

Abstract
Chordoma is a rare malignant bone tumor that can arise anywhere along the central neural axis and many involve head and neck sites, most commonly the skull base. The relative rarity of these tumors, combined with the complex anatomy of the head and neck, pose diagnostic challenges to pathologists. This article describes the pertinent clinical, pathologic, and molecular features of chordomas and describes how these features can be used to aid in formulating a differential diagnosis. Emphasis is placed on key diagnostic pitfalls and the importance of incorporating immunohistochemical information into the diagnosis.
Keywords: Chordoma, Chondrosarcoma, Skull base, Brachyury, Head and neck
Introduction
Chordoma is a rare malignant bone tumor that can arise anywhere along the central neural axis [1, 2]. Chordoma is a unique entity in that it is a malignant tumor derived from remnants of the notochord, an embryonic structure that is required for the induction of the neural plate in the embryonic disk. Among head and neck sites, the majority arise in the skull base with a small minority arising along the cervical spine [1–3]. Chordomas have also been reported in extra-axial locations in the head and neck [4, 5], including the nasopharynx, paranasal sinuses, lateral nasal wall, oropharynx, and the soft tissue of the neck [6–11]. Some extra-axial chordomas of the nasopharynx are associated with a sinus tract extending from the clivus [7, 10]. Males and females are affected equally and head and neck chordomas tend to present one decade earlier than other sites. Chordomas tend to be slow growing tumors and symptoms of head and neck chordomas are related to mass effect including headache, neck pain, diplopia or cranial nerve palsy [12]. Some chordomas in children may be associated with tuberous sclerosis complex although the behavior of these tumors does not appear to differ from their sporadic counterparts [13].
Pathologic Features
The typical chordoma appears grossly as a lobulated mass with a gelatinous or chondroid cut surface. When centered in bone, the tumor typically extends beyond the cortex into the surrounding soft tissue. Microscopically, the conventional chordoma is composed of cords of tumor cells embedded within a myxoid matrix and arranged in lobules separated by fibrous septa (Fig. 1a). The diagnostic “physaliferous” cells have abundant eosinophilic cytoplasm and intracytoplasmic vacuoles, although the relative proportion of these features varies from tumor to tumor (Fig. 1b). Although all chordomas are malignant by definition, cytologic atypia ranges from low grade, with uniform tumor cells and occasional mitoses, to high-grade with significant nuclear pleomorphism and frequent mitoses. Tumor heterogeneity with regards to atypia may be seen. Necrosis is frequent and may be extensive.
Fig. 1.
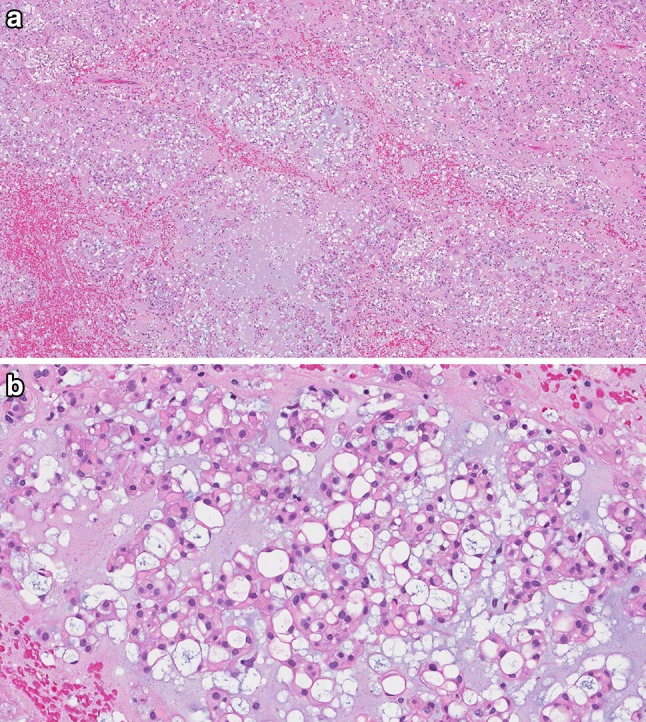
Conventional chordoma. a Conventional chordoma consists of cords of tumor cells arranged in lobules, separated by thin fibrous septae and embedded within a myxoid matrix. b The tumor cells have abundant eosinophilic cytoplasm and prominent cytoplasmic borders. The characteristic physaliferous cells have vacuolated cytoplasm
Pathologists need to be aware of several distinct histologic subtypes of chordoma that, on initial inspection, lack the typical features of a conventional chordoma. These subtypes include chondroid chordoma, cellular chordoma, poorly differentiated chordoma and dedifferentiated chordoma. Chondroid chordoma accounts for 7–63% of skull base chordomas [12, 14]. Like conventional chordoma, chondroid chordoma consists of cords of tumor cells arranged in lobules, with physaliferous cells; however the matrix resembles neoplastic hyaline cartilage (Fig. 2) [15–17]. Cellular chordoma is composed of sheets of tumor cells, including physaliferous cells, with little to no myxoid stroma (Fig. 3) [18]. Poorly differentiated chordomas are composed of tightly packed, small, atypical epithelioid cells arranged nests and sheets with a fibrous background and lack physaliferous cells [18]. Chordomas with rhabdoid cells have been described, and may represent a poorly differentiated chordoma [14, 18]. A conventional chordoma that demonstrates an abrupt transformation into an undifferentiated pleomorphic sarcoma or osteosarcoma is described as a dedifferentiated chordoma [15]. Historically, “atypical chordoma” has been described in the literature, however the criteria are not well established and this group likely contains both cellular and poorly differentiated chordomas and thus, the use of this term is discouraged [18].
