

Abstract
Objective
Neprilysin (NEP) is the dominant Aβ peptide-degrading enzyme in the brain. HIV-1 subtype B Tat protein is known to interfere with NEP function, but whether this is true of HIV-1C Tat, which has a defective chemokine motif, is not known. This study aimed to analyze the impact of HIV subtype on NEP-mediated cleavage of Aβ by comparing CSF and serum levels of NEP between HIV+ (27 patients with HIV-1B and 26 with HIV-1C), healthy HIV-controls (n=13); and patients with Alzheimer’s disease (AD,n=24).
Methods
NEP, as well as Aβ oligomers 38, 40, 42 levels were measured in CSF and serum by Immunoassays. Ratios between NEP and Aβ-38,40,42, and total were calculated in CSF and serum. Comparisons between HIV(+) and HIV(−) were adjusted by linear regression for gender and age; HIV subtype comparisons were adjusted for nadir CD4 and plasma viral load suppression.
Results
Levels of NEP and ratios in CSF were comparable for HIV-1C and B subtypes. The ratio of serum NEP/Aβ-40 was lower for HIV1-C than HIV1-B (p=0.032). The CSF/serum index of NEP/Aβ-40, NEP/Aβ-42, and NEP/Aβ- total were lower for HIV1-B than HIV1-C (p=0.008, 0.005 and 0.017 respectively); corroborating the findings for serum. CSF NEP was comparable for HIV+, HIV−, and AD.
Conclusion
HIV subtypes B and C differed in their impact on NEP and Aβ. The ratio of NEP/Aβ-40 in serum was lower in HIV1-C than in HIV1-B. These results extend previous findings of lower CSF Aß-42 levels in HIV1-C than HIV1-B, suggesting more abundant amyloid ß brain tissue deposition in HIV1-C than HIV1-B.
Keywords: Neprilysin, HIV, amyloid metabolism, CSF, biomarkers, neuronal injury, subtype, central nervous system
The interactions between HIV and aging are an increasingly important topic. Since the introduction of highly active antiretroviral therapy (HAART), patients with HIV infection (HIV+) have been living longer with an improved quality of life and decreased morbidity.1 We face an increase in the age of the HIV-1 infected population, which is not only due to effective antiretroviral therapy but also to new infections among older people. In addition, HIV infected patients show aging signs earlier, chronic HIV infection results in epigenetic increase in biological age.2
HIV-1 infected cells actively secrete the viral protein transactivator of transcription (Tat).3 Tat has been found in the brain of patients with HIV-1 infection, and amyloid β (Aβ) staining was significantly increased in human brain from individuals with HIV-1 infection compared to controls.4,5 Tat inhibits neprilysin (NEP)-mediated cleavage of Aβ peptide; the inhibitory function of NEP depends on the Tat “CC” dimotif.6,7 Tat in patients with HIV1-subtype C (HIV1-C) has defective chemokine activity8,9 due to replacement of the CC dimotif with serine, CS or SC.9 It is not known how the HIV1-C defective Tat interferes with NEP function; this was the first study to analyze NEP in patients with HIV1-C.
NEP becomes inactivated and down-regulated during both the early stages of AD and in normal aging10. Reduced CSF NEP activity levels have been shown to occur in early AD, suggesting that altered CSF NEP activity levels may also be associated with dementia and lowered levels of CSF Aβ-426,11.
The aim of this study was to analyze the impact of the HIV1-C subtype, as a proxy for the defective CC dimotif, on NEP activation levels, compared to HIV1-B. We also compared the CSF levels of neprilysin between HIV+, healthy HIV- controls; and patients with Alzheimer’s disease (AD). An in vitro study showed that the exposure to HIV-1 Tat subtype B decreased protein and mRNA levels of NEP and zonula occludens- (ZO-) 1 in human cerebral microvascular endothelial (HBEC-5i) cells culture, as well as increased transendothelial transfer of Aβ and intracellular reactive oxygen species (ROS) levels. These results suggest that the Ras signaling pathway is involved in HIV-1 Tat-induced changes in ZO-1 and NEP, as well as Aβ deposition in cell culture12.
We hypothesized that HIV-1 subtype B patients would have lower CSF NEP levels compared with HIV1-C; due to the fact that HIV1-B Tat inhibits NEP. HIV+ patients would have lower CSF NEP levels compared with HIV- subjects, with the lowest levels in patients with AD and highest levels in HIV- healthy controls.
METHODS
This study, which was a cross-sectional survey of stored CSF samples, was approved by the University of California-San Diego (UCSD, San Diego, CA, USA) Institutional Review Board (IRB), Hospital de Clínicas-Universidade Federal do Paraná (HC-UFPR, Curitiba, Paraná, Brazil) IRB, and the National Commission of Ethics in Research (CONEP).
Subjects
All participants signed consent forms approved by the IRBs in Brazil and USA. For patients with AD, the responsible caregiver signed the consent form. Samples of the CSF and serum were collected under a NIMH-funded protocol (R21 MH076651-01). The methods and the demographic and infection characteristics of the HIV+ patients were described previously13.
A total of 105 CSF samples and 92 paired serum samples were analyzed.
HIV+ participants
The HIV+ participants, n=68, were recruited at the HC-UFPR. Individuals with opportunistic CNS infections were excluded. All volunteers provided blood and CSF samples and underwent serological testing to confirm HIV status before enrollment in accordance with previously published guidelines.14
For participants with clinically resistant infection, the infecting HIV subtype was genotyped with pol sequences, while env sequences were used for all other participants. Genotyping indicated that 27 individuals were infected with HIV1-B and 40 were infected with non-B HIV1 subtypes (C, n = 26; BF, 10; BC, 1; CF, 1; and F, 2). In one participant, the HIV-1 subtype could not be genotyped.
HIV− controls
Because lumbar punctures in uninfected volunteers are not allowed in Brazil, we recruited a control group of 13 age-matched HIV− individuals at the HIV Neurobehavioral Research Center, University of California San Diego (HNRC-UCSD). They had no neurological comorbidities or cognitive complaints and tested negative on serological tests for hepatitis C and syphilis. The CSF cytochemical criteria for inclusion in the control group were white blood cell (WBC) count ≤ 5cells/mm3, total protein ≤ 45 mg/dL, and glucose ≥ 55 mg/dL.
AD participants
Twenty-four patients with AD were diagnosed by the Dementia Investigative team from the Cognitive Dysfunction Outclinic (Neurology Unit, HC-UFPR). All AD participants met the dementia criteria of the DSM-V15 and criteria for probable AD of the National Institute on Aging-Alzheimer’s Association.16 Diagnostic methods of the AD group were described in detail previously11.
