

Abstract
Activity of voltage-gated potassium (Kv) channels controls membrane potential, which subsequently regulates cytoplasmic free calcium concentration ([Ca2+]cyt) in pulmonary artery smooth muscle cells (PASMCs). Acute hypoxia inhibits Kv channel function in PASMCs, inducing membrane depolarization and a rise in [Ca2+ ]cyt that triggers vasoconstriction. Prolonged hypoxia inhibits expression of Kv channels and reduces Kv channel currents in PASMCs. The consequent membrane depolarization raises [Ca2+]cyt, thus stimulating PASMC proliferation. The present review discusses recent evidence for the involvement of Kv channels in initiation of hypoxic pulmonary vasoconstriction and in chronic hypoxia-induced pulmonary hypertension.
Keywords: hypoxia, intracellular calcium, membrane potential, smooth muscle
Introduction
The main function of the low-pressure, low-resistance pulmonary circulation is to oxygenate blood to be supplied to the rest of the body. In order to perform this function efficiently, the lung must match its regional ventilation and perfusion. Thus, in regions where alveolar oxygen levels are low the pulmonary arteries constrict, diverting blood flow to better ventilated areas of the lung where gas exchange can take place more efficiently. This important physiologic phenomenon is known as 'hypoxic pulmonary vasoconstriction' (HPV), and is unique to the pulmonary circulation; hypoxia generally causes dilatation in systemic arterial beds.
In the fetus, gas exchange occurs via the placenta and HPV acts to maintain a high pulmonary vascular resistance to divert blood flow away from the lungs through the ductus arteriosus. In the adult, HPV can act as a protective mechanism during acute exposure to hypoxia in order to maintain ventilation-perfusion matching (eg during an acute asthma attack). Chronic exposure to hypoxia (eg chronic obstructive pulmonary disease or living at high altitude) causes continuous alveolar hypoxia, which results in sustained HPV, leading to vascular remodeling, pulmonary hypertension, and possibly right heart failure and death.
The mechanism that is responsible for HPV has still not been fully elucidated. The present review concentrates specifically on summarizing the knowledge gained, during the past 10 years, regarding the role of Kv channels in initiation of the acute and chronic responses of pulmonary arteries to hypoxia. Also briefly summarized are some of the mechanisms that underlie current treatment of hypoxia-related cardiopulmonary disease states.
Acute hypoxic pulmonary vasoconstriction
In whole animals, or isolated lungs, acute exposure to hypoxia causes a sustained increase in pulmonary arterial pressure. Hypoxia also causes contraction in isolated pulmonary arteries [1,2,3] and single PASMCs [4,5]. In contrast, hypoxia causes relaxation of cerebral arterial smooth muscle cells (SMCs) [4]. These findings indicate that the mechanism of HPV is intrinsic to the vascular wall of pulmonary arteries, and imply that the effects of neural inputs, blood-borne agents, and lung parenchyma do not directly mediate HPV, but rather they modulate the response.
Removal of the endothelium does not abolish hypoxia-mediated contraction in isolated pulmonary arteries [3]. The observations of hypoxia-induced contraction in single PASMCs imply that the endothelium is not necessary for HPV, and that all of the mechanisms that are necessary for sensing and responding to hypoxia are intrinsic to the PASMC [4,5]. It is generally accepted that the endothelium does modulate HPV. This effect could be achieved through release of endothelium-derived contracting factors (eg endothelin-1 or thromboxane A2) or through reduction of vasodilator activity of endothelium-derived relaxing factors (eg nitric oxide [NO] and prostacyclin) and endothelium-derived hyperpolarizing factors.
Role of intracellular calcium
An increase in [Ca2+]cyt is necessary to elicit contraction in smooth muscle. In PASMCs, [Ca2+]cyt can be increased by calcium influx through sarcolemmal calcium channels and by its release from intracellular stores (mainly sarcoplasmic reticulum). There are at least three classes of calcium-permeable channels in the plasma membrane: voltage-dependent calcium channels (VDCCs), receptor-operated calcium channels, and store-operated calcium channels. Calcium influx through VDCCs is governed by the membrane potential (Em). VDCCs are normally closed at resting Em, but when the membrane is depolarized they open, allowing calcium influx into the cells [6,7].
McMurtry et al [8] demonstrated that blockers of calcium channels reduced the hypoxic response in isolated lungs, indicating that a rise in [Ca2+]cyt is a necessary step in HPV. Subsequent studies in isolated PASMCs, using fluorescence microscopy techniques, have shown that hypoxia does cause an increase in [Ca2+]cyt [9,10]. This increase in [Ca2+]cyt is smaller in the absence of extracellular calcium, implying contributions from calcium release from intracellular stores and calcium entry across the sarcolemma. The partial inhibition of HPV by the VDCC blocker verapamil [8] suggested that hypoxia must cause opening of VDCCs, implying that membrane depolarization would also appear to be a necessary step in the hypoxic response. Harder et al [1] and Madden et al [2] demonstrated that hypoxia does indeed induce membrane depolarization and elicits calcium-dependent action potentials in cat pulmonary arteries.
Taken together, the results of these studies indicate that initial depolarization is necessary for opening of VDCCs to allow influx of calcium into PASMCs and to elicit HPV. They also imply that one or more of the potassium channels involved in regulating Em in PASMCs must be sensitive to hypoxia, and could be involved in initiating HPV. These findings have led to a considerable body of research concerned with identifying the particular potassium channel(s) that are involved and the mechanism by which changes in oxygen tension are sensed by the channel(s).