Fig. 2.

Chondroid chordoma. a The tumor cells of chondroid chordoma are identical to conventional chordoma, with cords of tumor cells arranged in lobules and physaliferous cells, however the matrix resembles neoplastic hyaline cartilage. b At high power, scattered vacuolated physaliferous cells are seen
Fig. 3.

Cellular chordoma. a Cellular chordoma consists of sheets of typical chordoma tumor cells, including physaliferous cells, but lack either a myxoid or chondroid matrix, and thus can be mistaken for carcinoma. b At high power, the vacuolated physaliferous cells are seen scattered throughout the tumor
Immunohistochemistry
A recommended immunohistochemical panel to aid in the differential diagnosis for head and neck chordomas outlining the most common immunohistochemical profiles is shown in Table 1. All chordomas, including the specific histologic subtypes described above, express cytokeratins (Fig. 4a) and most are immunoreactive for epithelial membrane antigen (EMA) (Fig. 4b) and S100 protein. The most specific marker of chordoma is brachyury, a nuclear protein associated with notochord differentiation (Fig. 4c) [19–22]. While expression of brachyury is highly specific for chordoma, poorly differentiated tumors and dedifferentiated areas may demonstrate loss of brachyury immunoreactivity [14, 19, 20, 22]. Importantly, immunoreactivity for brachyury may also be lost following decalcification and effort should always be made to procure tumoral tissue for immunohistochemistry prior to decalcification. On occasion, chondroid chordoma may demonstrate only focal cytokeratin expression [14]. Interestingly, some studies have reported a loss of SMARCB1/INI1 staining in poorly differentiated chordomas although the significance of this finding is not yet established [23–25].
Table 1.
Immunohistochemical panel to aid in the differential diagnosis for chordoma demonstrating the most frequent immunoprofiles
| Entity | Recommended immunohistochemical panel | Other | |||
|---|---|---|---|---|---|
| Cytokeratin | EMA | S100 | Brachyury | ||
| Chordoma | Positive | Positive | Positive | Positive | |
| Conventional Chondrosarcoma | Negative | Negative/ focal | Positive | Negative | |
| Chordoid meningioma | Variable | Variable | Variable | Negative | PR |
| Chordoid glioma | Variable | Variable | Positive | Negative | GFAP |
| Mixed tumour/myoepithelial-rich tumours | Positive | Positive | Positive | Negative | |
| Extra-skeletal myxoid chondrosarcoma | Negative | Positive | Negative | Negative | NR4A3 rearrangements |
| Metastatic adenocarcinoma | Positive | Positive | Negative | Negative | Site specific immunohistochemical markers |
EMA epithelial membrane antigen, PR progesterone receptor, GFAP glial fibrillary acid protein
Fig. 4.

Conventional chordoma, demonstrating diffuse staining with a cytokeratin AE1/3, b S100, and c brachyury
Differential Diagnosis
Chordoma must be differentiated from a variety of other lesions including chondrosarcoma (Fig. 5a), meningioma (Fig. 5b), myoepithelioma/myoepithelial carcinoma (Fig. 5c), and glioma; and metastatic carcinomas such as mucinous adenocarcinoma and clear cell renal cell carcinoma. Other than involvement restricted to the clivus, seen only in chordoma, the clinical distinction between skull-base chordoma and chondrosarcoma is difficult and typically requires a biopsy, since both demonstrate similar clinical, CT and MRI findings [26, 27]. Moreover, the histologic similarities between chondroid chordoma and chondrosarcoma may pose a diagnostic challenge, especially on limited and crushed biopsy material that is typical of skull base lesions [14].
Fig. 5.