Laboratory Methods
CSF biomarkers
Neprilysin, activated as well as unactivated forms, was measured in the CSF and serum with an enzyme-linked immunosorbent assay (MyBioSource, San Diego, USA) with a quantitation range of 6.25-400pg/mL. The minimum detectable dose of NEP was less than 2.51pg/mL. Serum and CSF Amyloid beta isoforms (Aβ-38, 40, and 42) were assayed by electrochemiluminescence (MULTI-ARRAY, Meso Scale Diagnostics, LLC, Rockville, MD, USA); CSF Aβ-38, 40, and 42 levels were described previously13.
All samples were assayed concurrently in duplicate according to the manufacturers’ instructions. The acceptable coefficient of variation between duplicates was less than 20%. When the results were under the minimum low detection limit determined by the manufacturers, the low-detection-limit value was considered in the statistical analysis. To enhance the specificity of the neuronal injury biomarkers,17 combinations of these biomarkers in ratios and indexes were calculated in to a single value. Ratios between NEP and Aβ-38,40,42 or total were calculated in CSF and serum, as well as CSF/serum indexes of these ratios; NEP index was calculated, NEPIndex=(NEPCSF × Albserum)/(NEPserum × AlbCSF).
Blood-CSF barrier function
CSF and serum albumin were quantified by the nephelometry (Dade Behring BNII, Deerfield, IL). The functional integrity of the blood-CSF barrier was assessed by the CSF albumin/serum albumin quotient (QAlb=AlbCSF/Albserum). The upper limit of the reference range for QAlb was calculated for each participant according to the age18.
Specimen collection and storage
CSF was collected by lumbar punctures with atraumatic spinal needles under aseptic conditions by a trained neurologist. The samples were collected in polypropylene tubes to avoid adherence of the proteins to the tube walls. CSF total protein, glucose, and WBC counts were measured with standard laboratory methods. All HIV+ and AD samples were collected at the same time interval of the day to limit diurnal variability. CSF and serum aliquots were frozen and stored at −80°C at HC-UFPR.
Data analyses
Demographic and clinical variables were compared between the groups with Kruskal-Wallis test for continuous variables and Fisher’s exact test for binary and categorical variables. CSF, serum levels, ratios and CSF/serum indexes of biomarkers were log10− transformed to normalize their distributions prior to the statistical analyses. A hierarchy of comparisons was performed with the AD versus HIV+ groups as the primary comparison, HIV− versus AD groups as the secondary comparison, and HIV− 1 versus HIV+ groups as the exploratory comparison, without adjustment for multiple comparisons. Age and gender were included as covariates in multivariable linear regression models if the p value was less than 0.2 in the adjusted model. If the age effect was significantly nonlinear, a smooth age effect was used within a generalized additive model. The p values within each class of biomarkers (CSF, serum, and CSF/serum) were then corrected for multiple testing with the Benjamini-Hochberg (BH) procedure.
The CSF and serum biomarkers, ratios, and CSF/serum indexes were then compared between the HIV1-B and HIV1-C groups. A multivariable model was applied to control for plasma HIV viral load (VL) suppression and nadir CD4 counts. As described above, the p values for the biomarker effects were corrected for multiple testing with the BH procedure.
The analysis results were considered statistically significant at the 5% alpha level. The differences between groups are presented as Cohen’s d effect sizes (and 95% confidence intervals). The statistical analyses were performed with R (version 3.2.3).
RESULTS
The demographic, clinical and laboratorial characteristics of the groups studied are summarized in Tables 1 and 2.
Table 1.
Demographic and clinical characteristics, and co-morbidities of HIV participants, uninfected volunteers and Alzheimer’s disease group
| HIV+ | HIV− | AD | P | |
|---|---|---|---|---|
| N | 68 | 13 | 24 | - |
|
Demographics Age, years |
43 (35; 48) |
39 (37.5; 52.5) |
76.5(67;79.5) |
<0.0001 |
| Education, years | 8 (5;11) | 12 (10.5;16) | 4 (2;6) | <0.0001 |
| Gender, n male (%) | 33 (49) | 10 (77) | 8(33) | 0.0405 |
| Caucasians, n (%) | 66 (97) | 13 (100) | 22 (92) | 0.3688 |
| Clinical | ||||
| Durationa,b, months | 89 (31; 135)a | - | 36 (24;60)b | - |
| MEEM | - | - | 14(9.5;20) | - |
| MoCA, (n=8) | 11.5(10.5;12.5) | - | ||
| FAQ | - | - | 23.5(15;27.5) | - |
| GDS | 0.65 (0.30; 1.05) | 0.0(0.0; 0.2) | - | - |
| Depression | 13 (8, 25)c | - | 1(0.5;3)d | - |
|
Co-morbidities HCVe, n (%) |
12 (18) |
0 |
0 |
- |
| Log Plasma HCV RNA | 2.9 (1.7; 5.9) | 0 | 0 | - |
Data are median (IQR) or number of cases (%).Participants co-infected with HCV were not on treatment with interferon-gamma. Clinical duration:
of infection on HIV-positive;
of symptoms on AD. Cognitive impairment evaluated by: global deficit score (GDS) on HIV-positive; mini–mental state examination (MMSE); Montreal Cognitive Assessment (MoCA); Functional Activities Questionnaire (Pfeffer’s FAQ) on AD. Major depression disorder (MDD) diagnosed by
Beck depression inventory (BDI) on HIV-positive;
Geriatric Depression scale (GD scale) on AD.
Hepatitis C virus (HCV) status was assessed by antibody testing (Abbott-Architect).
Table 2.
Biochemical, cytological, and virological characteristics of the CSF in HIV-positive, uninfected volunteers and Alzheimer’s disease participants.
| HIV+ | HIV− | AD | P | |
|---|---|---|---|---|
| N | 68 | 13 | 24 | – |
| WBC, cells/mm³ | 2.1 (0.6; 7.2) | 1.0 (1.0;2.0) | 0.6(0.3;1.4) | 0.0025 |
| WBC count > 5 cells/mm3, n(%) | 20 (29) | 0 | 0 | - |
| Glucose, mg/dL | 57.0 (53.0; 62.0) | 61.0 (58.5; 71.0) | 60.5 (54.0; 72.5) | 0.0085 |
| Total protein, mg/dL | 40.0 (32.0; 46.0) | 30.0 (25.0; 36.5) | 37.35(30.7; 49.0) | 0.0140 |
| Total protein > 45 mg/dL, n(%) | 20 (29) | 0 | 7 (29) | 0.0302 |
| Albumin, mg/dL | 22.4 (16.4 ; 28.9) | 15.0 (13.0 ; 23.0) | - | 0.0408 |
| Albumin quotient, QAlb | 0.0064 (0.0049 ; 0.0097) | 0.004 (0.003 ; 0.005) | - | 0.0002 |
| Lactic acid, mmol/L | 1.6 (1.5 ; 1.8) | - | 1.7(1.5 ; 1.8) | 0.6499 |
| RBC, cells/mm³ | 0.5 (0.0; 7.5) | 2.0 (0.5;3.5) | 3.4 (0.9; 53.0) | 0.0149 |
| Log CSF HIV RNA | 1.7 (1.7; 2.8) | - | - | - |
| Viral load < 50 copies/mL, n(%) | 36 (53) | - | - | - |
| HIV RNA CSF > blood, n(%) | 12 (18) | - | - | - |
Data are median (IQR) or number of cases (%).
