

Abstract
The cellular and molecular mechanisms that are involved in airway hyper-responsiveness are unclear. Current studies suggest that tumor necrosis factor (TNF)-α, a cytokine that is produced in considerable quantities in asthmatic airways, may potentially be involved in the development of bronchial hyper-responsiveness by directly altering the contractile properties of the airway smooth muscle (ASM). The underlying mechanisms are not known, but growing evidence now suggests that most of the biologic effects of TNF-α on ASM are mediated by the p55 receptor or tumor necrosis factor receptor (TNFR)1. In addition, activation of TNFR1 coupled to the tumor necrosis factor receptor-associated factor (TRAF)2-nuclear factor-κB (NF-κB) pathway alters calcium homeostasis in ASM, which appears to be a new potential mechanism underlying ASM hyper-responsiveness.
Keywords: airway hyper-responsiveness, airway remodeling, airway smooth muscle, tumor necrosis factor-α, tumor necrosis factor receptor 1, thapsigargin
Introduction
Asthma, which is characterized by airway inflammation, exaggerated airway reactivity to contractile agonists, and a decrease in β-adrenoceptor-mediated airway relaxation, remains a common cause of pulmonary morbidity and mortality. Although the mechanisms that underlie changes in airway responsiveness are unknown, recent reports support the notion that inflammatory mediators, which are present in high levels in asthmatic airways, directly modulate ASM function. Using cultured human ASM cells that retain their physiologic responsiveness to agonists [1], investigators have shown that TNF-α markedly stimulates the synthetic function of ASM, defined as secretion of cytokines and chemokines and stimulation of expression of the adhesion molecules, intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule-1 (for review [2]). In addition, new evidence shows that TNF-α also induces a hyper-contractile phenotype by enhancing agonist-induced calcium signals, as well as agonist-induced force generation (for review [3]). These data represent important findings because changes in ASM phenotype induced by cytokines may play a role in the airway remodeling and bronchial hyper-responsiveness that is observed in asthma. A better understanding of the mechanisms that modulate ASM function may lead to the development of new therapeutic interventions for the treatment of asthma.
The present review examines recent studies that investigated the potential mechanisms activated by TNF-α that modulate ASM responsiveness.
Intracellular calcium regulates TNF-alpha; signaling in various cell types
Growing evidence suggests that intracellular calcium plays an important role in mediating the biologic effects of cytokines. Many studies have shown that cytokines, by activating specific receptors coupled to a variety of signaling pathways, generate or modulate the calcium responses and/or alter regulatory proteins that are involved in calcium signaling. For example, studies suggest that TNF-α generates calcium responses in different cell types such as 3T3 cells, skin fibroblasts [4,5], astroglial cells [6], U937 cell line [7] and human neutrophils [8]. TNF-α-induced calcium signals may be stimulated by several distinct pathways, including an indirect emptying of intracellular calcium stores through the generation of second messengers. Accordingly, reports show that TNF-α can directly activate inositol phosphate turnover in some but not all cell types, as a result of phospholipase C stimulation [4,9,10].
Additionally, stimulation of calcium influx due to direct activation of plasma membrane-associated calcium channels represents an alternative mechanism by which TNF-α increases cytosolic calcium concentration [11]. Interestingly, TNF-α activates crucial proteins that are involved in maintaining calcium homeostasis. TNF-α stimulates the expression of a calcium-binding protein, calbindin-D28K, in neuronal cells [12], and increases activity of proteins with calcium pumping properties in fetal pancreatic islets [13]. The effects of TNF-α on calcium homeostasis probably mediate some cellular functions that are important in regulating local inflammation. For example, TNF-α-induced modulation of cytosolic calcium induces cell death in the transformed cell line L929 [9], cell toxicity in cardiac myocytes [14], impairment of function in neuronal cells [6], and perturbation of insulin secretion by Langerhans' islets [13]. Collectively, these studies suggest that changes in calcium homeostasis represent a new mechanism by which TNF-α may regulate cellular responses.
TNF receptor 1 induces airway smooth muscle cell hyper-responsiveness
Studies now show that TNF-α alters ASM contractile function in a manner that mimics that observed in vivo in asthmatic patients. In a sensitized guinea pig model, inhibiting TNF-α completely abrogated the development of bronchial hyper-responsiveness and airway inflammation [15]. ASM exposed to TNF-α either in vitro or in vivo become hyper-responsive to many contractile agonists (for review [3]). We also found that murine tracheal rings preincubated with TNF-α for 24 h have an increased contractile response to carbachol (Fig.1). The underlying mechanism remains unclear, but our data show that ASM cells pretreated with TNF-α also potentiated calcium signals induced by a variety of contractile agonists. These studies suggest that TNF-α is able to "prime" ASM cells for a nonspecific increase in calcium responsiveness, because TNF-α alone had no effect on cytosolic calcium levels [16,17].