Differential diagnosis. Differential diagnosis for chordoma include (a) chondrosarcoma, (b, c) chordoid meningioma, and (d) myoepithelioma. a Conventional hyaline chondrosarcoma may have “vacuoles” within the stroma, which at low power may mimic physaliferous cells; however, the tumor cells reside within lacunae, rather than having corded appearance. Although chondrosarcomas are S100 positive and may be EMA positive, they are negative for cytokeratin and brachyury. A chordoid meningioma (b) can look identical to a conventional chordoma at low power with vacuolated tumor cells set in a myxoid stroma, but foci of typical meningioma (c) are often seen. The typical meningioma areas may be absent on biopsy, but chordoid meningiomas are negative for brachyury. A myoepithelioma (d), with its corded neoplastic cells set in a chondromyxoid stroma, can be confused with a chordoma however physaliferous are not present. Although myoepithelioma are typically immunoreactive for cytokeratins, S100 and EMA, they are negative for brachyury
Ancillary studies are frequently required for skull base biopsies and the most discriminatory immunostains are combined cytokeratin and brachyury to distinguish chondroid chordoma from chondrosarcoma [14]. EMA is a sensitive marker for chordoma; however, it is not entirely specific since chondrosarcomas may also demonstrate immunoreactivity [14, 28]. A recent study has shown that both D2-40 and SOX-9, a homeobox transcription factor involved in chondrogenesis, can be positive in chordoma and are not helpful in distinguishing chondrosarcoma from chordoma [14].
Although expression of cytokeratins is helpful to distinguish chordoma from chondrosarcoma, it is not helpful in distinguishing conventional chordoma from metastatic mucinous or clear cell carcinomas without immunoreactivity for brachyury and S100 protein [19–21]. Poorly differentiated chordomas may be particularly challenging to diagnose as they consist of small atypical epithelioid cells immunoreactive for cytokeratins and lack the characteristic physaliferous cells and myxoid matrix. Addition of brachyury and S100 immunostains for any skull base tumor is highly recommended.
Co-expression of cytokeratins and S100 protein, along with EMA, will not help distinguish chordomas from myoepithelial tumors, including mixed tumor, myoepithelioma, parachordoma and myoepithelial carcinoma [4, 5, 29]. Distinguishing chordoma from myoepithelial tumors requires immunohistochemical evidence of brachyury expression, in addition to cytokeratins and S100 [4, 5].
Extraskeletal myxoid chondrosarcoma shares overlapping histologic features with soft tissue conventional chordoma. Both are soft tissue neoplasms with tumor cells arranged in cords and set in a myxoid background. Although both express S100 protein and extraskeletal myxoid chondrosarcoma may express EMA [29], extraskeletal myxoid chondrosarcoma is negative for cytokeratins and brachyury. In addition, extraskeletal myxoid chondrosarcoma demonstrate rearrangements involving NR4A3 gene, most commonly NR4A3-EWSR1 fusion.
Distinguishing chordoma from chordoid meningioma and chordoid glioma can be challenging. Chordoid meningioma and chordoid glioma are so named because both microscopically resemble chordoma. Both tumors consist of cords of epithelioid tumor cells with well-defined cell borders, embedded within a myxoid or mucinous stroma, and tumor cells of chordoid meningioma may be vacuolated. In chordoid meningioma, traditional meningioma areas are intermixed, aiding with the diagnosis; however, this may be absent on a small biopsy. Cytokeratins, EMA and S100 positivity may be seen in all three tumors; however, only chordoma is positive for brachyury [30]. Chordoid gliomas are strongly positive for glial fibrillary acidic protein (GFAP), however conventional chordoma and chondroid chordoma may demonstrate focal GFAP immunoreactivity [14, 30].
Molecular Alterations
As a relatively rare tumor, few studies to date have investigated the molecular landscape of chordomas and the studies that have been performed were small. For example a recent study of nine chordomas using ultra-deep sequencing analysis identified hot-spot mutations in the genes TP53, KIT, and KDR and all three of these genes have been shown previously to be involved in chordoma oncogenesis [31]. Decreased expression of PTEN, overexpression of PDGFR-α, EGFR, Raf-1, components of the mTOR pathway and activation of the STAT3 pathway have been identified in chordoma and are associated with tumor progression and/or poor prognosis [32–35]. Recent studies have shown down-regulation of various microRNAs implicated in tumor growth, progression and prognosis [36–38]. Recurrent clival chordomas have demonstrated MGMT promoter methylation [39]. Additional studies are required to determine if these markers have the potential to serve as prognostic markers and therapeutic targets in chordoma patients.
Genetic alterations in the gene encoding for brachyury, a transcription factor encoded by T gene, have been identified in both sporadic and familial cases. Notably, duplication of brachyury has been described in familial chordomas [40]. In contrast, the much more common sporadic chordoma may demonstrate T gene amplification and single nucleotide polymorphism in the coding region of the T gene [41, 42]. Cytogenetic studies of chordomas have demonstrated several anomalies, including losses of 1p and 3p, and gains in chromosomes 7 and 19 [43–45].
IDH1 and IDH2 mutations, which are frequently seen in conventional chondrosarcomas, are not detected in chordomas [46, 47].
