolymers for the preparation of paclitaxel loaded polymeric micelle formulations. Adv Drug Deliv Rev. 2002;54:191–202. doi: 10.1016/s0169-409x(02)00016-9. [DOI] [PubMed] [Google Scholar]
- Lim J, Simanek EE. Synthesis of water-soluble dendrimers based on melamine bearing 16 paclitaxel groups. Org Lett. 2008;10:201–204. doi: 10.1021/ol7024907. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008;68:6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Liong M, Sherman S, Xia T, Kovochich M, Nel AE, Zink JI, Tamanoi F. Mesoporous silica nanoparticles for cancer therapy: energy-dependent cellular uptake and delivery of paclitaxel to cancer cells. Nanobiotechnology. 2007;3:89–95. doi: 10.1007/s12030-008-9003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H. The tumor blood vessel as an ideal target for macromolecular anticancer agents. J Controlled Release. 1992;19:315–324. [Google Scholar]
- Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- Milane L, Duan ZF, Amiji M. Biodistribution and pharmacokinetic analysis of combination lonidamine and paclitaxel delivery in an orthotopic animal model of multidrug-resistant breast cancer using EGFR-targeted polymeric nanoparticles. Nanomed Nanotechnol Biol Med. 2011 Jan 8; doi: 10.1016/j.nano.2010.12.009. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko T, Patil ML, Zhang M, Khandare JJ, Saad M, Chandna P, Taratula O. LHRH-targeted nanoparticles for cancer therapeutics. Methods Mol Biol. 2010;624:281–294. doi: 10.1007/978-1-60761-609-2_19. [DOI] [PubMed] [Google Scholar]
- Mitra A, Lin S. Effect of surfactant on fabrication and characterization of paclitaxel-loaded polybutylcyanoacrylate nanoparticulate delivery systems. J Pharm Pharmacol. 2003;55:895–902. doi: 10.1211/0022357021341. [DOI] [PubMed] [Google Scholar]
- Nardin C, Bolikal D, Kohn J. Nontoxic block copolymer nanospheres: design and characterization. Langmuir. 2004;20:11721–11725. doi: 10.1021/la0490285. [DOI] [PubMed] [Google Scholar]
- Ohtsu T, Sasaki Y, Tamura T, Miyata Y, Nakanomyo H, Nishiwaki Y, Saijo N. Clinical pharmacokinetics and pharmacodynamics of paclitaxel: a 3-hour infusion versus a 24-hour infusion. Clin Cancer Res. 1995;1:599–606. [PubMed] [Google Scholar]
- Ooya T, Lee J, Park K. Hydrotropic dendrimers of generations 4 and 5: synthesis, characterization, and hydrotropic solubilization of paclitaxel. Bioconjug Chem. 2004;15:1221–1229. doi: 10.1021/bc049814l. [DOI] [PubMed] [Google Scholar]
- Potineni A, Lynn DM, Langer R, Amiji MM. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive biodegradable system for paclitaxel delivery. J Controlled Release. 2003;86:223–234. doi: 10.1016/s0168-3659(02)00374-7. [DOI] [PubMed] [Google Scholar]
- Reul R, Renette T, Bege N, Kissel T. Nanoparticles for paclitaxel delivery: a comparative study of different types of dendritic polyesters and their degradation behavior. Int J Pharm. 2011;407:190–196. doi: 10.1016/j.ijpharm.2011.01.028. [DOI] [PubMed] [Google Scholar]
- Rivera E. Liposomal anthracyclines in metastatic breast cancer: clinical update. Oncologist. 2003;8(Suppl. 2):3–9. doi: 10.1634/theoncologist.8-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- Sheihet L, Chandra P, Batheja P, Devore D, Kohn J, Michniak B. Tyrosine-derived nanospheres for enhanced topical skin penetration. Int J Pharm. 2008;350:312–319. doi: 10.1016/j.ijpharm.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Sheihet L, Dubin RA, Devore D, Kohn J. Hydrophobic drug delivery by self-assembling triblock copolymer-derived nanospheres. Biomacromolecules. 2005;6:2726–2731. doi: 10.1021/bm050212u. [DOI] [PubMed] [Google Scholar]
- Sheihet L, Piotrowska K, Dubin RA, Kohn J, Devore D. Effect of tyrosine-derived triblock copolymer compositions on nanosphere self-assembly and drug delivery. Biomacromolecules. 2007;8:998–1003. doi: 10.1021/bm060860t. [DOI] [PubMed] [Google Scholar]
- Shenoy D, Little S, Langer R, Amiji M. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs. Part 2: In vivo distribution and tumor localization studies. Pharm Res. 2005;22:2107–2114. doi: 10.1007/s11095-005-8343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Yu B, Wei Y, Wientjes MG, Au JL. Low-dose suramin enhanced paclitaxel activity in chemotherapy-naive and paclitaxel-pretreated human breast xenograft tumors. Clin Cancer Res. 2004;10:6058–6065. doi: 10.1158/1078-0432.CCR-04-0595. [DOI] [PubMed] [Google Scholar]
- Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, Desai N. Comparative preclinical and clinical pharmacokinetics of a Cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol) Clin Cancer Res. 2005;11:4136–4143. doi: 10.1158/1078-0432.CCR-04-2291. [DOI] [PubMed] [Google Scholar]
- Sparreboom A, van Zuylen L, Brouwer E, Loos WJ, de Bruijn P, Gelderblom H, Pillay M, Nooter K, Stoter G, Verweij J. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res. 1999;59:1454–1457. [PubMed] [Google Scholar]
- Stolnik S, Dunn SE, Garnett MC, Davies MC, Coombes AG, Taylor DC, Irving MP, Purkiss SC, Tadros TF, Davis SS, et al. Surface modification of poly(lactide-co-glycolide) nanospheres by biodegradable poly(lactide)-poly(ethylene glycol) copolymers. Pharm Res. 1994;11:1800–1808. doi: 10.1023/a:1018931820564. [DOI] [PubMed] [Google Scholar]
- Sui M, Huang Y, Park BH, Davidson NE, Fan W. Estrogen receptor alpha mediates breast cancer cell resistance to paclitaxel through inhibition of apoptotic cell death. Cancer Res. 2007;67:5337–5344. doi: 10.1158/0008-5472.CAN-06-4582. [DOI] [PubMed] [Google Scholar]
- Tabuchi Y, Matsuoka J, Gunduz M, Imada T, Ono R, Ito M, Motoki T, Yamatsuji T, Shirakawa Y, Takaoka M, Haisa M, Tanaka N, Kurebayashi J, Jordan VC, Naomoto Y. Resistance to paclitaxel therapy is related with Bcl-2 expression through an estrogen receptor mediated pathway in breast cancer. Int J Oncol. 2009;34:313–319. [PubMed] [Google Scholar]
- ten Tije AJ, Verweij J, Loos WJ, Sparreboom A. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin Pharmacokinet. 2003;42:665–685. doi: 10.2165/00003088-200342070-00005. [DOI] [PubMed] [Google Scholar]
- Toppmeyer DL, Gounder M, Much J, Musanti R, Vyas V, Medina M, Orlando T, Pennick M, Lin Y, Shih W, Goodin S, Rubin E. A phase I and pharmacologic study of the combination of marimastat and paclitaxel in patients with advanced malignancy. Med Sci Monit. 2003;9:PI99–PI104. [PubMed] [Google Scholar]
- Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Controlled Release. 2001;73:137–172. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- Tsallas A, Jackson J, Burt H. The uptake of paclitaxel and docetaxel into ex vivo porcine bladder tissue from polymeric micelle formulations. Cancer Chemother Pharmacol. 2010 Nov 11; doi: 10.1007/s00280-010-1499-2. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Vassileva V, Grant J, De Souza R, Allen C, Piquette-Miller M. Novel biocompatible intraperitoneal drug delivery system increases tolerability and therapeutic efficacy of paclitaxel in a human ovarian cancer xenograft model. Cancer Chemother Pharmacol. 2007;60:907–914. doi: 10.1007/s00280-007-0449-0. [DOI] [PubMed] [Google Scholar]
- Wang YC, Tang LY, Sun TM, Li CH, Xiong MH, Wang J. Self-assembled micelles of biodegradable triblock copolymers based on poly(ethyl ethylene phosphate) and poly(ε-caprolactone) as drug carriers. Biomacromolecules. 2008;9:388–395. doi: 10.1021/bm700732g. [DOI] [PubMed] [Google Scholar]
- Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, Baker JR, Jr, Van Echo DA, Von Hoff DD, Leyland-Jones B. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8:1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- Xu ZK, Nie FQ, Qu C, Wan LS, Wu J, Yao K. Tethering poly(ethylene glycol)s to improve the surface biocompatibility of poly(acrylonitrile-co-maleic acid) asymmetric membranes. Biomaterials. 2005;26:589–598. doi: 10.1016/j.biomaterials.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Zahr AS, Pishko MV. Encapsulation of paclitaxel in macromolecular nanoshells. Biomacromolecules. 2007;8:2004–2010. doi: 10.1021/bm070177m. [DOI] [PubMed] [Google Scholar]
- Zhao M, Liang C, Li A, Chang J, Wang H, Yan R, Zhang J, Tai J. Magnetic paclitaxel nanoparticles inhibit glioma growth and improve the survival of rats bearing glioma xenografts. Anticancer Res. 2010;30:2217–2223. [PubMed] [Google Scholar]