

Abstract
Phosphoinositide 3-kinases (PI-3Ks) are enzymes that generate lipid second messenger molecules, resulting in the activation of multiple intracellular signalling cascades. These events regulate a broad array of cellular responses including survival, activation, differentiation and proliferation and are now recognised to have a key role in a number of physiological and pathophysiological processes in the lung. PI-3Ks contribute to the pathogenesis of asthma by influencing the proliferation of airways smooth muscle and the recruitment of eosinophils, and affect the balance between the harmful and protective responses in pulmonary inflammation and infection by the modulation of granulocyte recruitment, activation and apoptosis. In addition they also seem to exert a critical influence on the malignant phenotype of small cell lung cancer. PI-3K isoforms and their downstream targets thus provide novel therapeutic targets for intervention in a broad spectrum of respiratory diseases.
Keywords: airways smooth muscle; lung; phosphatidylinositol 3,4,5-trisphosphate; phosphoinositide 3-kinase; small cell lung cancer
Introduction
Although characterised only in the late 1980s, a vast literature now exists detailing the critical roles of the ubiquitous phosphoinositide 3-kinase (PI-3K) enzyme family in mitogenesis, cell survival, differentiation and activation, cytoskeletal remodelling and vesicular trafficking. PI-3Ks are lipid kinases - enzymes that phosphorylate membrane-associated lipids of the phosphoinositide family - and the resulting 3-phosphorylated lipids recruit and activate downstream targets to initiate a novel set of signalling cascades, culminating in the varied cellular responses listed above (see Figure 1). Although great progress has been made in elucidating the structure and mechanism of action of the PI-3Ks themselves, the identity and function of the downstream targets and their interactions with other signalling cascades within the cell are only just being unravelled.
Figure 1.
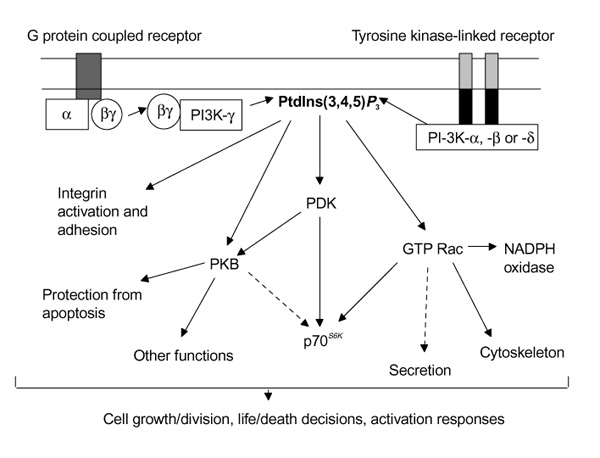
The PI-3K signalling network. Binding of ligand to receptors linked to G protein or tyrosine kinase activates PI-3K-γ and PI-3K-α, PI-3K-β or PI-3K-δ respectively. The resultant accumulation of phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5) P3] activates downstream signalling cascades leading to adhesion, proliferation, survival and activation responses. Putative pathways or those demonstrated in only a limited number of cell types are depicted by broken arrows.
Three classes of PI-3K are recognised on the basis of their structure, substrate specificity and regulation. Class I PI-3Ks are heterodimers comprising a catalytic (p110) and a regulatory (p50, p55, p85 or p101) subunit; in the resting unstimulated cell they are predominantly cytosolic and require activation (usually by a mechanism driven by cell-surface receptors) to display significant activity. These enzymes preferentially phosphorylate the constitutive plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate to generate the critical second messenger phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3]. PtdIns(3,4,5) P3 is metabolised by enzymes called phosphatases to phosphatidylinositol 3,4-bisphosphate, which itself can also act as a second messenger, and thence to phosphatidylinositol 3-phosphate. Two subfamilies of Class I PI-3K have been distinguished. Class IA comprises either an α, β or δ p110 catalytic subunit plus one of a family of regulatory subunits (p85α, p85β, p55γ and their splice variants), and are sensitive to activation by tyrosine kinase-linked receptor transduction systems (such as those initiated by the binding of growth factors to their receptors). The only Class IB PI-3K so far identified consists of a p110γ catalytic subunit and a unique p101 regulatory subunit. This enzyme is activated by βγ subunits derived from activated G-protein-coupled receptors (eg chemokine receptors) and, together with the Class 1A p110δ, seems to be expressed only in haematopoietic cells. All Class I PI-3K catalytic subunits contain a PI-kinase domain, a protein kinase domain and a Ras-binding domain. Recent crystallographic studies [1*] have shown that p110γ has a central helical spine, with the catalytic domain positioned to interact with phospholipid membranes and the Ras-binding domain placed adjacent to the catalytic domain, where it most probably drives the allosteric activation of the enzyme. The monomeric Class II PI-3Ks 3-phosphorylate phosphatidylinositol 4-phosphate and PtdIns, but their role in mammalian systems is unclear. Class III PI-3Ks use only PtdIns as a substrate; they do not seem to be regulated acutely by cell-surface receptors and have been implicated in cellular 'housekeeping' functions, particularly protein and vesicular trafficking.