Significant differences are highlighted in bold.
Concerning the HIV(+) group, 55 cases (81%) had AIDS; median (IQR) of the current CD4, 369 (201; 534) cells/mm3; Nadir CD4, 92 (37; 267) cells/mm3; Log plasma HIV RNA, 1.7 (1.7; 3.5); plasma HIV RNA < 50 copies/mL in 38 cases (56%). Combination anti-retroviral therapy (CART), mostly protease inhibitors (PIs), was prescribed to 55 participants (81%). Median (IQR) ARV CNS Penetration Effectiveness Rank (CPE)19 was 8(6; 9); Adherence (AIDS Clinical Trials Group, ACTG, adherence questionnaire) 51(93%). The demographic, clinical and laboratorial characteristics of the groups HIV1-B and C are described in Table 3. HIV subtype B- and C-infected individuals were similar in age, gender, and education. CSF samples of HIV subtype B- and C-infected individuals were comparable in total protein, WBC, number of cases with pleocytosis (WBC > 5 cell/mm³); albumin; QAlb; CSF HIV RNA Log10; and Log CSF HIV RNA <1.7.
Table 3.
Demographic, clinical, laboratorial characteristics and co-morbidities of HIV-1 subtypes B and C participants
| HIV1-B | HIV1-C | P | |
|---|---|---|---|
| N | 27 | 26 | |
|
Demographics Age, years |
44(36.5; 50) |
43 (34.5; 47.5) |
0.45 |
| Education, years | 8(5; 12) | 7 (5; 11.5) | 0.55 |
| Gender, n male (%) | 14 (51.9) | 11 (42.3) | 0.59 |
| Caucasians, n (%) | 27 (100) | 26 (100) | - |
| Clinical | |||
| Duration of infection, months | 91.03 (61.63; 144.00) | 81.37(27.82; 131.7) | 0.450 |
| AIDS | 22 (81) | 19 (73) | 0.526 |
| Current CD4, cells/mm3 | 457 (255; 614) | 359.5(176,5; 472,5) | 0.20 |
| Nadir CD4, cells/mm3 | 82 (26; 253.5) | 159 (16.5; 359.5) | 0.29 |
| CART, n (%) | 24 (88.9) | 18 (69.2) | 0.099 |
| CPE | 8 (6; 9) | 6 (5.5; 9) | 0.339 |
| Log Plasma HIV RNA | 1.7(1.7; 1.97) | 2.8 (1.7; 3.8) | 0.012 |
| plasma HIV RNA < 50 copies/mL | 20 (74.1) | 9 (34.6) | 0.006 |
| Log CSF HIV RNA | 1.7 (1.7; 2.2) | 2.2 (1.7; 2.9) | 0.084 |
| CSF Viral load < 50 copies/mL, n(%) | 16 (59.3) | 10 (38.5) | 0.173 |
| HIV RNA CSF > blood, n(%) | 3 (11) | 3 (11.5) | 1.00 |
| GDS | 0,95 (0,275; 1,725) | 0,50 (0,225; 0,875) | 0.126 |
| HCVa, n (%) | 6 (22.0) | 2 (7.7) | 0.250 |
Data are median (IQR) or number of cases (%)
CART- Combination anti-retroviral therapy
CPE- ARV CNS Penetration Effectiveness Rank19
GDS- Global deficit score.
Hepatitis C virus (HCV) status was assessed by antibody testing (Abbott-Architect).
Significant differences are highlighted in bold.
The AD participants were probable AD; moderate dementia, Clinical Dementia Rating median [Interquartile Range (IQR)] was 2 (2, 2.5); severely decreased daily activity; and no associated depression (Table 1).
NEP levels in HIV1-B and HIV1-C patients
Levels of CSF NEP (Table 4) were comparable for HIV1-C and B subtypes; serum NEP was numerically lower for HIV1-C than HIV1-B (p= 0.060) (Figure 1A). The ratio of NEP/Aβ-40 in serum was lower for HIV1-C than HIV1-B (p= 0.032) (Figure 1A); however, this difference was not significant after correction for multiple testing with the Benjamini-Hochberg (BH) procedure.
Table 4.
HIV-1 subtype B and C levels of neprilysin, ratios and indexes in cerebrospinal and serum.
| Biomarker | HIV1-B | HIV1-C | Diff (95% CI) | pa | pb |
|---|---|---|---|---|---|
| CSF (n) | 27 | 26 | |||
| NEP | 1.20 (0.17) | 1.17 (0.17) | 0.21(−0.33, 0.75) | 0.99 | 0.99 |
| NEP/Aβ-38 | −2.11 (0.25) | −2.09 (0.26) | −0.07(−0.61, 0.47) | 0.36 | 0.45 |
| NEP/Aβ-40 | −2.43 (0.23) | −2.41 (0.22) | −0.08 (−0.62, 0.46) | 0.31 | 0.45 |
| NEP/Aβ-42 | −1.47 (0.26) | −1.42 (0.23) | −0.21(−0.75, 0.33) | 0.17 | 0.45 |
| NEP/Aβ-total | −2.63 (0.23) | −2.61 (0.23) | −0.09 (−0.62, 0.45) | 0.31 | 0.45 |
| Serum (n) | 21 | 26 | |||
| NEP | 2.43 (0.32) | 2.30 (0.16) | 0.5 (−0.05, 1.04) | 0.060 | 0.083 |
| NEP/Aβ-38 | 1.50 (0.78) | 1.42 (0.58) | 0.15 (−0.39, 0.69) | 0.63 | 0.63 |
| NEP/Aβ-40 | 0.54 (0.52) | 0.31 (0.25) | 0.56 (0.01, 1.11) | 0.032 | 0.083 |
| NEP/Aβ-42 | 1.82 (0.56) | 1.60 (0.30) | 0.5 (−0.05, 1.05) | 0.067 | 0.083 |
| NEP/Aβ-total | 0.45 (0.55) | 0.23 (0.27) | 0.5 (−0.04, 1.05) | 0.055 | 0.083 |
| CSF/Serum | |||||
| NEP | −1.23 (0.33) | −1.13 (0.26) | −0.33 (−0.87, 0.22) | 0.12 | 0.15 |
| NEP/Aβ-38 | −3.66 (0.79) | −3.51 (0.68) | −0.20 (−0.74, 0.34) | 0.41 | 0.41 |
| NEP/Aβ-40 | −2.99 (0.51) | −2.72 (0.34) | −0.63 (−1.18, −0.07) | 0.008 | 0.021 |
| NEP/Aβ-42 | −3.32 (0.53) | −3.01 (0.30) | −0.7 (−1.25, −0.14) | 0.005 | 0.021 |
| NEP/Aβ-total | −3.11 (0.54) | −2.84 (0.36) | −0.57 (−1.12, −0.02) | 0.017 | 0.021 |
| NEP Indexc | 0.90 (0.43) | 1.02 (0.49) | −0.26 (−0.86, 0.35) | 0.22 | 0.22 |
Values were log10-transformed and presented in mean (SD); Diff: Group differences presented as Cohen’s d; CI: confidence interval.