Figure 1.
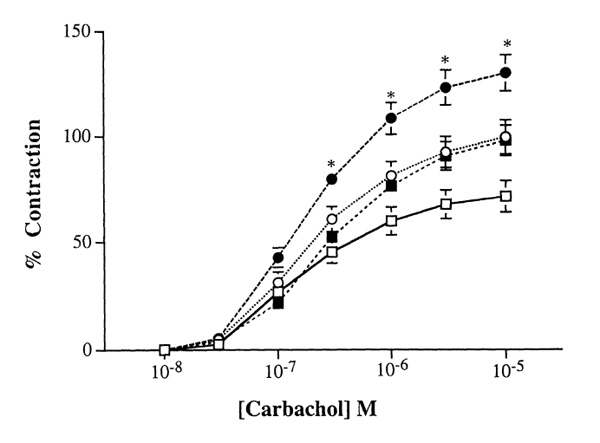
TNF-α enhances the contractile response to carbachol in murine tracheal rings. Murine tracheal rings were harvested and placed in culture overnight in an equal mixture of Ham F-12 and DMEM (vol/vol, 10% fetal bovine serum) in the presence or absence of TNF-α . Cumulative concentration-response curves to carbachol were measured on the cultured tracheal rings and compared among fresh controls (□ n = 18), or in the presence of 10 ng/ml (■ n = 12) or 50 ng/ml TNF-α (● n = 12), or in the absence of TNF-α (○ n = 7) for 24 h. Isometric measurements of tracheal reactivity were calculated as changes in milligram tensions per milligram weight (mg/mg) and expressed as percentage of 10-5 mol/l carbachol-induced tensions. Although TNF-α appeared to augment carbachol-induced force generation in cultured murine tracheal rings, it is interesting to note that carbachol-induced increases in force generation were also increased as compared with those obtained in freshly harvested tissues. Results are expressed as mean± standard error of the mean. *P <0.05.
In other studies we reported that TNF-α significantly enhances phosphoinositide turnover in response to bradykinin [18]. The use of agonistic antibodies or recombinant proteins of TNF-α that specifically activate either TNFR1 or TNFR2 receptor led us to conclude that TNF-α mediates most of its cellular effects by activating the TNFR1 receptor [17]. The effect of TNF-α on calcium responses therefore appears to involve a modulatory effect on G-protein-mediated signal transduction via its TNFR1 receptor. The specific target could be at the level of either G-protein or phospholipase C.
In parallel studies, TNF-α increased cytosolic calcium levels induced by NaF [18], an agent that directly activates G-protein in ASM cells, which strongly suggests that TNF-α effects occur downstream from the receptor, possibly at the level of the G-proteins.Consistent with this hypothesis are the recent findings of Hotta et al [19]. Those investigators showed that TNF-α increases expression of Gq and Gi proteins in human ASM cells. It remains to be defined whether both phenomena, that is, increased receptor-mediated calcium responses and increased smooth muscle responsiveness, are linked.
Alteration in the receptor-G protein-phospholipase C signaling pathway by TNF-α is probably not the only mechanism by which TNF-α modulates contractile agonist responses. By simultaneously measuring the intracellular calcium levels and isometric tensions in response to acetylcholine, Nakatani et al [20] recently showed that brief exposure to TNF-α for 30 min enhanced contractile responses to acetylcholine by increasing the calcium sensitivity of contractile elements in bovine tracheal smooth muscle. Such brief exposure of cells to TNF-α probably has little effect on protein synthesis, the other mechanism that has been shown to mediate the effects of TNF-α on calcium homeostasis [18]. Surprisingly, TNF-α did not affect the calcium signals induced by acetylcholine. Similar results were also described in guinea pig tracheal smooth muscle, in which TNF-α also increased calcium sensitivity [21]. Together, these studies suggest that brief treatment with TNF-α may modulate ASM contractile response by directly inducing calcium sensitization of intracellular contractile elements, rather than by modulating agonist-evoked calcium mobilization induced by longer pretreatment with TNF-α. Fig.2 summarizes potential mechanisms by which TNF-α modulates ASM responsiveness.
Figure 2.