Regulation of membrane potential in pulmonary artery smooth muscle cells
Em in vascular SMCs is predominantly controlled by potassium permeability, which is determined by sarcolemmal potassium channel conductance and transmembrane potassium gradient. Intracellular potassium concentration (140 mmol/l) is greater than that of extracellular potassium (5 mmol/l), favoring movement of potassium out of the cell when potassium channels are open, leading to hyperpolarization of the membrane. Closure of these channels would cause membrane depolarization, leading to opening of VDCCs and entry of calcium into the cell. To date, three types of potassium channels have been described in PASMCs: Kv channels [7,11*,12*], calcium-activated potassium (KCa) channels [13,14], and ATP-sensitive potassium (KATP) channels [6]. Potassium currents through all three of these channel types contribute to the regulation of Em [6]. Using the Kv channel blocker 4-aminopyridine, it has been shown [7,9,11*,12*,15] that potassium currents through Kv channels (IK(V)) are the major determinants of resting Em in PASMCs.
Effect of hypoxia on IK(V)
Post et al [16] first demonstrated that hypoxia reduced potassium currents in canine PASMCs, and others have since shown a similar effect of hypoxia on potassium currents in PASMCs from other animals [17,18**,19**]. There is now wide agreement that the current inhibited by acute hypoxia is IK(V) [12*,17,18**,19**]. Fig. 1 summarizes the effects of acute hypoxia on whole-cell IK(V), Em, and [Ca2+]cyt in PASMCs. Reduction in perfusate oxygen tension from 135-145 mmHg to 10-35 mmHg significantly reduces IK(V) in PASMCs (Fig. 1a and 1b). The resultant membrane depolarization leads to opening of VDCCs, allowing calcium influx into the cells to increase [Ca2+]cyt (Fig. 1c and 1d). This hypoxic inhibition of IK(V) is selective to PASMCs, because acute hypoxia has negligible effects on IK(V) in mesenteric arterial SMCs (Fig. 1b).
Figure 1.
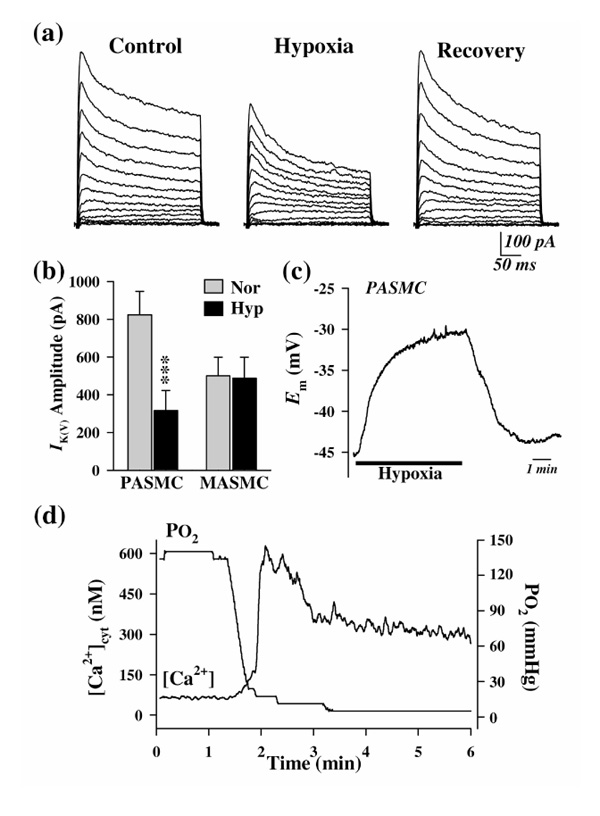
Effects of acute hypoxia on whole-cell IK(V), Em, and [Ca2+]cyt in PASMCs. (a) Representative families of currents elicited by depolarizing the cell to test potentials ranging from -50 to +80 mV in 10-mV increments (holding potential -70 mV); these were recorded before, during, and after reduction of partial oxygen tension (PO2)in the extracellular solution from 149 to 8 mmHg. (b) Summarized data showing the effect of acute hypoxia (Hyp) on IK(V), elicited by a test pulse of +60 mV, in PASMCs and mesenteric artery smooth muscle cells (MASMCs). Data are expressed as mean ± standard error. ***P < 0.001 versus normoxia (Nor). (c) Change in Em recorded in a PASMC on switching from normoxic to hypoxic bath solutions. (d) [Ca2+]cyt measured (using Fura-2) in a peripheral cytoplasmic region of a PASMC on switching from normoxic to hypoxic bath solutions. The PO2 level was measured using an oxygen meter positioned in the tissue chamber. Modified with permission from Yuan et al [19**].
Molecular identity of Kv channels
At the molecular level, Kv channels are homo- or heteromultimeric tetramers that are composed of two structurally distinct subunits: the pore-forming α-subunits and the regulatory β-subunits. The genes that encode functional Kv channel α-subunits fall into four subfamilies: Shaker (Kv1), Shab (Kv2), Shaw (Kv3), and Shal (Kv4). At least 25 vertebrate genes that encode functional Kv channel α-subunits have been isolated from mammals, and more than 15 genes from humans. Each of these Kv channel α-subunits produces functionally distinct Kv channels. Recently, four new subfamilies of electrically silent Kv channel α-subunits have been cloned: Kv5, Kv6, Kv8, and Kv9. Expression of these modulatory α-subunits per se does not produce potassium channel activity; however, coexpression of these modulatory α-subunits with other functional Kv channel α-subunits (eg Kv2.1) significantly modulates biophysical properties (eg activation and inactivation kinetics, voltage dependence) and expression levels of the functional Kv channels [20**].
Three subfamilies of Kv channel β-subunits - Kvβ1, Kvβ2, and Kvβ3 - have been identified in mammalian tissues [21,22]. Although expression of α-subunits alone is sufficient to generate Kv channels that possess many features of the corresponding channels in situ, studies on native channels have confirmed that the biophysical properties of Kv channels encoded by certain α-subunits can be dramatically altered by their association with β-subunits. For example, association of β-subunits with α-subunits can confer fast inactivation on slowly inactivating or noninactivating delayed rectifier Kv channels [22], and determines the response of the α-subunits to activated protein kinases. In addition to changing kinetic properties of Kv channels, β-subunits can also reduce IK(V) as an open-channel blocker.