p adjusted for plasma VL suppression and CD4 nadir count
p adjusted for plasma VL suppresion and CD4 nadir count, corrected for multiple testing with the Benjamini-Hochberg (BH) method. Significant differences are in bold typeface.
NEPIndex=(NEPCSF × Albserum)/(NEPserum × AlbCSF)
Figure 1.
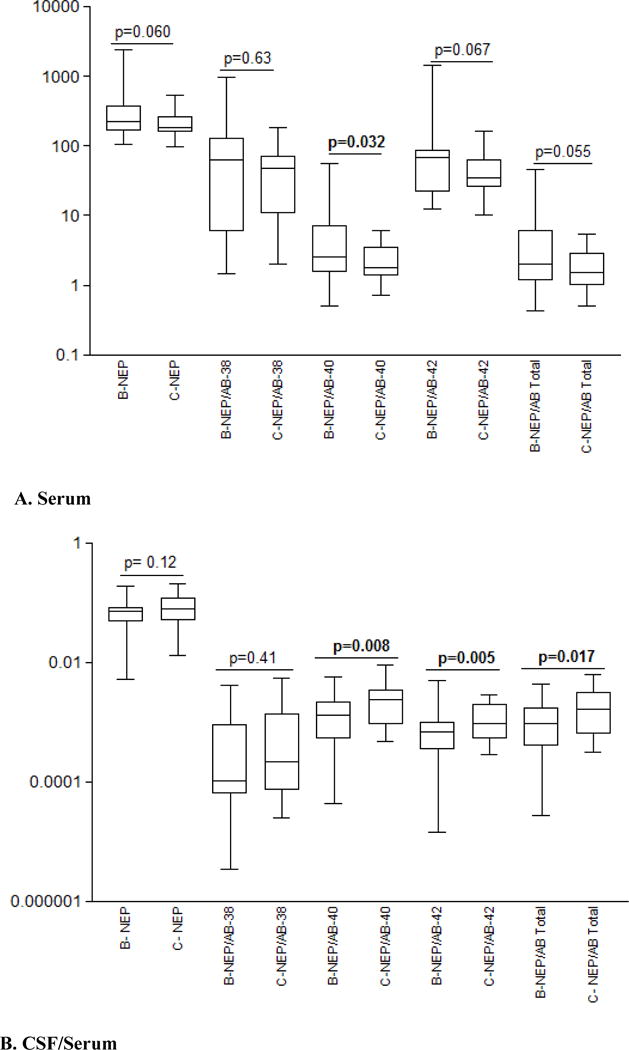
HIV-1 subtype B and C: (A). Neprilysin (NEP) pg/mL, and neprilysin/Aβ ratios in serum. (B). CSF/serum ratio for neprilysin and CSF/serum indexes for neprilysin/Aβ ratios.
Levels of serum Aβ-38, 40, 42 and total were lower in HIV1-B group than HIV1-C, although do not reach significance (p >0.05). Median (IQR) of serum Aβ 38, 40, 42 and total (pg/mL), in HIV1-B group were 2.48(2.48; 24.75), 64.30 (43.45; 147.90), 3.24 (2.06; 6.83), and 78.59(51.79;162.30) respectively; in HIV1-C group were 4.58 (2.48;17.69), 99.55 (70.95;133.60), 5.67 (3.11; 7.93), and 118 (78.66; 147). Values of CSF Aβ-38, 40, 42 and total were published previously13.
The ratios of NEP/Aβ-42 and NEP/Aβ- total in serum trended to significance (Table 4; figure 1A). The CSF/Serum index of NEP/Aβ-40, NEP/Aβ-42, and NEP/Aβ-total was lower for HIV1-B than HIV1-C (p-values 0.008, 0.005 and 0.017 respectively); corrected for multiple testing (BH) yielded significant p-values and Cohen’s d effect sizes were moderate to high; (Table 4; figure 1B). The results of CSF/Serum indexes corroborated the findings in serum.
NEP levels in HIV+, HIV-, and AD patients
Levels of CSF and serum NEP for the different groups are shown in Table 5; CSF NEP was comparable for the three groups studied. In serum, NEP levels were higher in HIV+ than AD and HIV− (0.007 and 0.056 respectively).
Table 5.
HIV-positive; HIV-negative and Alzheimer´s disease cerebrospinal fluid and serum levels of neprilysin, ratios, and indexes.