Management and Prognosis
The primary treatment modality for chordomas typically consists of surgical resection followed by adjuvant radiotherapy. Chordomas of the skull base are always resected in a piecemeal fashion, which precludes the pathologic assessment of margins. As a result, post-operative imaging is the preferred method for documenting the presence or absence of residual tumor. Unfortunately, complete resection is only achieved in 29% of cases and most tumors recur locally despite adjuvant radiotherapy to the tumor bed [12]. Although skull base chordomas and chondrosarcomas are treated similarly, distinction between the two is important since chondrosarcomas carries a better prognosis [12, 26, 48–51]. The local recurrence rate for skull base chondrosarcoma is 1.5% (up to 53% without adjuvant radiotherapy), whereas the local recurrence rate for skull base chordoma is 68%. The 5-year and 10-year overall survivals are more favourable for skull base chondrosarcoma (82–88.5 and 50%, respectively), compared to skull base chordoma (63–78.4 and 16–32%, respectively) [12, 26, 48–51].
Traditionally, chordomas are not sensitive to chemotherapy. Early studies assessing the efficacy of EGFR inhibitors, as well as imatinib, have been promising, particularly in locally advanced and metastatic chordoma [52–56].
Chondroid chordoma has been reported by some to have a better prognosis [57–59], however most recent studies have demonstrated that conventional and chondroid chordomas have similar outcomes [22, 26, 28, 60–63]. Children under the age of 5 years are diagnosed with a disproportionately high frequency of poorly differentiated chordomas, which is associated with an overall worse prognosis for this age group [18, 25, 62, 64–68]. Indeed, conventional and chondroid chordomas in pediatric patients appears to have similar or better survival when compared to adults [18, 57, 62].
Overall, poorly differentiated and dedifferentiated chordomas are more aggressive tumors with rapid tumor growth and higher rates of metastases. Chordomas most commonly metastasize to the lung, followed by the lymph nodes, bone, liver, and subcutaneous tissues [15, 64, 65]. In adults, sacral and vertebral chordomas are more likely to metastasize than skull base ones with metastatic rates ranging from 3% to almost 50% [15, 66, 69]. Chordomas in children under the age of 5 are much more likely to metastasize (up to 58% of cases) compared to children 5 years of age or older (only 8.5% of cases), reflecting the poorly differentiated histology seen more commonly in very young children [18, 64, 65]. Other adverse prognosticators include older age (≥ 50 years) with conventional or chondroid histology and larger tumor size at presentation (≥ 4.0 cm) [12].
Conclusion
Chordoma is a malignant tumor of notochord origin that can arise in various sites throughout the head and neck. While rare, chordomas are important to recognize as they can easily be confused for other more common entities with very different treatments and prognoses. Pathologists also need to be aware of histologic subtypes of chordoma that can mimic other high-grade sarcomas or carcinomas. Despite optimal medical management, most chordomas recur and future strategies will need to focus on targeted molecular therapies that eliminate residual tumor cells. More large studies are needed to the molecular alterations driving chordoma oncogenesis and growth.
Compliance with Ethical Standards
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12:1–11. doi: 10.1023/A:1008947301735. [DOI] [PubMed] [Google Scholar]
- 2.Smoll NR, Gautschi OP, Radovanovic I, Schaller K, Weber DC. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013;119:2029–2037. doi: 10.1002/cncr.28032. [DOI] [PubMed] [Google Scholar]
- 3.Mukherjee D, Chaichana KL, Gokaslan ZL, Aaronson O, Cheng JS, McGirt MJ. Survival of patients with malignant primary osseous spinal neoplasms: results from the Surveillance, Epidemiology, and End Results (SEER) database from 1973 to 2003. J Neurosurg Spine. 2011;14:143–150. doi: 10.3171/2010.10.SPINE10189. [DOI] [PubMed] [Google Scholar]