Molecular identity of the Kv channels that are responsible for the hypoxia-sensitive IK(V)
The potential candidate Kv channel α-subunits that could form oxygen-sensitive, homomeric or heteromeric channels include Kv1.2 [23*,24**,25**], Kv1.5 [25**,26], Kv2.1 [20**,24**,26], Kv3.1 [27*], Kv4.2 [28*], and Kv9.3 [20**,24**]. It has recently been reported that the Kvβ1 subunits, which specifically bind to the Shaker (Kv1) channel α-subunits, belong to an NAD(P)H-dependent oxidoreductase superfamily, suggesting that the β-subunits might play a critical role in sensing changes in redox status [22] and oxygen tension [28*].
A number of studies have determined the properties, including sensitivity to hypoxia, of channels formed by recombinant Kv channel subunits expressed in heterologous systems. Expression of Kv2.1 or coexpression of Kv2.1 with Kv9.3 (an electrically silent Kv α-subunit) in COS cells causes Em hyperpolarization [20**]. Hypoxia decreases the IK(V) generated by either Kv2.1 or Kv2.1/Kv9.3 channels transfected in COS or mouse L cells [20**,24**]. The current generated by either Kv2.1 or Kv2.1/Kv9.3 channels is also decreased by reduction in intracellular ATP [20**]. Because the Kv channels contain phosphorylation sites, these effects on IK(V) may be related to phosphorylation of the channel (Kv1.2 and/or Kv9.3) protein [20**]. Hypoxia also significantly decreases IK(V) in the following: COS cells transiently transfected with Kv3.1b channels [27*]; PC12 cells or Xenopus oocytes transfected with Kv1.2 [23*]; and HEK293 cells cotransfected with Kv4.2 and Kvβ1.2 [28*].
Using reverse transcription polymerase chain reaction and Western blotting, Yuan et al [15] demonstrated the expression of Kv1.1, Kv1.2, Kv1.4, Kv1.5, Kv1.6, Kv2.1, Kv4.3, Kv9.3, Kvβ1.1, Kvβ1.2, and Kvβ2.1 in rat PASMCs. Other groups have confirmed many of these findings and have further identified the presence of Kv1.3 and Kv3.1 in PASMCs isolated from rats [20**,26,27*]. Intracellular application of the antibody against Kv2.1 reduces whole-cell IK(V), depolarizes Em, and raises [Ca 2+]cyt in PASMCs, whereas the antibody against Kv1.5 reduces IK(V) and selectively inhibits the hypoxia-induced increase in [Ca2+]cyt and pulmonary vasoconstriction [26]. Taken together, these observations suggest that the homomeric Kv channels formed by Kv1.2, Kv1.5, Kv2.1, or Kv3.1b, and the heteromeric channels formed by Kv1.2/Kv1.5, Kv2.1/Kv9.3, or Kv4.2/Kvβ1.2 are potential molecular components in hypoxia-induced membrane depolarization and HPV. Table 1 summarizes some of the biophysical properties of these channels.
Table 1.
Biophysical properties of Kv channel α- and β-subunits that are potentially involved in oxygen sensing
| Subunit | Channel conductance (pS) | Threshold of activation (mV) | Sensitivity to hypoxia | References |
| Kv1.2 | 18 | -20 | Yes | [24**] |
| -40 | Yes | [23*] | ||
| Kv1.5 | 7.1 | -50 | - | [49] |
| -30 | No | [27*] | ||
| -60 | No | [24**] | ||
| - | Yes | [26] | ||
| Kv2.1 | 8.5 | -10 | Yes | [20**] |
| -45 | No | [23*] | ||
| -40 | Yes | [24**] | ||
| Kv3.1b | 14-31 | -20 | Yes | [27*] |
| Kv4.2 | - | -40 | Yes | [28*] |
| Kv1.2/Kv1.5 | - | -50 | Yes | [24**] |
| Kv2.1/Kv9.3 | 14.5 | -50 | Yes | [20**] |
| -50 | Yes | [24**] | ||
| Kv4.2/Kvβ1.2 | - | -40 | Yes | [28*] |
Many of the Kv channel α- and β-subunits identified in PASMCs, which are potentially involved in forming oxygen-sensitive Kv channels, including Kv1.2, Kv1.3, Kv1.4, Kv1.5, Kv2.1, Kv9.3, Kvβ1.1, Kvβ1.2, and Kvβ2.1, are also expressed in mesenteric arterial SMCs [29]. Qualitative differences in Kv channel α-subunits between PASMCs and systemic artery SMCs have not thus far been observed, but their divergent responses to hypoxia have been well documented. Therefore, why hypoxia only inhibits the aforementioned Kv channels in PASMCs, but not in systemic artery smooth muscle requires further investigation.
Possible mechanisms involved in oxygen sensing
Kv channels have also been implicated in sensing hypoxia in other oxygen-sensitive cells, including cells of the carotid and aortic bodies and neuroepithelial cells [30]. Multiple pathways may exist for hypoxia-induced Kv channel inhibition, and the mechanisms of hypoxia-induced inhibition of potassium currents in oxygen-sensitive cells may be similar. Acute hypoxia may inhibit Kv channel function by the following mechanisms: inhibiting oxidative phosphorylation [31]; changing redox status [17,28*,31]; altering a membrane-delimited oxygen-sensitive regulatory moiety that is adjacent or coupled to the channel protein [30,32]; activating mitochondrial NAD(P)H oxidase and increasing superoxide production [33,34]; inhibiting cytochrome P450 [7]; decreasing intracellular pH [35]; releasing calcium from intracellular stores [8,12*]; and affecting directly the Kv channel protein [30].