| Biomarker | HIV+ | AD | CTRL | Diff (95% CI)a | pa | Diff (95% CI)b | pb | Diff (95% CI)c | pc |
|---|---|---|---|---|---|---|---|---|---|
| CSF (n) | 68 | 24 | 13 | ||||||
| NEP | 1.19 (0.17) | 1.14 (0.34) | 1.08 (0.29) | 0.22(−0.31, 0.76) | 0.58 | −0.57(−1.2, 0.06) | 0.077 | −0.19(−0.86, 0.49) | 0.59 |
| NEP/Aβ-38 | −2.11 (0.26) | −2.11 (0.42) | −2.25 (0.30) | −0.01(−0.48, 0.45) | 0.82 | −0.52(−1.12, 0.08) | 0.089 | −0.37(−1.15, 0.41) | 0.53 |
| NEP/Aβ-40 | −2.42 (0.23) | −2.47 (0.39) | −2.57 (0.29) | 0.18(−0.35, 0.71) | 0.58 | −0.63(−1.32, 0.06) | 0.071 | −0.28(−0.99, 0.43) | 0.53 |
| NEP/Aβ-42 | −1.44 (0.26) | −1.32 (0.45) | −1.70 (0.29) | −0.38(−1, 0.23) | 0.58 | −0.98(−1.78, −0.17) | 0.009 | −0.95(−1.88, −0.01) | 0.41 |
| NEP/Aβ-total | −2.62 (0.24) | −2.65 (0.40) | −2.78 (0.29) | 0.09(−0.4, 0.58) | 0.65 | −0.62(−1.31, 0.07) | 0.071 | −0.35(−1.12, 0.43) | 0.53 |
| Serum (n) | 61 | 24 | 07 | ||||||
| NEP | 2.35 (0.23) | 2.18 (0.23) | 2.19 (0.22) | 0.71(0.23, 1.18) | 0.007 | −0.67(−1.3, −0.04) | 0.056 | 0.04(−0.64, 0.71) | 0.93 |
| NEP/Aβ-38 | 1.5 (0.67) | 0.76 (0.85) | 1.3 (0.53) | 0.97(0.45, 1.47) | <0.001 | −0.27(−0.86, 0.33) | 0.28 | 0.69(−0.11, 1.48) | 0.26 |
| NEP/Aβ-40 | 0.40 (0.39) | −0.11 (0.59) | 0.06 (0.22) | 1.14(0.58, 1.71) | <0.001 | −0.92(−1.64, −0.19) | 0.026 | 0.35(−0.43, 1.13) | 0.26 |
| NEP/Aβ-42 | 1.67 (0.42) | 1.08 (0.63) | 1.25 (0.20) | 1.22(0.56, 1.87) | <0.001 | −1.08(−1.89, −0.27) | 0.018 | 0.31(−0.47, 1.08) | 0.26 |
| NEP/Aβ-total | 0.32 (0.41) | −0.22 (0.61) | −0.01 (0.22) | 1.15(0.58, 1.72) | <0.001 | −0.84(−1.51, −0.17) | 0.026 | 0.42(−0.36, 1.2) | 0.26 |
| CSF/Serum | 61 | 24 | 07 | ||||||
| NEP | −1.16 (0.29) | −1.04 (0.46) | −1.11 (0.37) | −0.36(−0.83, 0.11) | 0.21 | 0.16(−0.45, 0.76) | 0.69 | −0.17(−0.85, 0.5) | 0.70 |
| NEP/Aβ-38 | −3.59 (0.73) | −2.87 (0.90) | −3.50 (0.29) | −0.93(−1.45, −0.4) | <0.001 | 0.12(−0.49, 0.73) | 0.57 | −0.85(−1.72, 0.03) | 0.20 |
| NEP/Aβ-40 | −2.84 (0.43) | −2.36 (0.69) | −2.60 (0.31) | −0.94(−1.46, −0.42) | <0.001 | 0.58(−0.14, 1.29) | 0.17 | −0.41(−1.12, 0.31) | 0.48 |
| NEP/Aβ-42 | −3.13 (0.43) | −2.41 (0.68) | −2.91 (0.28) | −1.43(−2.1, −0.76) | <0.001 | 0.54(−0.18, 1.25) | 0.17 | −0.88(−1.75, 0) | 0.20 |
| NEP/Aβ-total | −2.96 (0.46) | −2.43 (0.69 | −2.74 (0.28) | −1.01(−1.59, −0.43) | <0.001 | 0.51(−0.2, 1.22) | 0.17 | −0.53(−1.29, 0.23) | 0.43 |
| NEP Indexd | 0.99 (0.43) | – | 1.28 (0.41) | – | 0.67 (−0.15, 1.49) | 0.038 | 0.67 (−0.15, 1.49) | 0.038 |
Values were log10-transformed and presented in mean (SD)
HIV(+)xAD
HIV(+)xCTRL
ADxCTRL.Values in median (IQR); Diff: Group differences presented as Cohen’s d; CI: confidence interval; all p values adjusted for multiple testing with the Benjamini-Hochberg (BH) method, (a) and (c) adjusted for gender or age and BH method. Significant differences are in bold typeface.
NEPIndex=(NEPCSF × Albserum)/(NEPserum × AlbCSF)
DISCUSSION
This is the first study to analyze NEP in patients with HIV1 non-B subtypes. CSF and serum NEP were comparable for HIV1-C and B subtypes. However, the ratio of NEP/Aβ40 in serum was lower in HIV1-C than B. Lower NEP/Aβ40 in subtype C is consistent with less cleavage of Aβ40 with greater tissue deposition of Aβ40. This is also supported by previous findings of lower CSF Aβ42 in subtype C, reported by our research group13.
However, the findings overall are counterintuitive. We expected the defective Tat in subtype C to yield less inhibition of NEP with correspondingly lower tissue deposition of Aβ and therefore higher levels of Aβ in CSF. Previous studies on which this hypothesis was based were experimental 6,7. Our analysis of human samples did not confirm the in vitro findings. The Tat sequences used in the prior in vitro studies may have differed from those in our clinical samples. Other factors that could explain these differences include the use of antiretroviral medications, which may affect amyloid metabolism. More studies need to be done to elucidate the impact of HIV subtypes on NEP metabolism. The comparisons of these markers could provide very useful information that will lead to expanded studies on neuropathology associated with different HIV subtypes, especially in the aging population.
Based on the present results, we conclude that NEP was driving the results, as the values of serum NEP trend to be lower in HIV1-C than B, and were supported by the differences between the ratios in serum and CSF/serum indexes. Although numerical values of serum Aβ isoforms were lower in HIV1-B group than HIV1-C, probably they do not determine the results of ratios and indexes; as they are not statistically significant.
CSF NEP was comparable for the HIV+, HIV(−), and AD; in serum NEP levels were higher on HIV than AD and CTRL.
Our findings did not confirm the hypothesis that CSF NEP levels in HIV would be in-between those in AD (low) and in HIV-controls (high). There are several potential explanations for this finding. First, there is disagreement in the literature on NEP levels in AD. For example, one study showed reduced CSF NEP activity levels in early (prodromal) AD20 while another demonstrated that brain tissue NEP levels were increased in AD21. Thus NEP levels may change during the course of AD. The patients we studied had well-established AD, more advanced than in the Maruyama et al. (2005) study20. Our failure to find higher levels of NEP in HIV+ compared to AD therefore might be related to the stage of AD in our patients. Alternatively, CSF NEP levels may not correlate well with NEP levels and activity within brain tissue.