- 4.Lauer SR, Edgar MA, Gardner JM, Sebastian A, Weiss SW. Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol. 2013;37:719–726. doi: 10.1097/PAS.0b013e31827813e7. [DOI] [PubMed] [Google Scholar]
- 5.Tirabosco R, Mangham DC, Rosenberg AE, Vujovic S, Bousdras K, Pizzolitto S, De Maglio G, den Bakker MA, Di Francesco L, Kalil RK, Athanasou NA, O’Donnell P, McCarthy EF, Flanagan AM. Brachyury expression in extra-axial skeletal and soft tissue chordomas: a marker that distinguishes chordoma from mixed tumor/myoepithelioma/parachordoma in soft tissue. Am J Surg Pathol. 2008;32:572–580. doi: 10.1097/PAS.0b013e31815b693a. [DOI] [PubMed] [Google Scholar]
- 6.Gladstone HB, Bailet JW, Rowland JP. Chordoma of the oropharynx: an unusual presentation and review of the literature. Otolaryngol Head Neck Surg. 1998;118:104–107. doi: 10.1016/S0194-5998(98)70384-5. [DOI] [PubMed] [Google Scholar]
- 7.Kataria SP, Batra A, Singh G, Kumar S, Sen R. Chordoma of skull base presenting as nasopharyngeal mass. J Neurosci Rural Pract. 2013;4:S95-97. doi: 10.4103/0976-3147.116426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khurram SA, Biswas D, Fernando M. A parapharyngeal soft tissue chordoma presenting with synchronous cervical lymph node metastasis: an unusual presentation. Head Neck Pathol. 2016;10:400–404. doi: 10.1007/s12105-016-0712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynn-Macrae A, Haines GK, 3rd, Altman KW. Primary chordoma of the lateral nasal wall: case report and review. Ear Nose Throat J. 2005;84:593–595. [PubMed] [Google Scholar]
- 10.Nguyen RP, Salzman KL, Stambuk HE, Ahuja AT, Harnsberger HR. Extraosseous chordoma of the nasopharynx. AJNR Am J Neuroradiol. 2009;30:803–807. doi: 10.3174/ajnr.A1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tao ZZ, Chen SM, Liu JF, Huang XL, Zhou L. Paranasal sinuses chordoma in pediatric patient: a case report and literature review. Int J Pediatr Otorhinolaryngol. 2005;69:1415–1418. doi: 10.1016/j.ijporl.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 12.Bohman LE, Koch M, Bailey RL, Alonso-Basanta M, Lee JY. Skull base chordoma and chondrosarcoma: influence of clinical and demographic factors on prognosis: a SEER analysis. World Neurosurg. 2014;82:806–814. doi: 10.1016/j.wneu.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Lee-Jones L, Aligianis I, Davies PA, Puga A, Farndon PA, Stemmer-Rachamimov A, Ramesh V, Sampson JR. Sacrococcygeal chordomas in patients with tuberous sclerosis complex show somatic loss of TSC1 or TSC2. Genes Chromosomes Cancer. 2004;41:80–85. doi: 10.1002/gcc.20052. [DOI] [PubMed] [Google Scholar]
- 14.Oakley GJ, Fuhrer K, Seethala RR, Brachyury SOX-9, and podoplanin, new markers in the skull base chordoma vs chondrosarcoma differential: a tissue microarray-based comparative analysis. Mod Pathol. 2008;21:1461–1469. doi: 10.1038/modpathol.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fletcher CDM, World Health Organization., International Agency for Research on Cancer . WHO classification of tumours of soft tissue and bone. Lyon: IARC Press; 2013. [Google Scholar]
- 16.Heffelfinger MJ, Dahlin DC, MacCarty CS, Beabout JW. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32:410–420. doi: 10.1002/1097-0142(197308)32:2<410::AID-CNCR2820320219>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg AE, Brown GA, Bhan AK, Lee JM. Chondroid chordoma–a variant of chordoma. A morphologic and immunohistochemical study. Am J Clin Pathol. 1994;101:36–41. doi: 10.1093/ajcp/101.1.36. [DOI] [PubMed] [Google Scholar]
- 18.Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol. 2006;30:811–818. doi: 10.1097/01.pas.0000209828.39477.ab. [DOI] [PubMed] [Google Scholar]
- 19.Jambhekar NA, Rekhi B, Thorat K, Dikshit R, Agrawal M, Puri A. Revisiting chordoma with brachyury, a “new age” marker: analysis of a validation study on 51 cases. Arch Pathol Lab Med. 2010;134:1181–1187. doi: 10.5858/2009-0476-OA.1. [DOI] [PubMed] [Google Scholar]