Chronic hypoxic pulmonary vasoconstriction: role of Kv channels
In lungs from chronically hypoxic rats, the pressor response to acute hypoxia is impaired, whereas the pressor response to vasoconstrictor agonists (eg, angiotensin II, prostaglandin F2α and norepinephrine) is enhanced [36]. These observations suggest that the reduced pressor response to acute hypoxia in chronically hypoxic rats may result from abnormalities in the mechanism that couples acute hypoxia to contraction of the pulmonary vascular smooth muscle. That is, chronic hypoxia-mediated pulmonary hypertension may involve the same mechanisms as those responsible for acute HPV. Accordingly, a reduction in the activity of Kv channels in PASMCs, with the resultant membrane depolarization, may be involved in the development of chronic hypoxia-mediated pulmonary hypertension by mediating pulmonary vasoconstriction and vascular remodeling through increased [Ca2+]cyt in PASMCs.
Fig. 2 illustrates the effects of prolonged hypoxia on expression of some of the Kv channel subunits in rat PASMCs. Chronic hypoxia downregulates the mRNA expression of pore-forming Kv channel α-subunits, including Kv1.1, Kv1.2, Kv1.4, Kv1.5, Kv2.1, Kv4.3, and Kv9.3, in PASMCs (Fig. 2a and 2b) [25**] (Yu et al, unpublished data). The inhibitory effect of hypoxia is specific to α-subunits because hypoxia has negligible effects on mRNA expression of the cytoplasmic regulatory β-subunits, including Kvβ1.1, Kvβ1.2, and Kvβ2.1 (Fig. 2b) [25**].
Figure 2.
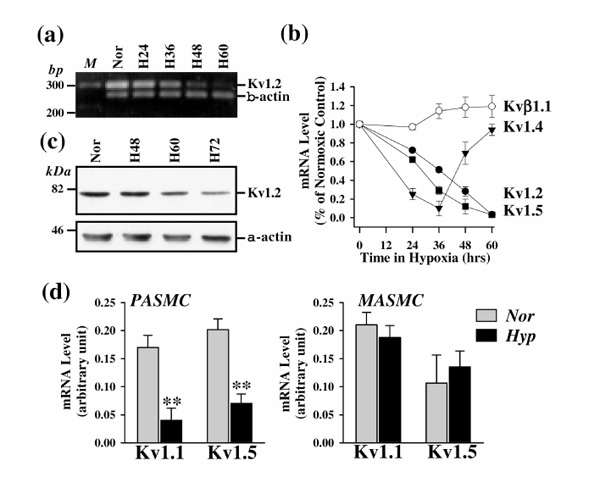
Effects of prolonged hypoxia on mRNA and protein levels of Kv channel α- and β-subunits, in PASMCs and mesenteric artery smooth muscle cells (MASMCs). (a) Polymerase chain reaction-amplified products for Kv1.2 (295 bp) and β-actin (244 bp) displayed in agarose gel. PASMCs were incubated in normoxia (Nor) and hypoxia (25-35 mmHg) for 24 h (H24), 36 h (H36), 48 h (H48), and 60 h (H60). (b) mRNA levels (mean ± standard error) of Kv1.2, Kv1.4, Kv1.5, and Kvβ1.1 in PASMCs, normalized to the amount of β-actin. (c) Western blot analysis of Kv1.2 channel proteins from PASMCs incubated in normoxia (Nor) and hypoxia (25-35 mmHg) for 48 h (H48), 60 h (H60), and 72 h (H72). (d) mRNA levels (mean± standard error), normalized to the respective amounts of β-actin, in PASMCs (left panel) and MASMCs (right panel) before (Nor) and after (Hyp) treatment with hypoxia. **P < 0.01 versus Nor. M, 100-bp DNA ladder. Modified with permission from Wang et al [25**].
Consistent with its inhibitory effect on Kv channel expression, prolonged hypoxia reduces IK(V) (Fig. 3a and 3b) and depolarizes PASMCs (Fig. 3d). The resultant increase in [Ca2+]cyt in PASMCs (Golovina and Yuan, unpublished data) may serve as an important trigger for pulmonary vasoconstriction and as a critical stimulus for PASMC proliferation during chronic hypoxia. Similar results have also been observed in freshly dissociated PASMCs isolated from chronically hypoxic animals [37**,38,39].
Figure 3.