Neprilysin, also known as CD10 or enkephalinase, is a type II transmembrane bound zinc metalloendopeptidase with relatively broad substrate specificity. Its molecular weight is 85.5 (85-110kDa) kD10,22 and the hydrodynamic radius (Rh) or Stokes radius is 56 Å. Because NEP is a high molecular weight molecule, diffusion does not occur through a functional blood–CSF barrier (BCSFB). An dysfunctional BCSFB, however, might result in diffusion of NEP from blood to CSF, Several studies have noted marked increases in diffuse amyloid plaques in in the HIV(+) group compared to HIV(−) age-matched controls4,6,23,24. This suggested that an inflammatory response in the brain to HIV-1 infection could lead to dysregulation of amyloid processing in the setting of HIV infection25, and facilitate amyloid plaque formation.
NEP is mainly synthesized in various types of cells outside brain, is widely distributed in the body and normally expressed by a variety of tissues 10. In brain it is expressed by activated astrocytes26 and microglia27. For proteins derived primarily from blood, as NEP, the CSF/serum quotients may be informative. To estimate NEP intrathecal synthesis, indexes between CSF and serum and the NEPindex were calculated28.
Cleavage by NEP greatly reduces the aggregation tendency and neurotoxicity of the resulting fragments29 as demonstrated for cleavage by the peptidase insulin30. NEP is the dominant Aβ peptide-degrading enzyme in the brain; chiefly Aβ-40, but also Aβ-42 and other Aβ isoforms31.
HIV-1-infected patients
HIV-1 markedly increased the endogenous Aβ levels and accumulation of exogenous Aβ in an HIV-1 exposure model of brain microvascular endothelial cells.32
HIV has great impact on Aβ metabolism, Aβ deposition differs in AD and HIV-1 brains. While extracellular amyloid plaques are the major amyloid pathology in AD, intraneuronal amyloid accumulation or diffuse perivascular amyloid depositions are more characteristic for HAND.33 The mechanisms underlying the interactions between Aβ and HIV-1 infection are not fully understood but several factors and/or pathways are likely to be involved. It has been hypothesized that aging, HIV-1 infection, and the secondary effects of ART may all contribute to brain Aβ accumulation in neurons and in perivascular space.4 HIV-1 can increase Aβ levels by increasing its synthesis, decreasing its degradation, or changing its transport mechanisms across the blood brain barrier, leading to its accumulation in the brain33,34.
Several HIV proteins are amyloidogenic, such as gp12024,35, gp4136, Tat6,7,24,37, or Nef.38 Mononuclear phagocytes, which are macrophages and microglia, that are infected by HIV play a pivotal role in Aβ degradation through the expression and execution of two endopeptidases, NEP and insulin-degrading enzyme (IDE).37 Additionally multiple studies have described the amyloidogenic interference of ART.39
Tat, which is the main HIV amyloidogenic protein, interferes with Aβ cleavage (chiefly via NEP) and amyloid peptide reuptake and clearance. HIV-1 Tat protein increases Aβ levels in cell culture.6 In addition, HIV-1 Tat, specifically HIV1-B, increases Aβ-40 levels in neuronal cell cultures by inhibiting NEP.6,7,24,37
HIV-1 infected cells actively secrete Tat, a viral transactivating transcription factor3. At micromolar concentrations, in vitro, HIV-1 Tat inhibits the major Aβ-degrading enzyme, neprilysin (NEP), a neuronal endopeptidase. These findings imply that premature accumulation of Aβ in the HIV-1-infected population will increase the risk of dementia compared to the normal aging population.
Tat release continues even in patients with durable virologic suppression, tat was detected in the CSF of individuals with undetectable HIV viral load in the blood and CSF40. In transgenic AD mice having HIV-1 Tat-expressing astrocytes, more neurodegeneration and Aβ deposition was observed compared with mice not expressing astrocytic Tat serine41.
Tat interference on Aß-42 clearance
Tat inhibited NEP activity by 80%, causing amyloid β (Aβ) accumulation6. Comparison of the overlapping Tat sequences, demonstrated that NEP maximal inhibition required the amino acid sequence KCCF (amino acids 22–37)7. Studies about CSF NEP on HIV are poorly reported on the literature. The only two in vitro studies that showed that cysteine-rich peptides derived from the Tat protein are inhibitors of NEP, were done with HIV clade B6,7. Although the cysteine-rich domain is described as highly conserved among different HIV-1 strains, in HIV clade C that infects the largest populations around the world, the cysteine residue in position 31 is mutated to a serine9.
The present study is not free of limitations. Samples from the HIV(−) controls were from a different study site. Although the HIV(+) and HIV(−) did not differ on race/ethnicity, Brazilian populations have a higher genetic heterogeneity than those in the United States. This limitation was not applicable to the comparison between subtypes B and C, as all the participants were from the same geographical region in Brazil and were similar in age and gender. The study was limited by its cross-sectional design. A longitudinal study might be able to predict the development of HAND in patients without clear symptoms. Vulnerability to AD is greatest in older individuals; our study did not include a substantial number of older HIV(+). We did not assess HIV(+) patients with known AD, although reports of patients with both conditions are rare. The HIV(−) group was younger than the AD group as this group was age- and gender-matched with the HIV(+) group. Future longitudinal studies that include older HIV(+) individuals are necessary to evaluate the effects of HIV on the NEP pathway. Finally, the assay used here quantified activated and unactivated NEP forms, but did not directly assess NEP activity.
This study had several strengths, this was the first study to examine HIV1-C patients and investigate HIV subtypes effects on NEP metabolism. The sample size was sufficient for power analysis, as absolute values of Cohen’s d effect sizes were medium to large. HIV+ and AD patients were from the same center and were analyzed at the same period of time; both healthy and disease controls were used. HIV positive and HIV- are age matched, this is important as NEP becomes inactivated and down-regulated with aging10.
In conclusion, the results of this study have shown that HIV infection affects serum NEP function on a subtype-dependent pattern, HIV subtypes C and B differed, suggesting less destruction of Aß; which might accumulate more in HIV1-C.
Acknowledgments
Part of this work was previously presented at the Conference on Retrovirus and Opportunistic Infections, CROI 2018, March 4-7, 2018, Boston, Massachusetts, USA.
