

ABSTRACT
The p53 tumor suppressor plays a critical role in protecting normal cells from malignant transformation. Development of small molecules to reactivate p53 in cancer cells has been an area of intense research. We previously identified an internal ribosomal entry site (IRES) within the 5′ untranslated region of p53 mRNA that mediates translation of the p53 mRNA independent of cap-dependent translation. Our results also show that in response to DNA damage, cells switch from cap-dependent translation to cap-independent translation of p53 mRNA. In the present study, we discovered a specific inhibitor of cap-dependent translation, 4EGI-1, that is capable of inducing the accumulation of p53 in cancer cells retaining wild-type p53. Our results show that 4EGI-1 causes an increase in p53 IRES activity, leading to increased translation of p53 mRNA. We also observed that 4EGI-1 induces cancer cell apoptosis in a p53-dependent manner. Furthermore, 4EGI-1 induces p53 in cancer cells without causing DNA double-strand breaks. In conclusion, we discovered a mechanistic link between inhibition of cap-dependent translation and enhanced p53 accumulation. This leads to apoptosis of cancer cells without causing collateral damage to normal cells, thus providing a novel and effective therapeutic strategy for cancer.
KEYWORDS: 4EGI-1, p53, eIF4E, IRES
INTRODUCTION
Protein synthesis is the most energy-consuming process in the cell. It is heavily regulated by multiple protein translation factors that predominantly affect the initiation step (1). In cancer cells, protein translation is deregulated, allowing cells to continuously move through the cell cycle without stopping at the proper checkpoints. Because of this, multiple signaling pathways and proteins that regulate protein translation have been identified as potential therapeutic targets. One such target that has received great attention is eukaryotic initiation factor 4E (eIF4E).
mRNAs in eukaryotic organisms are typically translated in a cap-dependent manner because the majority of them contain a cap structure, m7GpppN (where N is any nucleotide), at their 5′ termini. The initial and rate limiting step of translation initiation is the recognition and the binding of this cap structure by eIF4E. eIF4E then recruits and binds to eIF4G and eIF4A to assemble the eIF4F translation initiation complex (2), which facilitates the cap-dependent translation of eukaryotic mRNAs.
It is known that the activity of eIF4E and cap-dependent translation is elevated in cancer cells and the overexpression and hyperactivation of eIF4E cause malignant transformation and metastasis in many different types of cancers (3). Multiple lines of evidence suggest that inhibition of eIF4E results in stalled tumor growth and repressed metastasis without substantially affecting normal cellular and organismal function (4, 5). Therefore, eIF4E has become a promising target for cancer therapy (6). Cap-dependent protein translation is inhibited by eIF4E binding proteins (4E-BPs), including 4E-BP1. When 4E-BP1 is hypophosphorylated, it competes with eIF4G for binding to eIF4E. Once 4E-BP1 is bound to eIF4E, it inhibits cap-dependent translation by preventing the formation of the eIF4F complex (2). Recent studies have shown that inhibiting eIF4E function in cancer cells leads to cell apoptosis (7–9); however, the underlying mechanism for induction of apoptosis through inhibition of cap-dependent translation is unclear.
During stressful conditions such as DNA damage, an alternative mode of translation, termed cap-independent translation, occurs by utilizing the internal ribosomal entry sites (IRES) in the 5′ untranslated regions (5′ UTRs) of mRNAs rather than eIF4E (10). We discovered an IRES sequence within the 5′ UTR of p53 mRNA (10, 11). We also found that p53 IRES activity increases following DNA damage (11). The p53 tumor suppressor plays a key role in response to DNA damage or other cellular stress by halting cell cycle progression or inducing apoptosis (12–14). Under normal conditions, p53 levels are low, since p53 is induced only after DNA damage or other cellular stress (11, 15). While it was thought that p53 induction is regulated through posttranslational modifications which inhibit MDM2 from binding it for degradation, leading to an increased half-life of p53, it is now clear that the p53 IRES-mediated translation of p53 mRNA also contributes to the accumulation of p53 following DNA damage and other stress signals (16–20).
While p53 is the most commonly mutated gene in cancer, p53 mutation rates are much lower in certain types of cancer, including prostate cancer (8 to 17%), breast cancer (20%), and the majority of pediatric cancers (1 to 19%) (21–23). Searching for small molecules that can reactivate p53 tumor suppressor in cancer cells that retain wild-type p53 has been an intensive area of research. In our previous study, we discovered a switch from cap-dependent translation to IRES-mediated translation of p53 mRNA following DNA damage (11), which is accompanied by an increase of 4E-BP1 bound to eIF4E. Others have made similar findings in which a transition from cap-dependent translation to IRES-mediated synthesis of the p53 protein occurs in response to not only DNA damage but also other cellular stress, such as oncogene-induced senescence (16). Based on these observations, we hypothesized that halting cap-dependent translation in cancer cells by inhibiting eIF4E function would mimic conditions of DNA damage and cause IRES-mediated translation of p53 mRNA, leading to apoptosis.
In the present study, we tested several newly discovered small molecule cap-dependent inhibitors that employ distinct mechanisms to inhibit cap-dependent protein translation initiation (Fig. 1A) and found that 4EGI-1 is able to induce the accumulation of p53 tumor suppressor in cancer cells that still express wild-type p53. Our results show that 4EGI-1 causes an increase in p53 IRES activity, leading to increased translation of p53 mRNA without inducing DNA double-strand breaks. We also found that 4EGI-1 inhibits cancer cell proliferation by inducing apoptosis in a p53-dependent manner.
FIG 1.
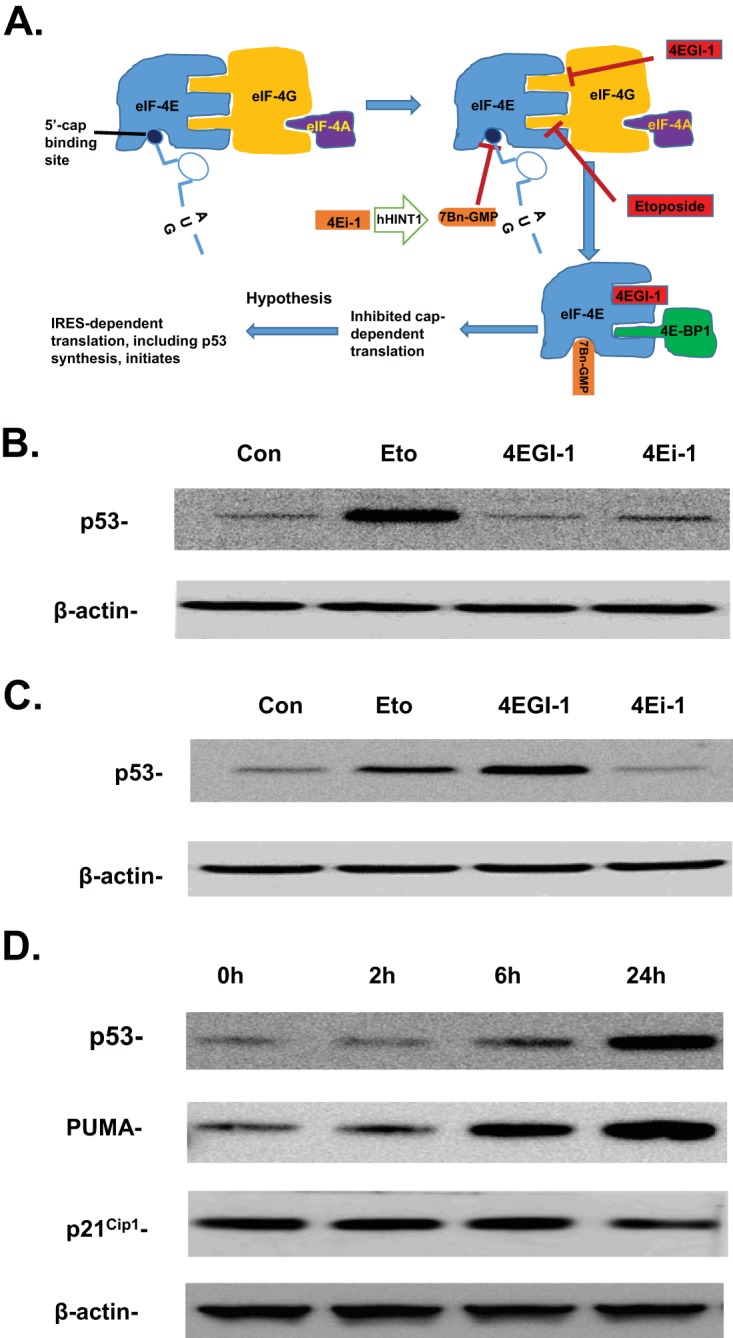
(A) A schematic diagram shows the distinct mechanisms used by multiple small molecule cap-dependent inhibitors to inhibit eIF4E function and cap-dependent translation initiation. (B and C) 4EGI-1 induces p53 in LNCaP cells. LNCaP cells were treated with 10 μM etoposide, 50 μM 4EGI-1, and 50 μM 4Ei-1 for 2 h (B) or 24 h (C). After treatment, cells were lysed. p53 and β-actin were detected following SDS-PAGE and Western blotting. (D) p53 induced by 4EGI-1 is accompanied by increased expression of PUMA. Subconfluent LNCaP cells were treated with 50 μM 4EGI-1 for the indicated time periods. After treatment, the cells were lysed. p53, PUMA, p21Cip1, and β-actin were detected following SDS-PAGE and Western blotting. The results in panels B to D are representative of three individual experiments.