The activation of Class I PI-3Ks results in the generation of membrane-associated PtdIns(3,4,5)P3, levels of which increase substantially (up to 50-fold) in appropriately stimulated cells. Proteins containing pleckstrin homology domains bind PtdIns(3,4,5) P3 with high affinity and thus are recruited to the plasma membrane, thereby bringing them into juxtaposition with their substrates and in some cases with upstream activating enzymes. This recruitment of pleckstrin homology domain-containing proteins in response to PtdIns(3,4,5) P3generation can be imaged directly in live cells by using fluorescently tagged target proteins. The binding of such proteins to PtdIns(3,4,5) P3 might result in direct allosteric activation, although definitive proof for this is currently lacking. Examples of proteins activated by PtdIns(3,4,5) P3 include phosphoinositide-dependent kinase-1 (PDK1), protein kinase B (PKB/AKT, implicated in cell survival), p70S6K (involved in mitogenesis), members of the protein kinase C family, phospholipase Cγ, and several small molecular signalling intermediates including Rac, Vav, Tiam-1 and centaurin-α . Techniques used to identify these PtdIns(3,4,5) P3-binding proteins and elucidate their functional roles include the use of selective PI-3K inhibitors (wortmannin and LY294002), development of constitutively active and dominant-negative forms of PI-3K and its targets, enzyme activity assays and the use of fluorescently labelled proteins. Although these and other methods have been applied principally in immortalised cell lines, more recent studies have used primary cell cultures. As exampled below, these investigations are now providing fascinating insights into the roles of the PI-3Ks and their accompanying signalling cascades in several tissues, including the lung.
PI3 kinase in proliferative responses
Airways smooth muscle
Although reversible airway narrowing leading to wheeze, cough and shortness of breath is a hallmark of asthma, patients with longstanding and severe disease can develop fixed airways obstruction that is associated with structural changes within the airway wall. The most prominent feature of the remodelled airway is an increase in airways smooth muscle (ASM). Heard and Hossain [2] demonstrated a threefold increase in both the cross-sectional area and number of smooth-muscle cells found within the bronchial wall of patients with fatal asthma in comparison with those dying from non-respiratory conditions. Subsequent mathematical modelling suggests that this characteristic increase in smooth-muscle bulk is the major cause of narrowing of airways in such patients. Furthermore, excessive ASM DNA synthesis has been demonstrated in at least two animal models of airways disease, including that associated with antigen challenge [3,4].
Although it has long been recognised that the signalling pathways based on extracellular signal-related kinase (ERK) and protein kinase C are involved in the mitogenic response of ASM, the importance of PI-3K has only recently been demonstrated. Scott et al [5] first showed that PI-3K activity was proportional to the mitogenic response in bovine ASM cells in culture and that the inhibition of PI-3K by wortmannin substantially (more than 90%) decreased DNA synthesis. They also provided data implicating p70S6Kas the probable downstream mediator of these effects. p70S6K is known to be activated by PI-3K (via PDK1 and possibly PKB/AKT and/or RAC) and is now known to be essential for the progression of cells from G1 to S phase in the cell cycle. Further studies [6] demonstrated a rapid activation of PI-3K and accumulation of PtdIns(3,4,5) P3 after stimulation of bovine ASM by thrombin and platelet-derived growth factor (PDGF), and showed that the magnitude of these effects was closely correlated with the mitogenic potential of these two growth factors. Krymskaya et al [7*] confirmed the requirement for PI-3K activity in human ASM cell mitogenesis, and again implicated p70S6K as an important mediator in this response. Rac 1 has also now been shown to be important as a downstream mediator of the PI-3K mitogenic effect [8], acting via the induction of cyclin D, which is required for cell cycle progression.