This work was supported by the following grants: National Institute of Health, NIH R21 MH76651 (Ellis, Ronald J; Almeida, Sergio M.), S10 RR31646 (Letendre, Scott), K24 MH097673(Letendre, Scott); University of California, San Diego, Center for AIDS Research (CFAR), an NIH-funded program (P30 AI036214), which is supported by the following NIH Institutes and Centers: NIAID, NCI, NIMH, NIDA, NICHD, NHLBI, NIA, NIGMS, and NIDDK. The HIV Neurobehavioral Research Center (HNRC) is supported by Center award P30MH062512 from NIMH. The San Diego HIV Neurobehavioral Research Center [HNRC] group is affiliated with the University of California, San Diego, the Naval Hospital, San Diego, and the Veterans Affairs San Diego Healthcare System, and includes: Director: Robert K. Heaton, Ph.D., Co-Director: Igor Grant, M.D.; Associate Directors: J. Hampton Atkinson, M.D., Ronald J. Ellis, M.D., Ph.D., and Scott Letendre, M.D.; Center Manager: Thomas D. Marcotte, Ph.D.; Jennifer Marquie-Beck, M.P.H.; Melanie Sherman; Neuromedical Component: Ronald J. Ellis, M.D., Ph.D. (P.I.), Scott Letendre, M.D., J. Allen McCutchan, M.D., Brookie Best, Pharm.D., Rachel Schrier, Ph.D., Debra Rosario, M.P.H.; Neurobehavioral Component: Robert K. Heaton, Ph.D. (P.I.), J. Hampton Atkinson, M.D., Steven Paul Woods, Psy.D., Thomas D. Marcotte, Ph.D., Mariana Cherner, Ph.D., David J. Moore, Ph.D., Matthew Dawson; Neuroimaging Component: Christine Fennema-Notestine, Ph.D. (P.I.), Monte S. Buchsbaum, M.D., John Hesselink, M.D., Sarah L. Archibald, M.A., Gregory Brown, Ph.D., Richard Buxton, Ph.D., Anders Dale, Ph.D., Thomas Liu, Ph.D.; Neurobiology Component: Eliezer Masliah, M.D. (P.I.), Cristian Achim, M.D., Ph.D.; Neurovirology Component: David M. Smith, M.D. (P.I.), Douglas Richman, M.D.; International Component: J. Allen McCutchan, M.D., (P.I.), Mariana Cherner, Ph.D.; Developmental Component: Cristian Achim, M.D., Ph.D.; (P.I.), Stuart Lipton, M.D., Ph.D.; Participant Accrual and Retention Unit: J. Hampton Atkinson, M.D. (P.I.), Jennifer Marquie-Beck, M.P.H.; Data Management and Information Systems Unit: Anthony C. Gamst, Ph.D. (P.I.), Clint Cushman; Statistics Unit: Ian Abramson, Ph.D. (P.I.), Florin Vaida, Ph.D. (Co-PI), Bin Tang, Ph.D., Anya Umlauf, M.S.
Study funding
Study Funded by CFAR (International Pilot Grant P30 AI036214 and CFAR Visiting Researcher Grant PTHMON7), and NIMH (R21 MH076651-01)
Footnotes
Author Disclosures
The authors declare that have no conflict of interest.
Author Contributions
Sérgio de Almeida, study concept and design, acquisition, analysis and interpretation of data.
Clea Ribeiro, acquisition of data
Indianara Rotta, acquisition of data, laboratorial support
Mauro Piovesan, acquisition of data
Scott Letendre, interpretation of data
Michael Potter, laboratory support
Bin Tang, statistical Analysis
Meiri Batistela, acquisition of data
Florin Vaida, statistical Analysis
Ronald Ellis, study concept and design, analysis and interpretation of data.
Bin Tang and Florin Vaida, conducted Statistical Analysis
The views expressed in this article are those of the authors and do not reflect the official policy or position of the Department of the Navy, Department of Defense, nor the United States Government.
References
- 1.Burgoyne RW, Tan DH. Prolongation and quality of life for HIV-infected adults treated with highly active antiretroviral therapy (HAART): a balancing act. J Antimicrob Chemother. 2008;6:469–473. doi: 10.1093/jac/dkm499. [DOI] [PubMed] [Google Scholar]
- 2.Gross AM, Jaeger PA, Kreisberg JF, et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol Cell. 2016;62:157–168. doi: 10.1016/j.molcel.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ensoli B, Buonaguro L, Barillari G, et al. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green DA, Masliah E, Vinters HV, et al. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS. 2005;19:407–411. doi: 10.1097/01.aids.0000161770.06158.5c. [DOI] [PubMed] [Google Scholar]
- 5.Achim CL, Adame A, Dumaop W, et al. Neurobehavioral Research Center Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J Neuroimmune Pharmacol. 2009;4:190–199. doi: 10.1007/s11481-009-9152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rempel HC, Pulliam L. HIV-1Tat inhibits neprilysin and elevates amyloid beta. AIDS. 2005;19:127–135. doi: 10.1097/00002030-200501280-00004. [DOI] [PubMed] [Google Scholar]
- 7.Daily A, Nath A, Hersh LB. Tat peptides inhibit neprilysin. J Neurovirol. 2006;12:153–160. doi: 10.1080/13550280600760677. [DOI] [PubMed] [Google Scholar]
- 8.Satishchandra P, Nalini A, Gourie-Devi M, et al. Profile of neurologic disorders associated with HIV/AIDS from Bangalore, South India (1989-96) Indian J Med Res. 2000;111:14–23. [PubMed] [Google Scholar]
- 9.Ranga U, Shankarappa R, Siddappa NB, et al. Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J Virol. 2004;78:2586–2590. doi: 10.1128/JVI.78.5.2586-2590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gayathiri K, Prabhavathi A, Tamilarasi R, et al. Role of Neprilysin in Various Diseases. International Journal of Pharmacological Research. 2014;4:91. www.ssjournals.com IJPR. [Google Scholar]
- 11.Sorensen KCN, Simonsen AH, Holmetoft UB, et al. Neprilysin-Like Activity Correlates with CSF-Tau and Phospho-Tau in Patients with Alzheimer’s Disease. Journal of Alzheimer’s Disease. 2013;xx:x–xx. doi: 10.3233/JAD-122410. [DOI] [PubMed] [Google Scholar]
- 12.Jiang W, Huang W, Chen Y, Zou M, Peng D, Chen D. HIV-1 Transactivator Protein Induces ZO-1 and Neprilysin Dysfunction in Brain Endothelial Cells via the Ras Signaling Pathway. Oxidative Medicine and Cellular Longevity. 2017 doi: 10.1155/2017/3160360. Article ID 3160360, 10 pages. https://doi.org/10.1155/2017/3160360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.deAlmeida SM, Ribeiro CE, Rotta I, Piovesan M, Tang B, Vaida F, Raboni SM, Letendre S, Potter M, Fernandes MSB, Ellis RJ, HIV Neurobehavioral Research Center (HNRC) Group Biomarkers of neuronal injury and amyloid metabolism in the cerebrospinal fluid of patients infected with HIV-1 subtypes B and C. J Neurovirol. 2017 doi: 10.1007/s13365-017-0591-3. https://doi.org/10.1007/s13365-017-0591-3. [DOI] [PMC free article] [PubMed]
- 14.Brasil, Ministério da Saúde. Programa Nacional de DST/AIDS. 2016 http://www.aids.gov.br/assistencia/manualdst/item12.htm.