RESULTS
To test our hypothesis, we examined several cap-dependent inhibitors in LNCaP cells. LNCaP is a prostate cancer cell line that contains wild-type p53 (24). 4EGI-1 is a small molecule that allosterically inhibits the interaction between eIF4E and eIF4G, preventing the formation of the eIF-4F complex without affecting 4E-BP1 binding (25). 4Ei-1, another small molecule translation inhibitor, is a prodrug that is converted to 7Bn-GMP by the enzyme hHINT1. 7Bn-GMP is able to compete with the 5′-mRNA cap for binding to eIF4E, preventing the recruitment of the 5′ mRNA cap structure to the eIF4F complex (26). While 4Ei-1 and 4EGI-1 are strong inhibitors of cap-dependent translation, both 4Ei-1 and 4EGI-1 actually stimulate cellular IRES activity (27, 28), which agrees with the hypothesis. Etoposide is a DNA-damaging agent which mimics IR and causes DNA double-strand breaks. Our previous results show that etoposide treatment inhibits cap-dependent translation and causes a transition from cap-dependent translation to IRES-mediated cap-independent translation of p53 mRNA (11).
We first treated LNCaP cells with 4EGI-1 and 4Ei-1 for 2 h with etoposide as the control. We observed that etoposide caused an increase in p53 accumulation (Fig. 1B). Next, we treated cells with these compounds at the same concentrations for 24 h. We observed an increase in p53 when cells were treated with 4EGI-1. Surprisingly, this increase was even higher than in cells treated with etoposide (Fig. 1C). We then performed a time course study and found that 4EGI-1 not only induced p53 accumulation but also caused increased expression of p53 downstream target PUMA (p53-upregulated modulator of apoptosis), which is known to be involved in p53-controlled apoptotic process (Fig. 1D). Interestingly, we did not observe a significant change of p21Cip1 expression, a regulator of cell cycle progression, under the same treatment conditions (Fig. 1D).
To further examine the effect of 4EGI-1 on cap-dependent translation, an eIF4E pulldown with m7-GTP-Sepharose beads was performed. Our results show that eIF4G bound to eIF4E decreased, but 4EGI-1 treatment also led to increased binding between 4E-BP1 and eIF4E (Fig. 2A). It was reported that 4EGI-1 not only inhibits the interaction between eIF4E and eIF4G but also causes enhanced 4E-BP1 binding to eIF4E in a dose-dependent manner (27). Our results further showed that 4EGI-1 leads to decreased binding between eIF4E and eIF4G and increased interaction between eIF4E and 4E-BP1 in a time-dependent manner (Fig. 2A), without significantly affecting the phosphorylation of 4E-BP1 (data not shown). This result agrees with a recent report which demonstrated that 4EGI-1 allosterically inhibits the binding of eIF4G to eIF4E (25).
FIG 2.
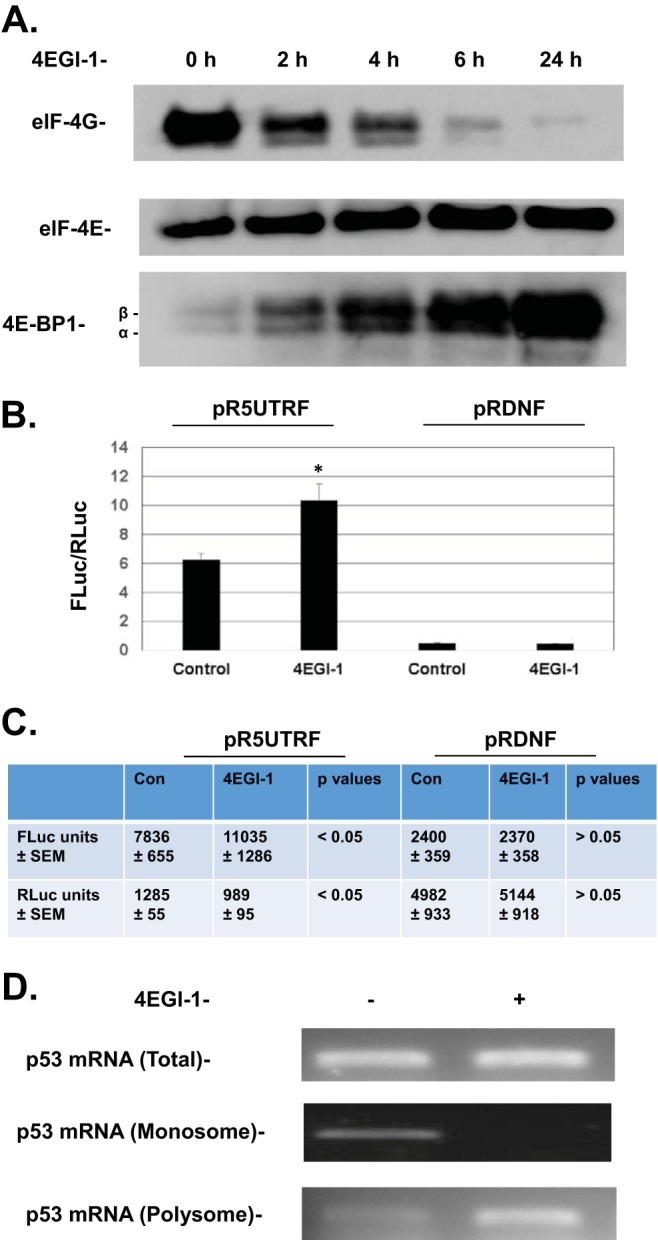
(A) 4EGI-1 treatment results in decreased association of eIF4G with eIF4E and increased interaction between 4E-BP1 and eIF4E. LNCaP cells were treated with 50 μM 4EGI-1 for the indicated time points. After treatment, the cells were lysed, and equal amounts of proteins were incubated with m7-GTP-Sepharose beads. After incubation, the beads were washed and mixed with SDS-PAGE loading buffer as described in Materials and Methods. The eluted proteins were then subjected to SDS-PAGE and transferred to a PVDF membrane. eIF4G, eIF4E, and 4E-BP1 were then detected. (B and C) 4EGI-1 significantly stimulates the activity (FLuc/RLuc) of p53 IRES. After transfection with pR5UTRF and pRDNF for 24 h, LNCaP cells were treated with 50 μM 4EGI-1 for another 24 h. The cells were then lysed, and a dual-luciferase assay was performed as described in Materials and Methods. The FLuc/RLuc ratios in panel B are averages ± the standard errors of the mean (SEM) from four repeats (*, P < 0.05 versus cells transfected with pR5UTRF but not treated with 4EGI-1). The corresponding average values of FLuc or RLuc ± the SEM in the presence or absence of 4EGI-1 along with P values between FLuc or RLuc units from 4EGI-1-treated or untreated cells are also shown in panel C. (D) p53 mRNA associates with polyribosomes in 4EGI-1-treated LNCaP cells. Cells were treated with 50 μM 4EGI-1 for 24 h and then lysed in a polysomal buffer. The fractionation of cytoplasmic polyribosomes and monoribosomes was performed as described in Materials and Methods. The RNAs in the polyribosomal fraction, monoribosomal fraction, and the cytoplasmic extracts were isolated and were subjected to reverse transcription and semiquantitative PCR for p53 mRNA as described in Materials and Methods.