Small cell lung cancer
PI-3K activity has been shown to be critical for the integrin-mediated invasive behaviour of breast and colon carcinoma cell lines [9], and the proto-oncogene PKB/AKT [recruited by PtdIns(3,4,5) P3 to the plasma membrane and phosphorylated by the PI-3K-dependent PDK1] has been demonstrated to be overexpressed in ovarian, breast and pancreatic cancer. Small cell lung cancer (SCLC) is the most aggressive and invasive form of lung cancer, with a highly metastatic phenotype and a 5-year survival of only 3-8%. Interest in the role of PI-3K in the malignant potential of this disease was stimulated by the observation [10] that p70S6K is constitutively phosphorylated and activated in SCLC cells, and that rapamycin, which inhibits the activation of p70S6K, blocks SCLC proliferation. These observations were extended by Moore et al [11**], who demonstrated a high constitutive activity of PI-3K, PKB/AKT and p70S6K in SCLC cell lines and showed that the proliferation of SCLC cells in liquid culture was inhibited by the PI-3K inhibitor LY294002. This inhibition resulted from a combination of decreased mitogenesis and enhanced apoptosis (see below). PI-3K inhibition also decreased SCLC cell colony formation in semi-solid media. Thus the high constitutive activity of PI-3K in these cells seems to promote growth and also anchorage-independence, contributing to the highly aggressive nature of this tumour. These observations have not yet been extended to other human lung cancer cell types; further developments are awaited.
PI3 kinase in activation responses
Neutrophils
Although not resident pulmonary cells, substantial numbers of neutrophils are recruited to the lungs in many respiratory disease states and have a critical role in the pathogenesis of the acute respiratory distress syndrome (ARDS), pulmonary fibrosis, bronchiectasis and fatal asthma. Neutrophils cause tissue damage by their capacity to release toxic oxygen radicals (generated by the NADPH oxidase complex), the exocytosis of granules containing highly histotoxic compounds such as elastase and collagenases, and the elaboration and release of additional pro-inflammatory cytokines. PI-3Ks have been shown to be key regulators of both neutrophil recruitment and activation. In mice lacking the catalytic subunit of the myeloid restricted PI-3K-γ, neutrophil migration to the inflamed peritoneum was severely compromised [12**, 13**] and, although not examined directly, a similar defect in granulocyte recruitment to the lungs is likely. The accumulation of PtdIns(3,4,5) P3 also seems to be correlated precisely with respiratory burst activity, in that neutrophil priming by agents such as tumour necrosis factor-α markedly enhanced both the size and the duration of the release of superoxide anions and the accumulation of PtdIns(3,4,5) P3 [14]. In these cells PI-3K inhibitors abolish the production of oxygen radicals induced by physiological agonists; neutrophils from PI-3K-γ knockout mice exhibit a diminished respiratory burst, with residual activity most probably attributable to the action of remaining Class IA PI-3K. The signalling intermediates linking PtdIns(3,4,5) P3 to activation of the oxidase are uncertain but most probably include the small GTPase Rac 2, which is both highly expressed in neutrophils and an essential component of the NADPH oxidase complex. The role of PI-3K in granule exocytosis is less clearly delineated as high concentrations of wortmannin only partialy inhibit this process, indicating that inputs from other signalling pathways might impinge on this response.
Eosinophils
Like neutrophils, eosinophils are non-resident pulmonary cells that accumulate in the bronchial tree and lung parenchyma in a number of disease states, including asthma and eosinophilic pneumonia. Toxic eosinophil-derived mediators such as eosinophil cationic protein, major basic protein and oxygen radicals (again products of the NADPH oxidase) can damage the airway epithelium and are thought to contribute significantly to airway hyper-responsiveness. In non-allergic subjects, eosinophils are scarce in peripheral blood and have therefore proved more difficult than neutrophils to study. Despite this, IL-5-stimulated eosinophil release from bone marrow has been shown to be inhibited by both wortmannin and LY294002 [15]; the migration of eosinophils to a number of chemoattractants also seems to be sensitive to wortmannin [16]. The role of PI-3Ks in eosinophil degranulation is not known, but these enzymes are required for activation of the eosinophil NADPH oxidase complex [17].