- 20.Miettinen M, Wang Z, Lasota J, Heery C, Schlom J, Palena C. Nuclear brachyury expression is consistent in chordoma, common in germ cell tumors and small cell carcinomas, and rare in other carcinomas and sarcomas: an immunohistochemical study of 5229 cases. Am J Surg Pathol. 2015;39:1305–1312. doi: 10.1097/PAS.0000000000000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sangoi AR, Karamchandani J, Lane B, Higgins JP, Rouse RV, Brooks JD, McKenney JK. Specificity of brachyury in the distinction of chordoma from clear cell renal cell carcinoma and germ cell tumors: a study of 305 cases. Mod Pathol. 2011;24:425–429. doi: 10.1038/modpathol.2010.196. [DOI] [PubMed] [Google Scholar]
- 22.Vujovic S, Henderson S, Presneau N, Odell E, Jacques TS, Tirabosco R, Boshoff C, Flanagan AM. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209:157–165. doi: 10.1002/path.1969. [DOI] [PubMed] [Google Scholar]
- 23.Chavez JA, Nasir Ud D, Memon A, Perry A. Anaplastic chordoma with loss of INI1 and brachyury expression in a 2-year-old girl. Clin Neuropathol. 2014;33:418–420. doi: 10.5414/NP300724. [DOI] [PubMed] [Google Scholar]
- 24.Hasselblatt M, Thomas C, Hovestadt V, Schrimpf D, Johann P, Bens S, Oyen F, Peetz-Dienhart S, Crede Y, Wefers A, Vogel H, Riemenschneider MJ, Antonelli M, Giangaspero F, Bernardo MC, Giannini C, Ud Din N, Perry A, Keyvani K, van Landeghem F, Sumerauer D, Hauser P, Capper D, Korshunov A, Jones DT, Pfister SM, Schneppenheim R, Siebert R, Fruhwald MC, Kool M. Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismal prognosis. Acta Neuropathol. 2016;132:149–151. doi: 10.1007/s00401-016-1574-9. [DOI] [PubMed] [Google Scholar]
- 25.Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R, Hayden MG, Cherry AM, Gokden M, Edwards MS, Fisher PG, Vogel H. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol. 2010;120:745–753. doi: 10.1007/s00401-010-0767-x. [DOI] [PubMed] [Google Scholar]
- 26.Almefty K, Pravdenkova S, Colli BO, Al-Mefty O, Gokden M. Chordoma and chondrosarcoma: similar, but quite different, skull base tumors. Cancer. 2007;110:2457–2467. doi: 10.1002/cncr.23073. [DOI] [PubMed] [Google Scholar]
- 27.Pamir MN, Ozduman K. Analysis of radiological features relative to histopathology in 42 skull-base chordomas and chondrosarcomas. Eur J Radiol. 2006;58:461–470. doi: 10.1016/j.ejrad.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 28.Rosenberg AE, Nielsen GP, Keel SB, Renard LG, Fitzek MM, Munzenrider JE, Liebsch NJ. Chondrosarcoma of the base of the skull: a clinicopathologic study of 200 cases with emphasis on its distinction from chordoma. Am J Surg Pathol. 1999;23:1370–1378. doi: 10.1097/00000478-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Folpe AL, Agoff SN, Willis J, Weiss SW. Parachordoma is immunohistochemically and cytogenetically distinct from axial chordoma and extraskeletal myxoid chondrosarcoma. Am J Surg Pathol. 1999;23:1059–1067. doi: 10.1097/00000478-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Sangoi AR, Dulai MS, Beck AH, Brat DJ, Vogel H. Distinguishing chordoid meningiomas from their histologic mimics: an immunohistochemical evaluation. Am J Surg Pathol. 2009;33:669–681. doi: 10.1097/PAS.0b013e318194c566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fischer C, Scheipl S, Zopf A, Niklas N, Deutsch A, Jorgensen M, Lohberger B, Froehlich EV, Leithner A, Gabriel C, Liegl-Atzwanger B, Rinner B. Mutation analysis of nine chordoma specimens by targeted next-generation cancer panel sequencing. J Cancer. 2015;6:984–989. doi: 10.7150/jca.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akhavan-Sigari R, Gaab MR, Rohde V, Abili M, Ostertag H. Expression of PDGFR-alpha, EGFR and c-MET in spinal chordoma: a series of 52 patients. Anticancer Res. 2014;34:623–630. [PubMed] [Google Scholar]
- 33.Chen K, Mo J, Zhou M, Wang G, Wu G, Chen H, Zhang K, Yang H. Expression of PTEN and mTOR in sacral chordoma and association with poor prognosis. Med Oncol. 2014;31:886. doi: 10.1007/s12032-014-0886-7. [DOI] [PubMed] [Google Scholar]
- 34.Yang C, Schwab JH, Schoenfeld AJ, Hornicek FJ, Wood KB, Nielsen GP, Choy E, Mankin H, Duan Z. A novel target for treatment of chordoma: signal transducers and activators of transcription 3. Mol Cancer Ther. 2009;8:2597–2605. doi: 10.1158/1535-7163.MCT-09-0504. [DOI] [PubMed] [Google Scholar]
- 35.Zhang K, Chen H, Zhang B, Sun J, Lu J, Chen K, Yang H. Overexpression of Raf-1 and ERK1/2 in sacral chordoma and association with tumor recurrence. Int J Clin Exp Pathol. 2015;8:608–614. [PMC free article] [PubMed] [Google Scholar]