To determine whether the p53 IRES activity increases during 4EGI-1 treatment when cap-dependent translation is halted, a bicistronic dual-luciferase reporter vector pR5UTRF (11), which contains the p53 5′ UTR sequence (located at nucleotide −131 before the first AUG of the p53 open reading frame [accession number NM_000546.4]), was used to determine p53 IRES activity. The vector pRDNF, which has an over 50% deletion of the p53 IRES sequence, was used as a control for the pR5UTRF vector (11). LNCaP cells were transfected with either pR5UTRF or pRDNF. p53 IRES activity was then measured as the ratio of firefly luciferase (Fluc; controlled by the p53 IRES) activity to Renilla luciferase (Rluc) activity (11). Rluc is controlled by eIF4E and cap-dependent protein translational machinery and was used as an internal control for Fluc. We found that in LNCaP cells transfected with pR5UTRF, the p53 IRES activity was significantly increased, as shown by an enhanced Fluc/Rluc ratio, following 4EGI-1 treatment (Fig. 2B). In contrast, the pRDNF has lost the majority of the p53 IRES activity, as shown by a dramatic decrease in the Fluc/Rluc ratio (similar to the results seen in reference 11), and the Fluc/Rluc ratio of pRDNF exhibited no significant change after the treatment with 4EGI-1 (Fig. 2B). Individual values of Fluc and Rluc of pR5UTRF (Fig. 2C) further showed that the enhanced p53 IRES activity of pR5UTRF is a combined result of both increased Fluc and decreased Rluc activities caused by 4EGI-1 treatment (Fig. 2C), indicating that 4EGI-1 indeed caused a transition from cap-dependent translation to IRES-mediated p53 translation of p53 mRNA.
To further confirm that p53 is translationally regulated by 4EGI-1, we examined whether the p53 mRNA is associated with polyribosomes following 4EGI-1 treatment. To do so, polyribosomal mRNA was isolated from cytoplasmic extracts of LNCaP cells treated with or without 4EGI-1. The purified polyribosomal RNA, monoribosomal RNA, and the total RNA in the cytosol were all subjected to reverse transcription-PCR (RT-PCR). Analysis of the PCR products (Fig. 2D) showed that the total p53 mRNA levels in the cytosol did not change when the cells were treated with or without 4EGI-1. However, 4EGI-1 treatment did lead to increased association between p53 mRNA and polyribosomes, along with decreased amount of p53 mRNA with monoribosomes (Fig. 2D). These results further demonstrate that the accumulation of p53 protein following 4EGI-1 treatment was accompanied by an increase in the translation of p53 mRNA.
We sought to further determine whether 4EGI-1 affects cell viability of LNCaP cells. We found that 4EGI-1 caused a decrease in cell viability in a concentration-dependent manner (Fig. 3A). Since p53 is a strong stimulator of cell apoptosis (29, 30), we examined the levels of poly-ADP-ribose polymerase (PARP), a substrate of caspase 3, in LNCaP cells. We found that at a concentration of 50 μM, 4EGI-1 caused an increase of cleaved PARP, indicating enhanced cellular apoptosis (Fig. 3B). This was also shown by a cell death enzyme-linked immunosorbent assay (ELISA) analysis, which indicates that 4EGI-1 caused enhanced fragmentation of DNA (Fig. 3C), another hallmark of apoptosis. The significant increase of apoptosis in LNCaP cells treated with 4EGI-1 was further confirmed by annexin V–7-aminoactinomycin D (7-AAD) assays, as shown by increased distribution of apoptotic cells in the total cell population (Fig. 3D and E).
FIG 3.

(A) 4EGI-1 inhibits the viability of LNCaP cells in a dose-dependent manner. Subconfluent cells were treated with 4EGI-1 at the indicated concentrations for 48 h. After treatment, an MTT assay was performed as described in Materials and Methods. The results are averages ± the SEM from three repeats. (B) 4EGI-1 induces PARP cleavage in LNCaP cells. Subconfluent cells were treated with the indicated concentrations of 4EGI-1 for 24 h. After treatment, both floating and attached cells were harvested and lysed. Equal amounts of protein were then subjected to SDS-PAGE and transferred to PVDF membranes. PARP, cleaved PARP, and β-actin were detected by immunoblotting. The results are representative of three repeats. (C) 4EGI-1 causes DNA fragmentation in LNCaP cells. Subconfluent cells were treated with 50 μM 4EGI-1 for 24 h. Both floating and attached cells were harvested and lysed after 4EGI-1 treatment. Fragmented DNA was measured by an ELISA cell death assay as described in Materials and Methods. The results are averages ± the SEM from three repeats (*, P < 0.05). (D and E) 4EGI-1 induces apoptosis in LNCaP cells: the cell apoptosis assay was performed using the Muse annexin V and dead cell assay kit (Millipore) according to the manufacturer's protocol. Briefly, LNCaP cells were grown to subconfluence and then treated with 50 μM 4EGI-1 for 24 h. After the treatment, both floating and attached cells were harvested and then labeled with Muse annexin V and dead cell reagent as described in Materials and Methods. After incubation, the tubes were read individually (as shown in panel D) using the Muse cell analyzer (Millipore). The averages of total apoptotic cells (early apoptotic and late apoptotic) ± the SEM from three individual experiments are presented in panel E (P < 0.05).
Since both 4EGI-1 and etoposide can induce p53 in cancer cells, we compared the capability of 4EGI-1 and etoposide in induction of apoptosis in LNCaP cells. Surprisingly, while 4EGI-1 readily induced apoptosis in LNCaP cells, etoposide did not cause increased apoptosis compared to control cells. Moreover, we found a much stronger increase in apoptosis when LNCaP cells were treated with both 4EGI-1 and etoposide versus cells treated with 4EGI-1 alone (Fig. 4A). This result suggests that 4EGI-1 can also sensitize LNCaP cells to etoposide, leading to enhanced apoptosis.
FIG 4.