Alveolar macrophages
Alveolar macrophages undertake a number of key host defence functions within the lung. These include the phagocytosis of inhaled particles and respiratory pathogens, antigen presentation, and the generation of inflammatory cytokines. Additionally, they might be important in the resolution of acute inflammation by the ingestion of apoptotic neutrophils. So far, although few studies have addressed the role of PI-3Ks in the alveolar macrophage, such data are available for monocyte-derived macrophages, macrophage cell lines and murine peritoneal macrophages. If we extrapolate these results to alveolar macrophages, it seems highly likely that PI-3Ks will again be shown to have a critical role in the response profile of these cells. Hence, murine PI-3K-γ -null macrophages show decreased migration towards a variety of chemotactic agents, and greatly diminished recruitment to the inflamed peritoneum [13**]. The induction of cytokine gene expression in monocytes stimulated by formylated peptide has also been shown to be sensitive to PI-3K inhibition [18]. Most importantly, the consequences of excessive PI-3K activation have also been explored; mice deficient in SHIP (SH2-containing inositol-5-phosphatase), an enzyme that hydrolyses PtdIns(3,4,5) P3, suffer from lethal infiltration of the lungs by myeloid cells, principally macrophages [19]. A remarkably similar phenotype is seen in mice with a deletion of the tyrosine phosphatase SHP-1 (Src homology 2 domain phosphatase-1); macrophages from these mice display a 10-15-fold increase in the 3-phosphorylated products of PI-3K, with enhanced integrin-dependent adhesive properties [20*]. Thus, as with neutrophils and eosinophils, it seems that one or more of the PI-3Ks is required for macrophage recruitment to the lung and for at least a subset of activation responses.
Although comparatively little is known about the function of the PI-3Ks in other pulmonary cells, several reports have emerged and indicate the global importance of this signalling pathway in other settings in the lung. For example, Liu et al [21] have demonstrated that PI-3K is a downstream mediator of PDGF-stimulated glycosaminoglycan synthesis in rat foetal lung fibroblasts, suggesting a role in the maintenance of the lung extracellular matrix. Similarly, PI-3K has been reported to mediate lung epithelial cell differentiation and surfactant protein expression fibroblast induced by growth factor-2 [22], although subsequent reports have suggested that PI-3K inhibits surfactant secretion from type II alveolar cells [23]. Future work will doubtless help to clarify the role of PI-3Ks in the differentiation and function of pulmonary epithelial cells.
PI-3K in cell survival
In addition to their central role in cell proliferation and activation, Class I PI-3Ks have also been implicated as having a key role in inhibiting apoptotic cell death. PKB/AKT, a downstream effector of PI-3K, is believed to promote cell survival by the phosphorylation and inactivation of both caspase-9 (a central regulator of apoptosis) and the pro-apoptotic factor BAD. Granulocyte apoptosis is now thought to be important in the resolution of pulmonary inflammation; in recent months several papers have emerged that implicate PI-3K as a mediator of cell survival in neutrophils [24*] and monocytes [25], but not eosinophils [26]. Finally, inhibition of PI-3K activity in SCLC cell lines results in enhanced apoptosis [11**], suggesting that the high basal PI-3K activity observed in these cells might contribute to the malignant phenotype by inhibiting apoptosis.
Conclusion
Although the role played by Class I PI-3Ks in the embryological development and everyday 'housekeeping' functions of the normal lung remains unclear, it is evident that these enzymes are of central importance in a broad spectrum of respiratory diseases (see Figure 2). The importance of this signalling pathway in ASM mitogenesis and eosinophil recruitment and activation suggests that PI-3Ks might have a key role in the pathogenesis of asthma. PI-3Ks are also critical for the recruitment, activation and survival of neutrophils and thereby influence a wide range of inflammatory and infective conditions within the lung, including ARDS, pulmonary fibrosis and bronchiectasis. Additionally, work on SCLC cell lines points to PI-3Ks as being constitutively active and contributing to the malignant phenotype of this tumour, perhaps via the activation of the proto-oncogene PKB/AKT or p70S6K. The development of inhibitors of specific PI-3K isoforms (particularly of the myeloid-restricted PI-3K-δ and PI-3K-γ) and of downstream signalling targets might lead to novel therapeutic strategies for a variety of respiratory diseases.
Figure 2.