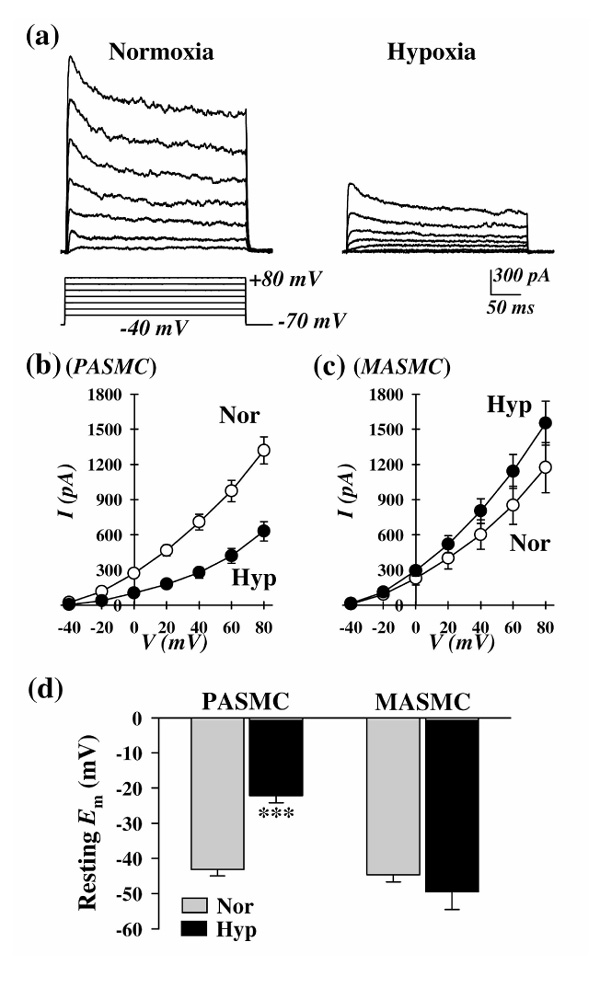
Effects of prolonged hypoxia (3 days) on whole-cell IK(V) and resting Emin PASMCs and mesenteric artery smooth muscle cells (MASMCs). (a) Representative families of currents, elicited by depolarizing the cell to a series of test potentials ranging from -40 to +80 mV (holding potential -70mV) in PASMCs incubated in normoxia or hypoxia (25-35 mmHg). (b) and (c) Current-voltage relationship (I-V) curves (mean ± standard error) of whole-cell IK(V) in (b) PASMCs and (c)MASMCs incubated in normoxia (white circle) or hypoxia (black circle). (d) Summarized data (mean ± standard error) showing resting Em in PASMCs and MASMCs incubated in normoxia (Nor) or hypoxia (Hyp, partial oxygen tension 25-35 mmHg, for 60 h). ***P < 0.001 versus Nor.
Chronic hypoxia has little effect on systemic arterial pressure, measured via a catheter positioned in the carotid artery (Yuan, unpublished data). In mesenteric arterial SMCs, chronic hypoxia did not significantly alter the expression of the Kv channel α- or β-subunits examined (Fig. 2d; Yu and Yuan, unpublished data). Also, in these cells IK(V) was slightly increased (Fig. 3c) and Em was hyperpolarized (Fig. 3d; Platoshyn and Yuan, unpublished data).
Cellular and molecular mechanisms mediating the effects of chronic hypoxia
As discussed above, it appears that hypoxia regulates the function of Kv channel α-subunits via an intrinsic mechanism that exists uniquely in PASMCs. The selectivity of hypoxia-induced downregulation of Kv channel expression and reduction in IK(V) in PASMCs suggests that the molecular mechanisms involved in the regulation of Kv channel gene expression are quite different between pulmonary and systemic arterial SMCs.
The common pathways that transduce hypoxic signals from the sensor to appropriate gene expression involve phosphorylation and/or redox modification of responsive transcription factors [40]. Hypoxia can modulate a variety of transcription factors and signal transduction signaling proteins, such as HIF-1, nuclear factor-κB, c-fos/c-jun, p53, KBF, c/EBP, FixL, and FixJ, suggesting that a large number of transcriptional systems contribute to the response to hypoxia [40,41,42].
Chronic hypoxia may inhibit Kv channel activity by directly or indirectly downregulating mRNA and protein expression of Kv channel α-subunits. In addition to the mechanisms by which acute hypoxia decreases Kv channel activity, the underlying mechanisms that are involved in chronic hypoxia-induced reduction in Kv channel expression may include the following: upregulation or downregulation of the transcription factors and signal transduction proteins [40] that can directly bind to the Kv channel gene promoters (eg Kv1.5 repressor element) and modulate the Kv channel gene transcription [41,42]; and induction of transcription factors that upregulate intermediate inhibitors of the Kv channel genes, such as endothelin-1 [38,39]. Preliminary results from our laboratory (Platoshyn et al, unpublished data) have shown that overexpression of c-jun significantly decreased whole-cell IK(V) in rat PASMCs.
Whether the hypoxia-sensitive transcription factors have direct and/or indirect modulatory effects on gene transcription of Kv channels in PASMCs is unclear and requires further study. Another possibility is that acute hypoxia decreases IK(V) by inhibiting Kv channel activity inPASMCs, with the resultant membrane depolarization attenuating Kv channel expression [43]. The precise mechanisms that are responsible for the differing effects of hypoxia on gene expression of Kv channels in pulmonary and mesenteric artery SMCs may underlie the upstream regulation of the Kv channel gene promoter.
Therapeutic approaches in hypoxia-mediated pulmonary hypertension
Acute HPV can be beneficial in vivo, but over a prolonged period it can be dangerous, leading to persistent elevation of pulmonary vascular resistance and vascular remodeling, pulmonary hypertension, right ventricular failure, and possibly death. Therefore, inhibition of HPV is a potentially useful therapeutic mechanism for patients with pulmonary hypertension secondary to hypoxic pulmonary diseases (eg chronic obstructive pulmonary diseases, Eisenmenger's syndrome, and mountain sickness).
Calcium channel blockers
McMurtry et al [8] first showed that the calcium channel blocker verapamil inhibited HPV in isolated rat lungs. Membrane depolarization-mediated influx of calcium into PASMCs during hypoxia is necessary for both vasoconstriction and cell proliferation. Calcium channel blockers, which have been shown to inhibit HPV in intact animals and isolated perfused lungs, have been used clinically (administered intravenously or orally) to reverse HPV, because they prevent calcium influx into cells and thus inhibit pulmonary vasoconstriction.