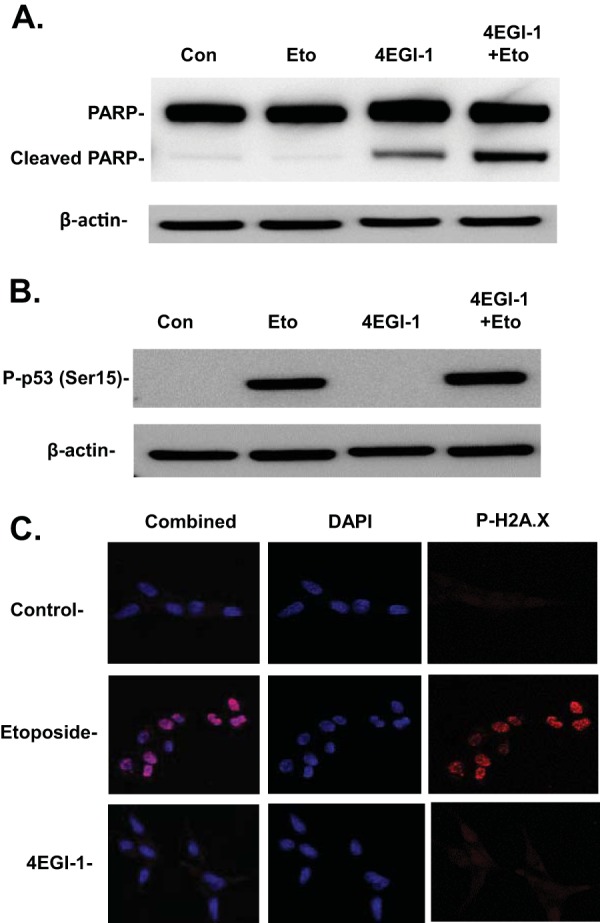
(A) Effect of 4EGI-1 and etoposide on cellular apoptosis in LNCaP cells. Subconfluent LNCaP cells were treated for 24 h with either 10 μM etoposide, 50 μM 4EGI-1, or both. Both floating and attached cells were collected and lysed following treatment. Equal amounts of protein were subjected to SDS-PAGE and transferred to a PVDF membrane. PARP, cleaved PARP, and β-actin were detected. (B) 4EGI-1 does not stimulate p53 phosphorylation at serine 15 in LNCaP cells. Subconfluent LNCaP cells were treated with either 10 μM etoposide, 50 μM 4EGI-1, or both for 4 h. After treatment, the cells were lysed. Phosphor-p53 at Ser15 and β-actin were detected following SDS-PAGE and Western blotting. (C) 4EGI-1 does not cause DNA double-strand breaks in LNCaP cells. Subconfluent LNCaP cells grown on chamber slides were treated with 10 μM etoposide or 50 μM 4EGI-1 for 4 h. After treatment, indirect immunofluorescence was performed with phosphor-histone H2A.X antibody as described in Materials and Methods, followed by DAPI staining. Images were taken using a confocal microscope at ×60 magnification.
p53 phosphorylation at serine 15 is an event that occurs following DNA damage, which leads to dissociation of MDM2 from p53 and subsequent increase of p53's half-life (10). In contrast to etoposide, 4EGI-1 does not appear to induce p53 phosphorylation at Ser15 (Fig. 4B), indicating that 4EGI-1 stimulates p53 in a manner different from MDM2-mediated p53 stabilization. To further determine whether 4EGI-1 induces p53 by causing DNA double-strand breaks, we looked at the focus formation of phosphorylated H2A.X, which recognizes DNA double-strand breaks. We treated LNCaP cells with 4EGI-1 using etoposide as the positive control (31). As seen in Fig. 4C, etoposide treatment induced the formation of a large amount of H2A.X foci within the nucleus of LNCaP cells, which were not seen in 4EGI-1-treated cells. These results indicate that 4EGI-1 is able to induce p53 without causing DNA double-strand breaks in the cells.
Next, we determined whether the apoptosis induced by 4EGI-1 is dependent on p53 by testing the effect of 4EGI-1 on isogenic human colon cancer cell lines HCT116-p53+/+ and HCT116-p53−/−. We again observed increased levels of p53 following treatment of HCT116-p53+/+ cells with 4EGI-1 (Fig. 5A). Similar to the results seen in LNCaP cells (Fig. 1D), we observed increased expression of PUMA in HCT116-p53+/+ cells treated with 4EGI-1, which was not seen in 4EGI-1-treated HCT116-p53−/− cells, whereas the levels of p21Cip1 exhibited no notable changes in both cell lines under the same experimental conditions (Fig. 5A). Our results also show that 4EGI-1 induced apoptosis in HCT116-p53+/+ cells at 24 h, whereas HCT116-p53−/− cells exhibited no significant increase in apoptosis at the same time span, as shown by both PARP cleavage (Fig. 5B) and annexin V/7-AAD assays (Fig. 5C). These results clearly demonstrated that 4EGI-1 induces apoptosis in cancer cells in a p53-dependent manner.
FIG 5.
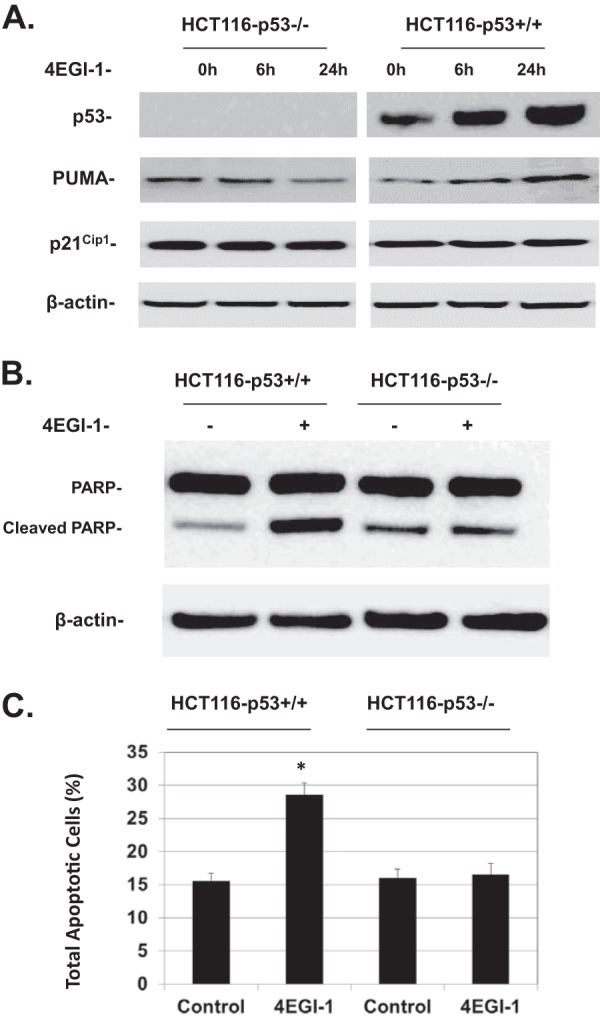
(A) 4EGI-1 induces p53 and PUMA but not p21Cip1 in HCT116-p53+/+ cells. HCT116-p53+/+ and HCT116-p53−/− cells were treated with 50 μM 4EGI-1 for the indicated time points. After treatment, the cells were lysed. p53, PUMA, p21Cip1, and β-actin were detected following SDS-PAGE and Western blotting. (B) 4EGI-1 induces PARP cleavage in HCT116-p53+/+ cells. Subconfluent HCT116-p53+/+ and HCT116-p53−/− cells were treated with 50 μM 4EGI-1 for 24 h. After treatment, both floating and attached cells were harvested and lysed. Equal amounts of protein were then subjected to SDS-PAGE and transferred to PVDF membranes. PARP, cleaved PARP, and β-actin were detected by immunoblotting. Results in panels A and B are representative of three repeats. (C) 4EGI-1 induces apoptosis in HCT116-p53+/+ cells. HCT116-p53+/+ and HCT116-p53−/− cells were grown to subconfluence and then treated with 50 μM 4EGI-1 for 24 h. Following the treatment, both floating and attached cells were harvested and then labeled with Muse annexin V and dead cell reagent as described in Materials and Methods. After incubation, the tubes were read using a Muse cell analyzer (Millipore). The averages of total apoptotic cells (early apoptotic and late apoptotic) ± the SEM from three individual experiments are presented (P < 0.05).