- 36.Bayrak OF, Gulluoglu S, Aydemir E, Ture U, Acar H, Atalay B, Demir Z, Sevli S, Creighton CJ, Ittmann M, Sahin F, Ozen M. MicroRNA expression profiling reveals the potential function of microRNA-31 in chordomas. J Neurooncol. 2013;115:143–151. doi: 10.1007/s11060-013-1211-6. [DOI] [PubMed] [Google Scholar]
- 37.Long C, Jiang L, Wei F, Ma C, Zhou H, Yang S, Liu X, Liu Z. Integrated miRNA-mRNA analysis revealing the potential roles of miRNAs in chordomas. PLoS ONE. 2013;8:e66676. doi: 10.1371/journal.pone.0066676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zou MX, Huang W, Wang XB, Li J, Lv GH, Wang B, Deng YW. Reduced expression of miRNA-1237-3p associated with poor survival of spinal chordoma patients. Eur Spine J. 2015;24:1738–1746. doi: 10.1007/s00586-015-3927-9. [DOI] [PubMed] [Google Scholar]
- 39.Marucci G, Morandi L, Mazzatenta D, Frank G, Pasquini E, Foschini MP. MGMT promoter methylation status in clival chordoma. J Neurooncol. 2014;118:271–276. doi: 10.1007/s11060-014-1445-y. [DOI] [PubMed] [Google Scholar]
- 40.Yang XR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li S, Goldstein AM, Parry DM, Kelley MJ. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet. 2009;41:1176–1178. doi: 10.1038/ng.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, Halai D, Berisha F, Cannon SR, Mead S, Kasperaviciute D, Palmen J, Talmud PJ, Kindblom LG, Amary MF, Tirabosco R, Flanagan AM. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet. 2012;44:1185–1187. doi: 10.1038/ng.2419. [DOI] [PubMed] [Google Scholar]
- 42.Presneau N, Shalaby A, Ye H, Pillay N, Halai D, Idowu B, Tirabosco R, Whitwell D, Jacques TS, Kindblom LG, Bruderlein S, Moller P, Leithner A, Liegl B, Amary FM, Athanasou NN, Hogendoorn PC, Mertens F, Szuhai K, Flanagan AM. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. J Pathol. 2011;223:327–335. doi: 10.1002/path.2816. [DOI] [PubMed] [Google Scholar]
- 43.Brandal P, Bjerkehagen B, Danielsen H, Heim S. Chromosome 7 abnormalities are common in chordomas. Cancer Genet Cytogenet. 2005;160:15–21. doi: 10.1016/j.cancergencyto.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 44.Le LP, Nielsen GP, Rosenberg AE, Thomas D, Batten JM, Deshpande V, Schwab J, Duan Z, Xavier RJ, Hornicek FJ, Iafrate AJ. Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS ONE. 2011;6:e18846. doi: 10.1371/journal.pone.0018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheil S, Bruderlein S, Liehr T, Starke H, Herms J, Schulte M, Moller P. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001;32:203–211. doi: 10.1002/gcc.1184. [DOI] [PubMed] [Google Scholar]
- 46.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 47.Arai M, Nobusawa S, Ikota H, Takemura S, Nakazato Y. Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol. 2012;29:201–206. doi: 10.1007/s10014-012-0085-1. [DOI] [PubMed] [Google Scholar]
- 48.Bloch OG, Jian BJ, Yang I, Han SJ, Aranda D, Ahn BJ, Parsa AT. A systematic review of intracranial chondrosarcoma and survival. J Clin Neurosci. 2009;16:1547–1551. doi: 10.1016/j.jocn.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho YH, Kim JH, Khang SK, Lee JK, Kim CJ. Chordomas and chondrosarcomas of the skull base: comparative analysis of clinical results in 30 patients. Neurosurg Rev. 2008;31:35–43. doi: 10.1007/s10143-007-0099-z. [DOI] [PubMed] [Google Scholar]
- 50.Di Maio S, Temkin N, Ramanathan D, Sekhar LN. Current comprehensive management of cranial base chordomas: 10-year meta-analysis of observational studies. J Neurosurg. 2011;115:1094–1105. doi: 10.3171/2011.7.JNS11355. [DOI] [PubMed] [Google Scholar]
- 51.Martin JJ, Niranjan A, Kondziolka D, Flickinger JC, Lozanne KA, Lunsford LD. Radiosurgery for chordomas and chondrosarcomas of the skull base. J Neurosurg. 2007;107:758–764. doi: 10.3171/JNS-07/10/0758. [DOI] [PubMed] [Google Scholar]
- 52.Hindi N, Casali PG, Morosi C, Messina A, Palassini E, Pilotti S, Tamborini E, Radaelli S, Gronchi A, Stacchiotti S. Imatinib in advanced chordoma: A retrospective case series analysis. Eur J Cancer. 2015;51:2609–2614. doi: 10.1016/j.ejca.2015.07.038. [DOI] [PubMed] [Google Scholar]