Nitric oxide and prostacyclin activate potassium channels in PASMCs
NO and prostacyclin are potent endothelium-derived relaxing factors and inhibitors of SMC proliferation. Inhalation of NO and intravenous administration of prostacyclin have been shown to be effective in inhibiting HPV. NO exerts its vasodilator effect in part by activating KCa and Kv channels, which subsequently causes membrane hyperpolarization, decreases resting [Ca2+]cyt, and elicits vasodilatation [44,45]. This activation of KCa and Kv channels can be directly mediated by the nitrosylation or oxidization effect on the channel protein, and/or indirectly mediated by the increase in cGMP production [44,45]. In PASMCs, NO can also decrease [Ca2+]cyt by the following mechanisms: inhibiting agonist-induced calcium release; blocking VDCCs; and enhancing calcium extrusion and sequestration into the sarcoplasmic reticulum. Prostacyclin may cause vasodilatation by partially activating KCa channels in PASMCs [46]. These results suggest that the therapeutic effects of NO and prostacyclin may be, at least in part, due to membrane hyperpolarization induced by enhancement of potassium currents through KCa and Kv channels, and the subsequent reduction in [Ca2+]cyt in PASMCs.
In animals, endothelin receptor antagonists have been shown to attenuate acute HPV and reverse or protect against the development of pulmonary hypertension and associated vascular remodeling [47]. BQ-123, a specific endothelin A receptor antagonist, showed selectivity for the pulmonary circulation, because it did not affect systemic arterial pressure [47]. The action of BQ-123 has been at least partly attributed to its ability to prevent endogenous endothelin from inhibiting potassium channels [39].
Potassium channel openers
Opening of potassium channels is a physiologic mechanism for reversing hypoxia-induced membrane depolarization, and thereby reversing depolarization-induced contractions. Synthetic potassium channel openers have been identified that open KATP or KCa channels. However, there are not yet specific potassium channel openers for Kv channels, which are the channels that are actually inhibited by hypoxia. Lemakalim has been shown to open KATP channels in PASMCs [6]. Recently, Peng et al [14] demonstrated that the novel KCa opener dehydroepiandrosterone, an endogenously produced hormone, reversed the chronic hypoxia-induced reduction in potassium currents through KCa channels in human PASMCs. Archer et al [48] demonstrated differential distribution of potassium channels in pulmonary arteries, with the smaller resistance arteries, which are most sensitive to hypoxia, having fewer KCa channels than the conduit pulmonary arteries. However, because KCa channels have a large unitary conductance (200-250 pS), opening of only a few KCa channels would have a significant effect on Em and would be a very efficient mechanism for reversing HPV. With the recent advances in molecular identification of specific potassium channel subunits in PASMCs, it should soon be possible to design new potassium channel openers that are selective for specific pulmonary potassium channels.
The current clinical therapies are aimed at reversing the pathophysiologic symptoms of hypoxia, which are principally vasoconstriction and cell proliferation. Current research is aimed at molecular identification of the oxygen sensor in the PASMC, and the cellular mechanisms of transduction of the hypoxic signal to effect HPV and PASMC proliferation. Identification of these mechanisms should enable the development of new, selective treatments for hypoxia-related cardiopulmonary disease states.
Conclusion
Ion channels are involved in regulation of vascular tone in physiologic and pathophysiologic conditions. Disruption of [Ca2+]cyt homeostasis, caused by defects in ion channels, appears to be involved in the pathology of many cardiovascular diseases, including hypoxic pulmonary hypertension. Hypoxia may inhibit Kv channels via multiple pathways in order to assure the efficacy and sensitivity of HPV. Acute hypoxia selectively inhibits Kv channel function and induces Em depolarization in PASMCs, and the resultant increase in [Ca2+]cyt triggers HPV. Chronic hypoxia selectively inhibits mRNA and protein expression of the Kv channel α-subunits (the pore-forming subunits) and decreases the number of functional Kv channels in PASMCs. The consequent reduction in IK(V) depolarizes PASMCs, raises [Ca2+]cyt, and stimulates cell proliferation.Ultimately, persistent pulmonary vasoconstriction and excessive vascular remodeling (via medial hypertrophy) increase pulmonary vascular resistance and cause pulmonary hypertension. Fig. 4 illustrates schematically the multiple pathways and modulators that are involved in effecting the physiologic and pathophysiologic responses to acute and chronic hypoxia. Further research into the mechanisms of HPV, the major contributing factor to the pulmonary hypertension that is present in a variety of cardiopulmonary diseases, should lead to more effective treatment of these diseases.
Figure 4.
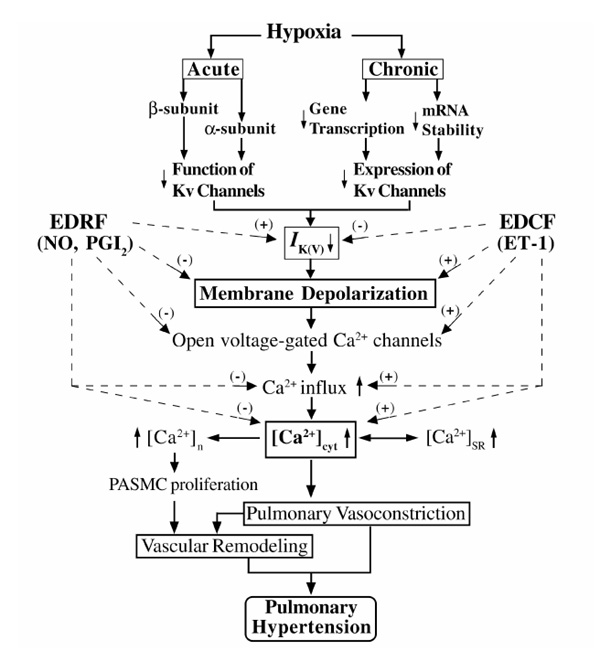
Proposed mechanisms involved in HPV and vascular remodeling. EDRF, endothelium-derived relaxing factor; EDCF, endothelium-derived constricting factor; PGI2, prostacyclin; ET-1, endothelin-1; [Ca2+]n, nuclear [Ca2+]; [Ca2+]SR, sarcoplasmic reticulum [Ca2+]; (+), increase; (-), decrease.