We also tested whether other cap-dependent inhibitors, either alone or in combination with 4EGI-1, can induce p53. Resveratrol was initially considered a regulator of sirtuins that regulate glucose homeostasis in the body. However, recent results have indicated that it is also an inhibitor of Akt and mTOR, thereby suppressing phosphorylation of 4E-BP1 (32, 33). Resveratrol can also induce apoptosis in cancer cells, but the underlying mechanism is still unclear (34). We also observed that resveratrol caused enhanced apoptosis in LNCaP cells (Fig. 6A and B). Our results further indicate that resveratrol could induce p53 accumulation in LNCaP cells (Fig. 6C). More interestingly, when LNCaP cells were treated with both 4EGI-1 and resveratrol, we found a much stronger induction of p53 compared to cells treated with 4EGI-1 or resveratrol alone (Fig. 6C). While either 4EGI-1 or resveratrol could induce apoptosis, cleavage of PARP was increased dramatically when cells were treated with both 4EGI-1 and resveratrol (Fig. 6D). These results suggest a synergistic effect of 4EGI-1 and resveratrol on induction of p53 and apoptosis. Furthermore, we found that resveratrol does not induce DNA double-strand breaks, as evidenced by the lack of focus formation of phosphorylated H2A.X (data not shown). These results suggest that strong inhibition of cap-dependent translation might be a general mechanism for induction of p53 and apoptosis.
FIG 6.
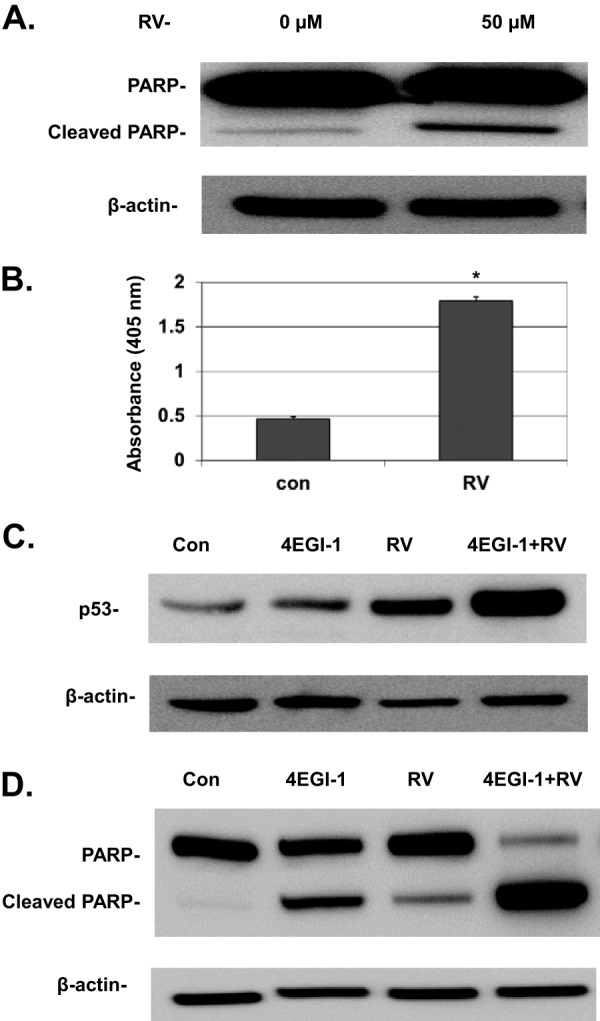
(A) Resveratrol (RV) induces PARP cleavage in LNCaP cells. Subconfluent cells were treated with 50 μM resveratrol for 24 h. After treatment, both floating and attached cells were harvested and lysed. Equal amounts of protein were then subjected to SDS-PAGE and transferred to PVDF membranes. PARP, cleaved PARP, and β-actin were detected. (B) Resveratrol causes DNA fragmentation in LNCaP cells. Subconfluent cells were treated with 50 μM resveratrol for 24 h, and then fragmented DNA was measured by an ELISA cell death assay as described in Materials and Methods. The results are averages ± the SEM from three repeats (*, P < 0.05). (C) 4EGI-1 and resveratrol treatment leads to a much stronger induction of p53 than 4EGI-1 or resveratrol alone. Subconfluent LNCaP cells were treated with either 50 μM 4EGI-1, 50 μM resveratrol, or both for 4 h. After treatment, the cells were lysed. p53 and β-actin were detected following SDS-PAGE and Western blotting. (D) 4EGI-1 and resveratrol work synergistically to induce apoptosis in LNCaP cells. Subconfluent cells were treated for 24 h with either 50 μM 4EGI-1, 50 μM resveratrol, or both. Both floating and attached cells were collected and lysed. Equal amounts of protein were subjected to SDS-PAGE and transferred to a PVDF membrane. PARP, cleaved PARP, and β-actin were detected. The results in panels A to D are representative of three individual repeats.
DISCUSSION
The majority of current studies aimed at reactivating p53 in cancer cells retaining wild-type p53 have focused on inhibiting the interaction between MDM2 and p53 to increase the half-life of p53 (23, 35). To date, these efforts have met with only limited success (35, 36). This is possibly due to the low frequencies of the MDM2 amplification in the majority of various types of cancer cells (37, 38). In contrast, our results provide a novel approach for stimulating p53 synthesis in cancer cells that harbor a wild-type p53 gene. We found that 4EGI-1 causes increased synthesis of p53, which in turn leads to the induction of apoptosis in cancer cells that express wild-type p53. We have also shown that the induction of p53 is accompanied by enhanced p53 IRES activity. Taking our results together, we have discovered a novel mechanism to induce p53 in cancer cells by inhibiting cap-dependent translation.
Cancer cells are addicted to growth factors and oncogenes such as cyclin D1 and c-myc. The mRNAs of these proteins contain “weak” 5′ untranslated regions (UTRs), which are long, complex structures that are not translated as efficiently as “strong” 5′ UTRs, which are relatively shorter, simpler structures (2). Because overexpression of eIF4E disproportionately upregulates oncogenes with complex 5′ UTRs relative to the less complex structures of housekeeping mRNAs, it is not surprising that cap-dependent inhibitors can specifically suppress the translation of multiple oncogenes and growth factors. This is supported by recent observations that eIF4E-haploinsufficient mice are physiologically normal yet resistant to tumor formation (39), suggesting that eIF4E dose is actually not a limiting factor for normal protein synthesis and cellular homeostasis, and high levels of eIF4E are critical for cancer development. It has also been shown that 4EGI-1 inhibits the proliferation of transformed cells to a much greater extent relative to nontransformed cells (27).
However, there is still no evidence to support the claim that inhibition of the translation of oncogenes and growth factors provides a mechanism leading to apoptosis. As stated earlier, we and others discovered a switch from cap-dependent translation to IRES-mediated translation of p53 mRNA when cells are exposed to DNA damage (11, 40). In addition, oncogene-induced senescence (OIS) is a rapid cellular response that permanently shuts down further proliferation to prevent malignant transformation. It was also shown that cap-dependent translation switches to IRES-mediated cap-independent translation of p53 mRNA during OIS (16). The p53 IRES exhibits enhanced activity following OIS, which leads to an increase in p53 accumulation as cap-dependent translation stops (16). In the present study, this switch has been further demonstrated in cancer cells treated with 4EGI-1, which induces IRES-mediated p53 synthesis through inhibition of cap-dependent translation. This also provides an explanation as to why 4EGI-1 can induce apoptosis while inhibiting cap-dependent translation.