- 15.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, DC: p. 2013. [Google Scholar]
- 16.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blennow K, Vanmechelen E. Combination of the different biological markers for increasing specificity of in vivo Alzheimer’s testing. J Neural Transm Suppl. 1998;53:223–235. doi: 10.1007/978-3-7091-6467-9_20. [DOI] [PubMed] [Google Scholar]
- 18.Reiber H, Peter JB. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci. 2001;184:101–22. doi: 10.1016/s0022-510x(00)00501-3. [DOI] [PubMed] [Google Scholar]
- 19.Letendre S, Ellis R, Deutsch R, et al. Correlates of time-to-loss-of-viral response in CSF and plasma in the CHARTER cohort; Program and abstracts of the 17th Conference on Retroviruses and Opportunistic Infections, 2010; San Francisco, CA. 16–19 February; (poster 430) [Google Scholar]
- 20.Maruyama M, Higuchi M, Takaki Y, Matsuba Y, Tanji H, Nemoto M, Tomita N, Matsui T, Iwata N, Mizukami H, Muramatsu S, Ozawa K, Saido TC, Arai H, Sasaki H. Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer's disease. Ann Neurol. 2005;57:832–842. doi: 10.1002/ana.20494. [DOI] [PubMed] [Google Scholar]
- 21.Miners JS, Baig S, Tayler H, Kehoe PG, Love S. Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J Neuropathol Exp Neurol. 2009;68:902–914. doi: 10.1097/NEN.0b013e3181afe475. [DOI] [PubMed] [Google Scholar]
- 22.Hersh LB, Rodgers DW. Neprilysin and amyloid beta peptide degradation. Curr Alzheimer Res. 2008;5(2):225–31. doi: 10.2174/156720508783954703. [DOI] [PubMed] [Google Scholar]
- 23.Esiri MM, Biddolph SC, Morris CS. Prevalence of Alzheimer plaques in AIDS. J Neurol Neurosurg Psychiatry. 1998;65:29–33. doi: 10.1136/jnnp.65.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aksenov MY, Aksenova MV, Mactutus CF, et al. HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci Lett. 2010;475:174–178. doi: 10.1016/j.neulet.2010.03.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pulliam L. HIV regulation of amyloid beta production. J Neuroimmune Pharmacol. 2009;4:213–217. doi: 10.1007/s11481-009-9151-9. [DOI] [PubMed] [Google Scholar]
- 26.Fisk L, Nalivaeva NN, Boyle JP, et al. Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurons and astrocytes. Neurochem Res. 2007;32:1741–8. doi: 10.1007/s11064-007-9349-2. [DOI] [PubMed] [Google Scholar]
- 27.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease. J Neurosci. 2008;28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leissring MA, Lu A, Condron MH, et al. Kinetics of amyloid beta-protein degradation determined by novel fluorescence- and fluorescence polarizationbased assays. J Biol Chem. 2003;278:37314–37320. doi: 10.1074/jbc.M305627200. [DOI] [PubMed] [Google Scholar]
- 29.Liao MQ. The correlation between neurotoxicity, aggregative ability and secondary structure studied by sequence truncated Abeta peptides. FEBS Lett. 2007;581:1161–1165. doi: 10.1016/j.febslet.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee A. Insulysin hydrolyzes amyloid beta peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J Neurosci. 2000;20:8745–8749. doi: 10.1523/JNEUROSCI.20-23-08745.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS. Neprilysin: An Enzyme Candidate to Slow the Progression of Alzheimer’s Disease. The American Journal of Pathology. 2008;172(5):1342–54. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.András IE, Eum SY, Huang W, et al. HIV-1-induced amyloid beta accumulation in brain endothelial cells is attenuated by simvastatin. Mol Cell Neurosci. 2010;43:232–243. doi: 10.1016/j.mcn.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Ikezu T. The comorbidity of HIV-associated neurocognitive disorders and Alzheimer’s disease: a foreseeable medical challenge in post-HAART era. J Neuroimmune Pharmacol. 2009;4:200–212. doi: 10.1007/s11481-008-9136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.András IE, Toborek M. Amyloid beta accumulation in HIV-1- infected brain: The role of the blood brain barrier. IUBMB Life. 2013;65:43–9. doi: 10.1002/iub.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Liu J, Katafiasz B, et al. HIV-1 gp120-induced axonal injury detected by accumulation of β-amyloid precursor protein in adult rat corpus callosum. J Neuroimmune Pharmacol. 2011;6:650–657. doi: 10.1007/s11481-011-9259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mankowski JL, Queen SE, Tarwater PM, et al. Accumulation of beta-Amyloid Precursor Protein in Axons Correlates with CNS Expression of SIV gp41. J Neuropathol Exp Neurol. 2002;61:85–90. doi: 10.1093/jnen/61.1.85. [DOI] [PubMed] [Google Scholar]
- 37.Lan X, Xu J, Kiyota T, et al. HIV-1 reduces Abeta-degrading enzymatic activities in primary human mononuclear phagocytes. J Immunol. 2011;186:6925–32. doi: 10.4049/jimmunol.1100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White JA, Manelli AM, Holmberg KH, et al. Differential effects of oligomeric and fibrillar amyloid-beta 1–42 on astrocyte-mediated inflammation. Neurobiol Dis. 2005;18:459–65. doi: 10.1016/j.nbd.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 39.Brown LAM, Jin J, Ferrell D, et al. Efavirenz Promotes b-Secretase Expression and Increased Ab1-40,42 via Oxidative Stress and Reduced Microglial Phagocytosis: Implications for HIV Associated Neurocognitive Disorders (HAND) PLoS ONE. 2014;9:e95500. doi: 10.1371/journal.pone.0095500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson TP, Patela K, Johnson KR, et al. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. PNAS. 2013;110:13588–93. doi: 10.1073/pnas.1308673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giunta B, Hou H, Zhu Y, et al. HIV-1 Tat contributes to Alzheimer’s disease-like pathology in PSAPP mice. Int J Clin Exp Pathol. 2009;2:433–43. [PMC free article] [PubMed] [Google Scholar]