Acknowledgments
Acknowledgments
We apologize to those whose work we have been unable to quote due to restrictions on space. We gratefully acknowledge the contributions of O Platoshyn, MS; J Wang, MD; Y Yu, MD, PhD; V Golovina, PhD; and J Zhang, PhD, to unpublished work cited in this review. This work was supported by grants from the National Institutes of Health (HL 54043 and HL 64549). Dr J Yuan is an Established Investigator of the American Heart Association.
References
- Harder DR, Madden JA, Dawson C. Hypoxic induction of Ca2+-dependent action potentials in small pulmonary arteries of the cat. J Appl Physiol. 1985;59:1389–1393. doi: 10.1152/jappl.1985.59.5.1389. [DOI] [PubMed] [Google Scholar]
- Madden JA, Dawson CA, Harder DR. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J Appl Physiol. 1985;59:113–118. doi: 10.1152/jappl.1985.59.1.113. [DOI] [PubMed] [Google Scholar]
- Yuan X-J, Tod ML, Rubin LJ, Blaustein MP. Contrasting effects of hypoxia on tension in rat pulmonary and mesenteric arteries. Am J Physiol. 1990;259:H281–H289. doi: 10.1152/ajpheart.1990.259.2.H281. [DOI] [PubMed] [Google Scholar]
- Madden JA, Vadula MS, Kurup VP. Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. . Am J Physiol. 1992;263:L384–L393. doi: 10.1152/ajplung.1992.263.3.L384. [DOI] [PubMed] [Google Scholar]
- Murray TR, Chen L, Marshall BE, Macarak EJ. Hypoxic contraction of cultured pulmonary vascular smooth muscle cells. Am J Respir Cell Mol Biol. 1990;3:457–465. doi: 10.1165/ajrcmb/3.5.457. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Yuan X-J. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res. 1995;77:370–378. doi: 10.1161/01.res.77.2.370. [DOI] [PubMed] [Google Scholar]
- McMurtry IF, Davidson AB, Reeves JT, Grover RF. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res. 1976;38:99–104. doi: 10.1161/01.res.38.2.99. [DOI] [PubMed] [Google Scholar]
- Gelband CH, Gelband H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. . Circulation. 1997;96:3647–3654. doi: 10.1161/01.cir.96.10.3647. [DOI] [PubMed] [Google Scholar]
- Salvaterra CG, Goldman WF. Acute hypoxia increases cytosolic calcium in cultured pulmonary arterial myocytes. Am J Physiol . 1993;264:L323–L328. doi: 10.1152/ajplung.1993.264.3.L323. [DOI] [PubMed] [Google Scholar]
- Evans AM, Osipenko ON, Gurney AM. Properties of a novel K+ current that is active at resting potential in rabbit pulmonary artery smooth muscle cells. J Physiol (Lond) 1996;496:407–420. doi: 10.1113/jphysiol.1996.sp021694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res. 1995;77:131–139. doi: 10.1161/01.res.77.1.131. [DOI] [PubMed] [Google Scholar]
- Albarwani S, Robertson BE, Nye PC, Kozlowski RZ. Biophysical properties of Ca2+- and Mg-ATP-activated K+channels in pulmonary arterial smooth muscle cells isolated from the rat. Pflugers Arch. 1994;428:446–454. doi: 10.1007/BF00374564. [DOI] [PubMed] [Google Scholar]
- Peng W, Hoidal JR, Farrukh IS. Role of a novel KCa opener in regulating K+ channels of hypoxic human pulmonary vascular cells. Am J Respir Cell Mol Biol. 1999;20:737–745. doi: 10.1165/ajrcmb.20.4.3390. [DOI] [PubMed] [Google Scholar]
- Yuan X-J, Wang J, Juhaszova M, Golovina VA, Rubin LJ. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am J Physiol. 1998;274:L621–L635. doi: 10.1152/ajplung.1998.274.4.L621. [DOI] [PubMed] [Google Scholar]
- Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. . Am J Physiol. 1992;262:C882–C990. doi: 10.1152/ajpcell.1992.262.4.C882. [DOI] [PubMed] [Google Scholar]
- Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res. 1993;73:1100–1112. doi: 10.1161/01.res.73.6.1100. [DOI] [PubMed] [Google Scholar]
- Osipenko ON, Evans AM, Gurney AM. Regulation of the resting potential of rabbit pulmonary artery myocytes by a low threshold,O2-sensing potassium current. Br J Pharmacol . 1997;120:1461–1470. doi: 10.1038/sj.bjp.0701075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X-J, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol. 1993;264:L116–L1123. doi: 10.1152/ajplung.1993.264.2.L116. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honore E. Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J. 1997;16:6615–6625. doi: 10.1093/emboj/16.22.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England SK, Uebele VN, Kodali J, Bennett PB, Tamkun MM. A novel K+ channel beta-subunit (hKvβ1.3) is produced via alternative mRNA splicing. J Biol Chem. 1995;270:28531–28534. doi: 10.1074/jbc.270.48.28531. [DOI] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- Conforti L, Bodi I, Nisbet JW, Millhorn DE. O2-sensitive K+ channels:role of the Kv1.2 -subunit in mediating the hypoxic response. . J Physiol (Lond) 2000;524:783–793. doi: 10.1111/j.1469-7793.2000.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Coppock EA, Felipe A, Martens JR, Tamkun MM. Oxygen sensitivity of cloned voltage-gated K+ channels expressed in the pulmonary vasculature. Circ Res. 1999;85:489–497. doi: 10.1161/01.res.85.6.489. [DOI] [PubMed] [Google Scholar]