DNA-damaging agents, such as etoposide, are commonly used in cancer chemotherapy. However, these agents also cause serious collateral damage to benign cells, leading to increased risks of future cancer development in treated individuals (38). While the majority of traditional chemotherapeutic reagents trigger a p53 response by inducing DNA damage (35), 4EGI-1 plays a similar role in induction of p53 without causing DNA double-strand breaks. Therefore, it is conceivable that 4EGI-1 replaces some of the DNA-damaging agents, thus preventing toxic side effects caused by traditional chemotherapy. In addition, one of the main reasons behind the resistance of cancer cells to DNA damage is because some of these compounds tend to cause cell cycle arrest rather than apoptosis, and the arrested cancer cells may later resume proliferation after completion of the chemotherapy (41). We have tested whether 4EGI-1 can induce cell cycle arrest and senescence in LNCaP cells and did not observe such effects (data not shown). These results are consistent with our observations that 4EGI-1 treatment does not lead to increased expression of p21Cip1 in both LNCaP and HCT116-p53+/+ cells. Therefore, the strong capability of 4EGI-1 in inducing apoptosis, along with its ability to sensitize cancer cells to etoposide, not only makes 4EGI-1 a potent therapeutic agent for cancer treatment but may also allow it to be combined with DNA-damaging agents, thereby decreasing cancer cells' drug resistance to traditional chemotherapy or radiation therapy.
More importantly, we also found that other cap-dependent inhibitors, such as resveratrol, not only can induce p53 and apoptosis by itself but also have a synergistic effect on the induction of p53 and apoptosis when combined with 4EGI-1. This may lead to the expansion of the usage of resveratrol not only as a preventive natural supplement but also as a chemotherapeutic agent for cancer. However, we did not observe a positive effect on p53 induction when treating LNCaP cells with 4Ei-1, despite its ability to stimulate cellular IRES activity (27, 28). Since the cap-dependent inhibition of 4Ei-1 is due to the competition between its intracellular metabolite (7-Bn-GMP) and the cap structure of eukaryotic mRNAs for binding to eIF4E, the affinity of 7-Bn-GMP to eIF4E is crucial for its efficacy against cap-dependent translation (2). While 4Ei-1 can sensitize cancer cells to gemcitabine or other chemotherapeutic agents, 4Ei-1 itself has limited cytotoxicity to cancer cells, possibly because of the relatively low binding affinity of 7-Bn-GMP for eIF4E (26). We are currently testing the efficacy of other analogs of 4Ei-1, such as 4Ei-10, whose metabolite in the cell has much stronger affinity for eIF4E, and have received promising results (D.-Q. Yang and C. R. Wagner, unpublished observations).
To determine whether our findings are applicable to other types of cancer, we also tested the effects of 4EGI-1 on multiple other cancer cell lines that still retain wild-type p53. These include a breast cancer cell line MCF-7 and multiple pediatric cancer cell lines, including SH-SY5Y (neuroblastoma) and CHLA-200 (pediatric glioma), and observed induction of p53 and apoptosis by 4EGI-1 in essentially all the cell lines tested (data not shown). Previous results have also shown that 4EGI-1 has strong cytotoxicity to multiple myeloma cell lines containing wild-type p53 gene (42). Interestingly, it was found that both Noxa and PUMA were increased when these cell lines were treated with 4EGI-1 (42). Another study reported that 4EGI-1 induces Noxa in a chronic lymphocytic leukemia cell line (43). While p53 induces apoptosis through upregulating the transcription of multiple downstream targets including PUMA, Noxa, and Bax (44–46), it was not clear at the time why 4EGI-1 could induce Noxa or PUMA since all previous results pointed to its ability to inhibit protein synthesis (42, 43). In the present study, we provide clear evidence that 4EGI-1 induces both p53 and its downstream target PUMA in cancer cells. Thus, our findings are the first to provide a novel mechanism, as well as an explanation for these previous observations, by linking the upregulation of Noxa and PUMA with the induction of p53 in cell lines treated with 4EGI-1.
In addition to binding canonical initiation factors such as eIF4G, the IRES structure serves as an anchoring point for auxiliary protein initiation factors known as IRES trans-acting factors (ITAFs) (47). These ITAFs modulate activity of the IRES, either positively or negatively (47, 48). Multiple ITAFs or putative ITAFs of the p53 IRES have been identified and have been shown to either stimulate (positive ITAFs) or inhibit (negative ITAFs) the synthesis of p53 (49). Furthermore, several proteins that can bind to the 3′ UTR of p53 mRNA and modulate its synthesis were also identified (50–52). Recently, we discovered that altered expression of p53 ITAFs is linked to defective p53 induction following DNA damage in cancer cells (20). It is thus conceivable that 4EGI-1 reactivates p53 in cancer cell lines with wild-type p53 by modulating the expression levels of p53 ITAFs or regulatory proteins bound to p53 3′ UTR, which results in enhanced p53 IRES activity and increased p53 synthesis (49).
In summary, the findings of the present work provide a mechanistic link between inhibition of cap-dependent protein translation with enhanced p53 accumulation and increased cellular apoptosis in cancer cells. The ability of these cap-dependent inhibitors to induce apoptosis through the upregulation of p53 and to specifically inhibit translation of oncogenic proteins makes these inhibitors a double-edged sword against cancer. While the detailed mechanism underlying 4EGI-1's role in inducing p53 synthesis requires further investigation, our results have introduced a new paradigm that links the inhibition of cap-dependent translation with p53 induction and may lead to the development of new treatment strategies and new therapeutic agents against cancer.
MATERIALS AND METHODS
Chemicals and reagents.
Etoposide and 4EGI-1 were purchased from Millipore. The antibody against human p53 was purchased from Santa Cruz. The antibody against phosphor-H2A.X was purchased from Millipore. The antibodies against PARP, eIF4E, eIF4G, phosphor-p53 at Ser15 and p21Cip1 were from Cell Signaling Technologies. The antibodies against 4E-BP1 and p53-upregulated modulator of apoptosis (PUMA) were from Bethyl Laboratories. 4Ei-1 was synthesized by Carston Wagner's laboratory at the University of Minnesota.
Cell culture, cell extract preparation, SDS-PAGE, and Western blotting.
LNCaP cells were purchased from ATCC. Colon cancer cell lines HCT116-p53+/+ and HCT116-p53−/− were generous gifts from Bert Vogelstein. Cancer cells were cultured in RPMI 1640 (LNCaP) or McCoy's 5A (HCT116-p53+/+ and HCT116-p53−/−) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (Millipore) containing protease inhibitor cocktails (Roche). The protein concentration was measured by the Lowry method. Equal amounts of protein were subjected to SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane. The proteins of interest were then probed with their respective antibodies.
MTT cell viability assay.
Cells were plated on a 24-well plate at 20,000 cells/well. After treatment with different reagents, the ratio of viable cells in each well was determined using a CellTiter cell proliferation kit (Promega) according to the manufacturer's instructions. MTS solution was placed in each well at a ratio of 20 μl/100 μl of cell culture medium, followed by incubation in a cell culture incubator for 3 h. Absorbance was then measured at a wavelength of 492 nm with a microplate reader.
Dual-luciferase assays.
Cells were lysed with passive lysis buffer (Promega). The dual-luciferase reporter assay (Promega) was then performed according to the manufacturer's instructions. Firefly and Renilla luciferase activities were determined with a Synergy 2 (BioTek) microplate reader.
eIF4E pulldown with M7-GTP-Sepharose beads.
Cell lysates containing equal amounts of protein were incubated with M7-GTP-Sepharose beads (GE Healthcare) overnight at 4°C with constant gentle agitation. After incubation, the mixture was centrifuged, and the beads were washed once with RIPA buffer and twice with cold phosphate-buffered saline. After washing, SDS sample buffer was added to the beads. Samples were boiled and then subjected to SDS-PAGE and immunoblotting.
Indirect immunofluorescence.