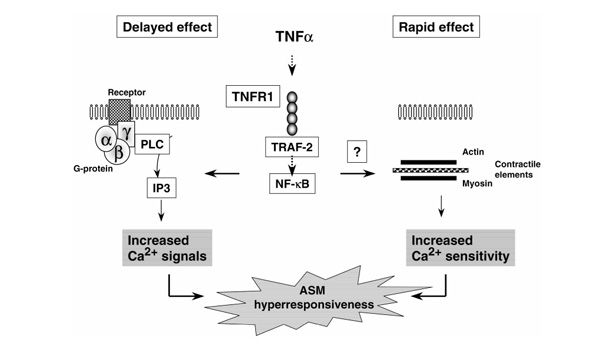
Potential intracellular mechanisms involved in the modulation of ASM hyper-responsiveness induced by TNF-α via TNFR1. Activation of TNFR1 coupled to the TRAF2-NF-κB pathway induces a delayed effect, in which long-term pretreatment with TNF-α enhances G-protein-coupled signal transduction, leading to increased calcium signals to contractile agonists. Activation of TNFR1 may also be involved in a rapid effect, in which short-term pretreatment with TNF-α enhances the calcium sensitization of intracellular contractile elements [20,21]. IP3, inositol-1,4,5-trisphosphate; PLC, phospholipase C.
Together, these studies support the notion that TNF-α may play a central role in modulating ASM responsiveness by inducing both early (less than 1 h) and late (greater than 4 h) alteration signals that modulate agonist-induced calcium homeostasis and/or force generation.
TNF receptor 1 signaling pathways that are activated in airway smooth muscle cells
TNF receptor-associated factor-2-nuclear factor κB pathway
TNF-α activates at least two cell-surface receptors -TNFR1 (55kDa) and TNFR2 (75kDa) - that are expressed in most cell types (for review [22]). Both receptors are expressed on cultivated human ASM cells [17] and on native ASM [23]. Evidence shows that the intracellular signals that couple TNFR1 include an intracellular signal protein termed `TNF receptor-associated death domain' [24]. Upon engagement of TNFR1, this intracellular signal protein acts as an adapter by recruiting the downstream transducer TRAF2, which stimulates NF-κB activation [24,25].
In addition to the effects of TNF-α on agonist-evoked calcium signals, we also reported that activation of TNFR1 by TNF-α regulates the expression of functional ICAM-1 on human ASM cells [23]. Using ASM cells transfected with an NF-κB reporter, our data also showed that TNF-α and htr-9, an activating antibody against TNFR1, both activated NF-κB [23,26]. Furthermore, overexpression of a dominant-negative TRAF2 construct, lacking the amino-terminal RING finger, completely abrogated both TNF-α-mediated and htr-9-mediated increases in NF-κB reporter activity [23]. Collectively, these data suggest that TRAF2 plays a role in TNFR1-mediated NF-κB activation in ASM cells. Interestingly, activation of NF-κB and expression of ICAM-1 by TNF-α in ASM cells were insensitive to dexamethasone pretreatment, a commonly used therapeutic agent in the treatment of asthma [26]. In many other cells, however, TNF-α-induced NF-κB activation is exquisitely sensitive to steroids [27].
Intracellular calcium stores
In order to define the role of intracellular calcium in TNF-α signaling, we studied the effect of thapsigargin, a specific inhibitor of intracellular calcium pumps (ie sarco-endoplasmic reticulum calcium-ATPase), on gene expression. In many cell types, thapsigargin has been shown to stimulate calcium signals by directly emptying intracellular stores without activating second messenger systems (for review [28]). Evidence supports the notion that thapsigargin-sensitive calcium pools are enhanced by TNF-α and are mediated by TNFR1 activation (for review [3]). Interestingly, thapsigargin-sensitive calcium stores were also found to be those activated by contractile agonists [29]. These data suggest that modulation of internal calcium stores may be involved in the potentiating effect of TNF-α on agonist-induced calcium signals.