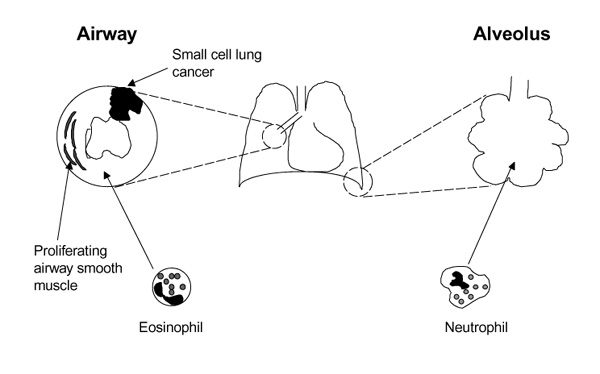
PI-3K in respiratory disease. Within the airway, activation of PI-3K is thought to contribute to the proliferation of smooth muscle and the accumulation of eosinophil characteristic of asthma, and to the mitogenesis and prolonged survival of small cell lung cancer cells. PI-3K-dependent neutrophil extravasation and activation have been implicated in the pathogenesis of multiple respiratory diseases including ARDS, pulmonary fibrosis, pulmonary vasculitides and bronchiectasis.
Acknowledgments
Acknowledgments
The authors' work is supported by the Wellcome Trust, MRC, National Asthma Campaign and British Lung Foundation. AC is a Wellcome Advanced Fellow.
References
- Walker EH, Perisic O, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. . Nature. 1999;402:313–320. doi: 10.1016/0022-328X(91)86245-L. [DOI] [PubMed] [Google Scholar]
- Heard BE, Hossain S. Hyperplasia of bronchial smooth muscle in asthma. J Pathol. 1973;110:319–332. [Google Scholar]
- Hershenson MB, Kelleher MD, Naureckas ET, Abe MK, Rubinstein VJ, Zimmermann A, Bendele AM, McNulty JA, Panettieri RA, Solway J. Hyperoxia increases airway cell S-phase traversal in immature rats in vivo. Am J Respir Cell Mol Biol. 1994;11:296–303. doi: 10.1165/ajrcmb.11.3.8086167. [DOI] [PubMed] [Google Scholar]
- Panettieri RA, Murray RK, Eszterhas AJ, Bilgen G, Martin JG. Repeated allergen inhalations induce DNA synthesis in airway smooth muscle and epithelial cell in vivo. Am J Physiol. 1998;274:L417–L424. doi: 10.1152/ajplung.1998.274.3.L417. [DOI] [PubMed] [Google Scholar]
- Scott PH, Belham CM, Al-Hafidh J, Chilvers ER, Peacock AJ. A regulatory role for cAMP in phosphatidylinositol 3-kinase/p70 ribosomal S6 kinase-mediated DNA synthesis in platelet-derived-growth-factor-stimulated bovine airway smooth-muscle cells. Biochem J. 1996;318:965–971. doi: 10.1042/bj3180965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TR, Moore SM, Lawson M F, Panettieri RA, Jr, Chilvers ER. Platelet-derived growth factor-BB and thrombin activate phosphoinositide 3-kinase and protein kinase B; role in mediating airway smooth muscle proliferation. Mol Pharmacol. 1998;54:1005–1015. doi: 10.1124/mol.54.6.1007. [DOI] [PubMed] [Google Scholar]
- Krymskaya VP, Penn RB, Orsini MJ, Scott PH, Plevin RJ, Walker TR, Eszterhas AJ, Amrani Y, Chilvers ER, Panettieri RA., Jr Phosphatidylinositol 3-kinase mediates mitogen-induced human airway smooth muscle cell proliferation. Am J Physiol. 1999;277:L65–L78. doi: 10.1152/ajplung.1999.277.1.L65. [DOI] [PubMed] [Google Scholar]
- Page K, Li J, Hodge JA, Liu PT, Vanden Hoek TL, Becker LB, Pestell RG, Rosner MR, Hershenson MB. Characterisation of a rac1 signalling pathway to cyclin D_ expression in airway smooth muscle cells. J Biol Chem. 1999;274:22065–22071. doi: 10.1074/jbc.274.31.22065. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Rabinovitz I, Wang HH-F, Toker A, Mercurio M. Activation of phosphoinositide 3-OH kinase by the α f6 β 4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Seufferlein T, Rozengurt E. Rapamycin inhibits constitutive p70S6K phosphorylation, cell proliferation and colony formation in small cell lung cancer cells. Cancer Res. 1996;56:3895–3897. [PubMed] [Google Scholar]