- 53.Launay SG, Chetaille B, Medina F, Perrot D, Nazarian S, Guiramand J, Moureau-Zabotto L, Bertucci F. Efficacy of epidermal growth factor receptor targeting in advanced chordoma: case report and literature review. BMC Cancer. 2011;11:423. doi: 10.1186/1471-2407-11-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheipl S, Barnard M, Cottone L, Jorgensen M, Drewry DH, Zuercher WJ, Turlais F, Ye H, Leite AP, Smith JA, Leithner A, Moller P, Bruderlein S, Guppy N, Amary F, Tirabosco R, Strauss SJ, Pillay N, Flanagan AM. EGFR inhibitors identified as a potential treatment for chordoma in a focused compound screen. J Pathol. 2016;239:320–334. doi: 10.1002/path.4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singhal N, Kotasek D, Parnis FX. Response to erlotinib in a patient with treatment refractory chordoma. Anticancer Drugs. 2009;20:953–955. doi: 10.1097/CAD.0b013e328330c7f0. [DOI] [PubMed] [Google Scholar]
- 56.Stacchiotti S, Longhi A, Ferraresi V, Grignani G, Comandone A, Stupp R, Bertuzzi A, Tamborini E, Pilotti S, Messina A, Spreafico C, Gronchi A, Amore P, Vinaccia V, Casali PG. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30:914–920. doi: 10.1200/JCO.2011.35.3656. [DOI] [PubMed] [Google Scholar]
- 57.Forsyth PA, Cascino TL, Shaw EG, Scheithauer BW, O’Fallon JR, Dozier JC, Piepgras DG. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases. J Neurosurg. 1993;78:741–747. doi: 10.3171/jns.1993.78.5.0741. [DOI] [PubMed] [Google Scholar]
- 58.Jian BJ, Bloch OG, Yang I, Han SJ, Aranda D, Tihan T, Parsa AT. Adjuvant radiation therapy and chondroid chordoma subtype are associated with a lower tumor recurrence rate of cranial chordoma. J Neurooncol. 2010;98:101–108. doi: 10.1007/s11060-009-0068-1. [DOI] [PubMed] [Google Scholar]
- 59.Mitchell A, Scheithauer BW, Unni KK, Forsyth PJ, Wold LE, McGivney DJ. Chordoma and chondroid neoplasms of the spheno-occiput. An immunohistochemical study of 41 cases with prognostic and nosologic implications. Cancer. 1993;72:2943–2949. doi: 10.1002/1097-0142(19931115)72:10<2943::AID-CNCR2820721014>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 60.Colli BO, Al-Mefty O. Chordomas of the skull base: follow-up review and prognostic factors. Neurosurg Focus. 2001;10:E1. doi: 10.3171/foc.2001.10.3.2. [DOI] [PubMed] [Google Scholar]
- 61.Gay E, Sekhar LN, Rubinstein E, Wright DC, Sen C, Janecka IP, Snyderman CH. Chordomas and chondrosarcomas of the cranial base: results and follow-up of 60 patients. Neurosurgery. 1995;36:887–896. doi: 10.1227/00006123-199505000-00001. [DOI] [PubMed] [Google Scholar]
- 62.Jian BJ, Bloch OG, Yang I, Han SJ, Aranda D, Parsa AT. A comprehensive analysis of intracranial chordoma and survival: a systematic review. Br J Neurosurg. 2011;25:446–453. doi: 10.3109/02688697.2010.546896. [DOI] [PubMed] [Google Scholar]
- 63.Romeo S, Hogendoorn PC. Brachyury and chordoma: the chondroid-chordoid dilemma resolved? J Pathol. 2006;209:143–146. doi: 10.1002/path.1987. [DOI] [PubMed] [Google Scholar]
- 64.Borba LA, Al-Mefty O, Mrak RE, Suen J. Cranial chordomas in children and adolescents. J Neurosurg. 1996;84:584–591. doi: 10.3171/jns.1996.84.4.0584. [DOI] [PubMed] [Google Scholar]
- 65.Coffin CM, Swanson PE, Wick MR, Dehner LP. Chordoma in childhood and adolescence. A clinicopathologic analysis of 12 cases. Arch Pathol Lab Med. 1993;117:927–933. [PubMed] [Google Scholar]
- 66.Kaneko Y, Sato Y, Iwaki T, Shin RW, Tateishi J, Fukui M. Chordoma in early childhood: a clinicopathological study. Neurosurgery. 1991;29:442–446. doi: 10.1227/00006123-199109000-00019. [DOI] [PubMed] [Google Scholar]
- 67.Matsumoto J, Towbin RB, Ball WS., Jr Cranial chordomas in infancy and childhood. A report of two cases and review of the literature. Pediatr Radiol. 1989;20:28–32. doi: 10.1007/BF02010629. [DOI] [PubMed] [Google Scholar]
- 68.Yadav R, Sharma MC, Malgulwar PB, Pathak P, Sigamani E, Suri V, Sarkar C, Kumar A, Singh M, Sharma BS, Garg A, Bakhshi S, Faruq M. Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro Oncol. 2014;16:372–381. doi: 10.1093/neuonc/not228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chambers PW, Schwinn CP. Chordoma. A clinicopathologic study of metastasis. Am J Clin Pathol. 1979;72:765–776. doi: 10.1093/ajcp/72.5.765. [DOI] [PubMed] [Google Scholar]