- Wang J, Juhaszova M, Rubin LJ, Yuan X-J. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest. 1997;100:2347–2353. doi: 10.1172/JCI119774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest. 1998;101:2319–2330. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipenko ON, Tate RJ, Gurney AM. Potential role for Kv3.1b channels as oxygen sensors. Circ Res. 2000;86:534–540. doi: 10.1161/01.res.86.5.534. [DOI] [PubMed] [Google Scholar]
- Perez-Garcia MT, Lopez-Lopez JR, Gonzalez C. Kvβ1.2 subunit co-expression in HEK293 cells confers O2 sensitivity to Kv4.2 but not to Shaker channels. J Gen Physiol. 1999;113:897–907. doi: 10.1085/jgp.113.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Lu Y, Tang G, Wang R. Expression of voltage-dependent K+ channel genes in mesenteric artery smooth muscle cells. . Am J Physiol. 1999;277:G1055–G1063. doi: 10.1152/ajpgi.1999.277.5.G1055. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Montoro RJ, Smani T, Garcia-Hirschfeld J, Urena J. K+ and Ca2+ channel activity and cytosolic [Ca2+] in oxygen-sensing tissues. . Respir Physiol. 1999;115:215–227. doi: 10.1016/S0034-5687(99)00016-X. [DOI] [PubMed] [Google Scholar]
- Yuan X-J, Tod ML, Rubin LJ, Blaustein MP. Deoxyglucose and reduced glutathione mimic effects of hypoxia on K+ and Ca2+ conductances in pulmonary artery cells. Am J Physiol. 1994;267:L52–L63. doi: 10.1152/ajplung.1994.267.1.L52. [DOI] [PubMed] [Google Scholar]
- Jiang C, Haddad GG. A direct mechanism for sensing low oxygen levels by central neurons. Proc Natl Acad Sci USA. 1994;91:7198–7201. doi: 10.1073/pnas.91.15.7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AR, Henderson L, Jones TO, Delpiano MA, Hentschel J, Acker H. Involvement of an AND(P)H oxidase as a pO2 sensor protein in the rat carotid body. Biochem J. 1990;272:743–747. doi: 10.1042/bj2720743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MG, Vandier C, Bonnet P, Jackson WF, Rusch NJ. Intracellular acidosis differentially regulates KV channels in coronary and pulmonary vascular muscle. Am J Physiol . 1998;275:H1351–H1359. doi: 10.1152/ajpheart.1998.275.4.H1351. [DOI] [PubMed] [Google Scholar]
- McMurtry IF, Petrun MD, Reeves JT. Lungs from chronically hypoxic rats have decreased pressor response to acute hypoxia. Am J Physiol. 1978;235:H104–H109. doi: 10.1152/ajpheart.1978.235.1.H104. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Robertson TP, Ward JP, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol. 1994;266:H365–H370. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Sylvester JT, Sham JS. Chronic hypoxia alters effects of endothelin and angiotensin on K+ currents in pulmonary arterial myocytes. Am J Physiol. 1999;277:L431–L439. doi: 10.1152/ajplung.1999.277.3.L431. [DOI] [PubMed] [Google Scholar]
- Li KX, Fouty B, McMurtry IF, Rodman DM. Enhanced ETA-receptor-mediated inhibition of KV channels in hypoxic hypertensive rat pulmonary artery myocytes. Am J Physiol. 1999;277:H363–H370. doi: 10.1152/ajpheart.1999.277.1.H363. [DOI] [PubMed] [Google Scholar]
- Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- Dhulipala PD, Kotlikoff MI. Cloning and characterization of the promoters of the maxiK channel alpha and beta subunits. Biochim Biophys Acta. 1999;1444:254–262. doi: 10.1016/S0167-4781(98)00276-0. [DOI] [PubMed] [Google Scholar]
- Valverde P, Koren G. Purification and preliminary characterization of a cardiac Kv1.5 repressor element binding factor. . Circ Res. 1999;84:937–944. doi: 10.1161/01.res.84.8.937. [DOI] [PubMed] [Google Scholar]
- Levitan ES, Gealy R, Trimmer JS, Takimoto K. Membrane depolarization inhibits Kv1.5 voltage-gated K+ channel gene transcription and protein expression in pituitary cells. J Biol Chem. 1995;270:6036–6041. doi: 10.1074/jbc.270.11.6036. [DOI] [PubMed] [Google Scholar]
- Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. . Proc Natl Acad Sci USA. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X-J, Tod ML, Rubin LJ, Blaustein MP. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage-gated K+channels. Proc Natl Acad Sci USA. 1996;93:10489–10494. doi: 10.1073/pnas.93.19.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapp LH, Turcato S, Hall S, Baloch M. Evidence that Ca2+-activated K+ channels play a major role in mediating the vascular effects of iloprost and cicaprost. Eur J Pharmacol. 1998;356:215–224. doi: 10.1016/S0014-2999(98)00549-4. [DOI] [PubMed] [Google Scholar]
- DiCarlo VS, Chen SJ, Meng QC, Durand J, Yano M, Chen YF, Oparil S. ETA-receptor antagonist prevents and reverses chronic hypoxia-induced pulmonary hypertension in rat. Am J Physiol. 1995;269:L690–L697. doi: 10.1152/ajplung.1995.269.5.L690. [DOI] [PubMed] [Google Scholar]
- Archer SL, Huang JM, Reeve HL, Hampl V, Tolarova S, Michelakis E, Weir EK. Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ Res. 1996;78:431–442. doi: 10.1161/01.res.78.3.431. [DOI] [PubMed] [Google Scholar]
- Clement-Chomienne O, Ishii K, Walsh MP, Cole WC. Identification, cloning and expression of rabbit vascular smooth muscle Kv1.5 and comparison with native delayed rectifier K+ current. J Physiol (Lond) 1999;515:653–667. doi: 10.1111/j.1469-7793.1999.653ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]