- Moore SM, Rintoul RC, Walker TR, Chilvers ER, Haslett C, Sethi T. The presence of a constitutively active phosphoinositide 3-kinase in small cell lung cancer cells mediates anchorage-independent proliferation via a protein kinase B and p70S6K-dependent pathway. . Cancer Res. 1998;58:5239–5247. [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM. Function of PI3Kγ in thymocyte development, T cell activation and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein coupled phosphoinositide 3-kinase in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Condliffe AM, Hawkins PT, Stephens LR, Haslett C, Chilvers ER. Priming of human neutrophil superoxide generation by tumour necrosis factor is signalled by enhanced phosphatidylinositol 3,4,5-trisphosphate but not inositol 1,4,5-trisphosphate accumulation. FEBS Lett. 1998;439:147–151. doi: 10.1016/S0014-5793(98)01358-1. [DOI] [PubMed] [Google Scholar]
- Palframen RT, Collins PD, Severs NJ, Rothery S, Williams TJ, Rankin SM. Mechanisms of acute eosinophil mobilization from bone marrow by interleukin 5: the role of specific adhesion molecules and phosphatidylinositol 3-kinase. J Exp Med. 1998;188:1621–1632. doi: 10.1084/jem.188.9.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunzendorfer S, Meirhofer C, Wiedermann CJ. Signaling in neuropeptide-induced migration of human eosinophils. J Leukoc Biol. 1998;64:828–834. doi: 10.1002/jlb.64.6.828. [DOI] [PubMed] [Google Scholar]
- Hofmann C, Dichmann S, Zimpfer U, Czech W, Herouy Y, Wagner E, Norgauer J. Metabolism and function of 3-D-phosphorylated phosphoinositides in C5a-stimulated eosinophils. Biochem Biophys Res Commun. 2000;269:816–821. doi: 10.1006/bbrc.2000.2365. [DOI] [PubMed] [Google Scholar]
- Pan ZK, Chen LY, Cochrane CG, Zuraw BL. fMet-leu-phe stimulates proinflammatory cytokine gene expression in human peripheral blood monocytes: the role of phosphatidylinositol 3-kinase. J Immunol. 2000;164:404–411. doi: 10.4049/jimmunol.164.1.404. [DOI] [PubMed] [Google Scholar]
- Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, Borowski A, Jirik F, Krystal G, Humphries RK. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach TI, Slater SE, White LS, Zhang X, Majerus PW, Brown EJ, Thomas ML. The protein tyrosine phosphatase SHP-1 regulates integrin-mediated adhesion of macrophages. Curr Biol. 1998;8:1035–1038. doi: 10.1016/s0960-9822(07)00426-5. [DOI] [PubMed] [Google Scholar]
- Liu J, Fitzli D, Liu M, Tseu I, Caniggia I, Rotin D, Post M. PDGF-induced glycosaminoglycan synthesis is mediated via phosphatidylinositol 3-kinase. Am J Physiol. 1998;274:L702–L713. doi: 10.1152/ajplung.1998.274.5.L702. [DOI] [PubMed] [Google Scholar]
- Matsui R, Brody JS, Yu Q. FGF-2 induces surfactant protein gene expression in foetal rat lung epithelial cells through a MAPK-independent pathway. Cell Signal. 1999;11:221–228. doi: 10.1016/S0898-6568(98)00070-9. [DOI] [PubMed] [Google Scholar]
- White MK, Stryer DS. Surfactant protein A regulates pulmonary surfactant secretion via activation of phosphatidylinositol 3-kinase in type II alveolar cells. Exp Cell Res. 2000;255:67–76. doi: 10.1006/excr.1999.4764. [DOI] [PubMed] [Google Scholar]
- Klein JB, Rane MJ, Scherzer JA. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-related kinase pathways. J Immunol. 2000;164:4286–4291. doi: 10.4049/jimmunol.164.8.4286. [DOI] [PubMed] [Google Scholar]
- Kelley TW, Graham MM, Doseff AI, Pomerantz RW, Lau SM, Ostrowski MC, Franke TF, Marsh CB. Macrophage colony-stimulating factor promotes cell survival through Akt/protein kinase B. J Biol Chem. 1999;274:26393–26398. doi: 10.1074/jbc.274.37.26393. [DOI] [PubMed] [Google Scholar]
- Miike S, Nakao A Hiraguri M, Kurasawa K, Saito Y, Iwamoto I. Involvement of JAK2, but not PI 3-kinase/Akt and MAP kinase pathways, in anti-apoptotic signals of GM-CSF in human eosinophils. J Leukoc Biol. 1999;65:700–706. doi: 10.1002/jlb.65.5.700. [DOI] [PubMed] [Google Scholar]