Chamber slides (eight wells; Lab-Tek) were treated with poly-l-lysine (Sigma) prior to seeding with LNCaP cells at 8,000 cells/well. Subconfluent cells were treated with different reagents. After treatment, cells were briefly fixed in 3% paraformaldehyde solution and then permeabilized with 0.2% Triton X-100 in Tris-buffered saline. After permeabilization, the cells were incubated with a phosphor-histone H2A.X antibody (Millipore) and then with a Texas Red-conjugated secondary antibody (Jackson Laboratories), followed by DAPI (4′,6′-diamidino-2-phenylindole) staining. Images were recorded using a Nikon Eclipse TE2000-E confocal microscope at ×60 magnification.
Cytoplasmic polysome fractionation.
Fractionation of cytoplasmic polysomes was performed following a method modified from previous reports (11, 53). LNCaP cells were grown to 80 to 85% confluence and then treated with or without 50 μM 4EGI-1. After treatment, cells (∼1.5 × 106 cells/treatment) were lysed using 400 μl of polysomal buffer (10 mM morpholinepropanesulfonic acid, pH 7.2; 250 mM NaCl; 2.5 mM magnesium acetate; 0.5% NP-40; 1 mM phenylmethylsulfonyl fluoride; 200 mg/ml heparin; 50 mg/ml cycloheximide) for 10 min (11, 53). Cellular debris and nuclei were removed from cell lysates by high-speed centrifugation at 17,000 × g for 20 min. A portion (25%, 100 μl) of the supernatant was saved for isolation of total cytoplasmic RNA. The polyribosomes in the rest of the supernatant (300 μl) were precipitated by ultracentrifugation at 115,000 × g for 60 min in a 90 Ti rotor (Beckman) at 4°C. The pellet from the ultracentrifugation was resuspended in 200 μl of polysomal buffer and used as the polyribosomal fraction. The supernatant (300 μl) was used as the monosomal fraction.
RNA isolation and RT-PCR.
The RNAs from the polysomal fraction (200 μl), the monosomal fraction (300 μl), and the total cytoplasmic extracts (100 μl) were isolated using TRI Reagent LS (Molecular Research Center, Inc.). The recovered RNAs were further purified by an RNeasy minikit (Qiagen) and treated with RNase-free DNase. Equal amounts of RNA from each fraction were then reverse transcribed by using a ProtoScript II first-strand cDNA synthesis kit (New England BioLabs) and oligo(dT) primers. The obtained cDNAs were then used as the templates for semiquantitative PCR for detecting p53 mRNA. The PCR products were resolved in a 1.0% agarose gel stained with ethidium bromide. The midpoint for linearity of the amplification of p53 mRNA for each fraction was determined individually (total cellular extract, 25 cycles; polysomal fraction, 28 cycles; monosomal fraction, 30 cycles).
Cell apoptosis ELISA.
Cells were plated on a 24-well plate at 200,000 cells/well, and subconfluent cells were treated with reagents. The generation of mononucleosomes and oligonucleosomes during apoptosis was detected with a cell death detection ELISA kit (Roche) according to the manufacturer's instructions. Both floating and attached cells were collected, and cells were lysed using the lysis buffer provided in the kit. The volume of buffer added was normalized to the amount of cells in each sample. After incubation, the cell lysate was collected, and the ELISA was performed. Absorbance was measured at the wavelength of 405 nm with a microplate reader.
Annexin V/7-AAD apoptosis assay.
The cytotoxic effects of 4EGI-1 on cancer cells were analyzed with the annexin V/7-AAD assays using the Muse annexin V and a dead cell assay kit from Millipore. The assay was performed according to the manufacturer's protocol. Cancer cells were grown to subconfluent and then treated with 4EGI-1. After the treatment, both attached and floating cells were precipitated and then resuspended in 100 μl of cell culture media with 1% FBS, followed by the addition of 100 μl of Muse annexin V and dead cell reagent to each tube. The tubes were mixed thoroughly, followed by incubation at room temperature for 20 min without light exposure. After incubation, the tubes were read individually using a Muse cell analyzer (Millipore). The same gate settings were used for all samples to distinguish the cells in the live, early-apoptotic, late-apoptotic, and dead categories.
ACKNOWLEDGMENTS
We thank Peter Bitterman and Douglas Yee (University of Minnesota) for helpful comments on the results presented in the manuscript.
Funding for this research was provided by two research grants (1RO3 CA177954 and 1R01 CA157012) from the National Cancer Institute of the National Institutes of Health, an institutional research grant (118198-IRG-58-001-52-IRG99) from the American Cancer Society, a pilot grant from the Prostate Cancer Translational Working group of UMN, and the Hormel Foundation.
REFERENCES
- 1.Holcik M, Sonenberg N. 2005. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 2.Jia Y, Polunovsky V, Bitterman PB, Wagner CR. 2012. Cap-dependent translation initiation factor eIF4E: an emerging anticancer drug target. Med Res Rev 32:786–814. doi: 10.1002/med.21260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Benedetti A, Graff JR. 2004. eIF-4E expression and its role in malignancies and metastases. Oncogene 23:3189–3199. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 4.Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, Schwier P, Capen A, Goode RL, Dowless MS, Chen Y, Zhang H, Sissons S, Cox K, McNulty AM, Parsons SH, Wang T, Sams L, Geeganage S, Douglass LE, Neubauer BL, Dean NM, Blanchard K, Shou J, Stancato LF, Carter JH, Marcusson EG. 2007. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest 117:2638–2648. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE, McCann DJ, Pelletier J, Furic L, Yingling JM, Stancato LF, Sonenberg N, Graff JR. 2011. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res 71:1849–1857. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- 6.Pelletier J, Graff J, Ruggero D, Sonenberg N. 2015. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res 75:250–263. doi: 10.1158/0008-5472.CAN-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Yang D-Q. 2010. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol Cancer Ther 9:113–125. doi: 10.1158/1535-7163.MCT-08-1189. [DOI] [PubMed] [Google Scholar]
- 8.Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, Manivel JC, Sonenberg N, Yee D, Bitterman PB, Polunovsky VA. 2004. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 5:553–563. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, Supko JG, Chorev M, Wagner G, Halperin JA. 2012. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 3:869–881. doi: 10.18632/oncotarget.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halaby MJ, Yang D-Q. 2007. p53 translational control: a new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 395:1–7. doi: 10.1016/j.gene.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 11.Yang DQ, Halaby MJ, Zhang Y. 2006. The identification of an internal ribosomal entry site in the 5′ untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 25:4613–4619. doi: 10.1038/sj.onc.1209483. [DOI] [PubMed] [Google Scholar]
- 12.Kastenhuber ER, Lowe SW. 2017. Putting p53 in context. Cell 170:1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 14.Vogelstein B, Lane D, Levine AJ. 2000. Surfing the p53 network. Nature 408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 15.Mosner J, Mummenbrauer T, Bauer C, Sczakiel G, Grosse F, Deppert W. 1995. Negative feedback regulation of wild-type p53 biosynthesis. EMBO J 14:4442–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellodi C, Kopmar N, Ruggero D. 2010. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J 29:1865–1876. doi: 10.1038/emboj.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim DY, Kim W, Lee KH, Kim SH, Lee HR, Kim HJ, Jung Y, Choi JH, Kim KT. 2013. hnRNP Q regulates translation of p53 in normal and stress conditions. Cell Death Differ 20:226–234. doi: 10.1038/cdd.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan D, Katoch A, Das A, Sharathchandra A, Lal R, Roy P, Das S, Chattopadhyay S, Das S. 2015. Reversible induction of translational isoforms of p53 in glucose deprivation. Cell Death Differ 22:1203–1218. doi: 10.1038/cdd.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halaby MJ, Li Y, Harris BR, Jiang S, Miskimins WK, Cleary MP, Yang DQ. 2015. Translational control protein 80 stimulates IRES-mediated translation of p53 mRNA in response to DNA damage. Biomed Res Int 2015:708158. doi: 10.1155/2015/708158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halaby MJ, Harris B, Miskimins WK, Cleary MP, Yang D-Q. 2015. Deregulation of IRES-mediated p53 translation in cancer cells with defective p53 response to DNA damage. Mol Cell Biol 35:4006–4017. doi: 10.1128/MCB.00365-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gasco M, Shami S, Crook T. 2002. The p53 pathway in breast cancer. Breast Cancer Res 4:70–76. doi: 10.1186/bcr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacGrogan D, Bookstein R. 1997. Tumour suppressor genes in prostate cancer. Semin Cancer Biol 8:11–19. doi: 10.1006/scbi.1997.0048. [DOI] [PubMed] [Google Scholar]
- 23.Van Maerken T, Rihani A, Van Goethem A, De Paepe A, Speleman F, Vandesompele J. 2013. Pharmacologic activation of wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett 344:157–165. doi: 10.1016/j.canlet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 24.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS. 2003. Molecular characterization of human prostate carcinoma cell lines. Prostate 57:205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 25.Papadopoulos E, Jenni S, Kabha E, Takrouri KJ, Yi T, Salvi N, Luna RE, Gavathiotis E, Mahalingam P, Arthanari H, Rodriguez-Mias R, Yefidoff-Freedman R, Aktas BH, Chorev M, Halperin JA, Wagner G. 2014. Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proc Natl Acad Sci U S A 111:E3187–E3195. doi: 10.1073/pnas.1410250111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S, Jia Y, Jacobson B, McCauley J, Kratzke R, Bitterman PB, Wagner CR. 2013. Treatment of breast and lung cancer cells with a N-7 benzyl guanosine monophosphate tryptamine phosphoramidate pronucleotide (4Ei-1) results in chemosensitization to gemcitabine and induced eIF4E proteasomal degradation. Mol Pharm 10:523–531. doi: 10.1021/mp300699d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G. 2007. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh B, Benyumov AO, Ghosh P, Jia Y, Avdulov S, Dahlberg PS, Peterson M, Smith K, Polunovsky VA, Bitterman PB, Wagner CR. 2009. Nontoxic chemical interdiction of the epithelial-to-mesenchymal transition by targeting cap-dependent translation. ACS Chem Biol 4:367–377. doi: 10.1021/cb9000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber JD, Zambetti GP. 2003. Renewing the debate over the p53 apoptotic response. Cell Death Differ 10:409–412. doi: 10.1038/sj.cdd.4401226. [DOI] [PubMed] [Google Scholar]
- 30.Horn HF, Vousden KH. 2007. Coping with stress: multiple ways to activate p53. Oncogene 26:1306–1316. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Xiong H, Yang D-Q. 2013. Functional switching of ATM: sensor of DNA damage in proliferating cells and mediator of Akt survival signal in post-mitotic human neuron-like cells. Chin J Cancer 31:364–372. doi: 10.5732/cjc.012.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ge J, Liu Y, Li Q, Guo X, Gu L, Ma ZG, Zhu YP. 2013. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomed Environ Sci 26:902–911. doi: 10.3967/bes2013.019. [DOI] [PubMed] [Google Scholar]
- 33.Lin JN, Lin VC, Rau KM, Shieh PC, Kuo DH, Shieh JC, Chen WJ, Tsai SC, Way TD. 2010. Resveratrol modulates tumor cell proliferation and protein translation via SIRT1-dependent AMPK activation. J Agric Food Chem 58:1584–1592. doi: 10.1021/jf9035782. [DOI] [PubMed] [Google Scholar]
- 34.Lin HY, Tang HY, Davis FB, Davis PJ. 2011. Resveratrol and apoptosis. Ann N Y Acad Sci 1215:79–88. doi: 10.1111/j.1749-6632.2010.05846.x. [DOI] [PubMed] [Google Scholar]
- 35.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. 2009. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 36.Stegh AH. 2012. Targeting the p53 signaling pathway in cancer therapy: the promises, challenges and perils. Expert Opin Ther Targets 16:67–83. doi: 10.1517/14728222.2011.643299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saiki AY, Caenepeel S, Cosgrove E, Su C, Boedigheimer M, Oliner JD. 2015. Identifying the determinants of response to MDM2 inhibition. Oncotarget 6:7701–7712. doi: 10.18632/oncotarget.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor AC, Shu L, Danks MK, Poquette CA, Shetty S, Thayer MJ, Houghton PJ, Harris LC. 2000. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol 35:96–103. doi:. [DOI] [PubMed] [Google Scholar]
- 39.Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, Ruggero D. 2015. Differential requirements for eIF4E dose in normal development and cancer. Cell 162:59–71. doi: 10.1016/j.cell.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellodi C, Krasnykh O, Haynes N, Theodoropoulou M, Peng G, Montanaro L, Ruggero D. 2010. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res 70:6026–6035. doi: 10.1158/0008-5472.CAN-09-4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitt E, Paquet C, Beauchemin M, Bertrand R. 2007. DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis. J Zhejiang Univ Sci B 8:377–397. doi: 10.1631/jzus.2007.B0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Descamps G, Gomez-Bougie P, Tamburini J, Green A, Bouscary D, Maiga S, Moreau P, Le Gouill S, Pellat-Deceunynck C, Amiot M. 2012. The cap-translation inhibitor 4EGI-1 induces apoptosis in multiple myeloma through Noxa induction. Br J Cancer 106:1660–1667. doi: 10.1038/bjc.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willimott S, Beck D, Ahearne MJ, Adams VC, Wagner SD. 2013. Cap-translation inhibitor, 4EGI-1, restores sensitivity to ABT-737 apoptosis through cap-dependent and -independent mechanisms in chronic lymphocytic leukemia. Clin Cancer Res 19:3212–3223. doi: 10.1158/1078-0432.CCR-12-2185. [DOI] [PubMed] [Google Scholar]
- 44.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321–328. doi: 10.1016/S1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 45.Yu J, Zhang L. 2005. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun 331:851–858. doi: 10.1016/j.bbrc.2005.03.189. [DOI] [PubMed] [Google Scholar]
- 46.Garrison SP, Phillips DC, Jeffers JR, Chipuk JE, Parsons MJ, Rehg JE, Opferman JT, Green DR, Zambetti GP. 2012. Genetically defining the mechanism of Puma- and Bim-induced apoptosis. Cell Death Differ 19:642–649. doi: 10.1038/cdd.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hellen CU, Sarnow P. 2001. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 48.Stoneley M, Willis AE. 2004. Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene 23:3200–3207. doi: 10.1038/sj.onc.1207551. [DOI] [PubMed] [Google Scholar]
- 49.Ji B, Harris BR, Liu Y, Deng Y, Gradilone SA, Cleary MP, Liu J, Yang DQ. 2017. Targeting IRES-mediated p53 synthesis for cancer diagnosis and therapeutics. Int J Mol Sci 18:E93. doi: 10.3390/ijms18010093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M. 2003. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci U S A 100:8354–8359. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galban S, Martindale JL, Mazan-Mamczarz K, Lopez De Silanes I, Fan J, Wang W, Decker J, Gorospe M. 2003. Influence of the RNA-binding protein HuR in pVHL-regulated p53 expression in renal carcinoma cells. Mol Cell Biol 23:7083–7095. doi: 10.1128/MCB.23.20.7083-7095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schumacher B, Hanazawa M, Lee MH, Nayak S, Volkmann K, Hofmann R, Hengartner M, Schedl T, Gartner A. 2005. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 120:357–368. doi: 10.1016/j.cell.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S. 2003. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol Cell 11:113–126. doi: 10.1016/S1097-2765(03)00012-1. [DOI] [PubMed] [Google Scholar]