Present evidence also shows that thapsigargin-sensitive calcium pools modulate TNF-α-induced gene expression in human ASM cells. Thapsigargin, in a dose-dependent manner, inhibited about 50% of the TNF-α-induced ICAM-1 expression [23]. The calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid acetoxymethyl-ester, which clamps the cytosolic calcium concentration, also inhibited ICAM-1 stimulated by TNF-α [23]. Surprisingly, thapsigargin suppressed TNF-α-mediated NF-κB-dependent gene transcription, an effect that was not due to cytokine-induced IκBα degradation or NF-κB nuclear translocation, which are crucial steps for NF-κB activation [23]. The mechanisms by which thapsigargin modulates gene expression remain unclear. In rat myocytes and NIH 3T3 fibroblasts, however, emptying internal calcium stores inhibits protein synthesis by acting at the level of translation initiation through the inactivation of the translation initiation factor eIF2α [30,31]. Additional studies support the notion that thapsigargin-sensitive calcium pools may modulate gene expression in an NF-κB-dependent manner. This hypothesis is also supported in other cell types, because a variety of agents that alter endoplasmic reticulum function are able to modulate NF-κB activation (for review [32]).
Thapsigargin may also affect pathways that modulate calcium homeostasis that are located in the nucleus that is believed to play a central role in stimulating gene expression. Recently, Adebanjo et al [33] showed the existence of a potential CD38/ADP ribosyl-cyclase pathway within the nucleosome that triggers nucleoplasmic calcium release via the production of cyclic ADP ribose. Further studies are needed to elucidate the precise mechanism that mediates thapsigargin inhibitory effects on gene expression.
Conclusion
Characterization of the cellular and biochemical events that regulate ASM function will likely lead to new therapeutic approaches in the management of asthma. In ASM cells, activation of TNFR1 coupled to the TRAF2-NF-κB pathway `primes' airway myocytes to augment calcium signals in response to thapsigargin and contractile agonists, an effect that may, in part, be due to the cross-talk between TNFR1 activation and internal calcium stores. A better understanding of TNFR1 signaling in ASM cells may offer new insight into the mechanisms that regulate both airway inflammation and bronchial hyper-responsiveness in asthma.
Acknowledgments
Acknowledgments
The work of the authors cited in the present review was supported by grants R01-HL64063 and R01-HL55301 from the National Institutes of Health. The authors thank Mary McNichol for assistance with preparation of the manuscript.
References
- Panettieri RA, Jr, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI. A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol (Cell Physiol) 1989;256/25:C329–C335. doi: 10.1152/ajpcell.1989.256.2.C329. [DOI] [PubMed] [Google Scholar]
- Johnson SR, Knox AJ. Synthetic functions of airway smooth muscle in asthma. Trends Pharmacol Sci. 1997;18:288–292. doi: 10.1016/S0165-6147(97)01092-4. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Panettieri RA., Jr Cytokines induce airway smooth muscle cell hyperresponsiveness to contractile agonists. Thorax. 1998;53:713–716. doi: 10.1136/thx.53.8.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchelouche PN, Bendtzen K, Bak S, Nielsen OH. Recombinant human tumour necrosis factor increases cytosolic free calcium in murine fibroblasts and stimulates inositol-phosphate formation in L-M and arachidonic acid release in 3T3 cells. Cell Signal. 1990;2:479–487. doi: 10.1016/0898-6568(90)90044-B. [DOI] [PubMed] [Google Scholar]
- Corkey BE, Geschwind JF, Deeney JT, Hale DE, Douglas SD, Kilpatrick L. Ca2+ responses to interleukin-1 and tumor necrosis factor in cultured human skin fibroblasts. Possible implications for Reye syndrome. J Clin Invest. 1991;87:778–786. doi: 10.1172/JCI115081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller H, Thiem K, Siebler M. Tumour necrosis factor-alpha increases intracellular Ca2+ and induces a depolarization in cultured astroglial cells. Brain. 1996;119:2021–2027. doi: 10.1093/brain/119.6.2021. [DOI] [PubMed] [Google Scholar]
- Peiretti F, Fossat C, Anfosso F, Alessi MC, Henry M, Juhan-Vague I, Nalbone G. Increase in cytosolic calcium upregulates the synthesis of type I plasminogen activator inhibitor in the human histiocytic cell line U937. Blood. 1996;87:162–173. [PubMed] [Google Scholar]
- Schumann MA, Gardner P, Raffin TA. Recombinant tumor necrosis factor alpha induces calcium oscillation and calcium-activated chloride current in human neutrophils. The role of calcium/calmodulin-dependent protein kinase. J Biol Chem. 1993;268:2134–2140. [PubMed] [Google Scholar]
- Beyaert R, Heyninck K, De Valck D, Boeykens F, Van Roy F, Fiers W. Enhancement of tumor necrosis factor cytotoxicity by lithium chloride is associated with increased inositol phosphate accumulation. . J Immunol. 1993;151:291–300. [PubMed] [Google Scholar]
- Rapuano BE, Bockman RS. Protein kinases A and C positively regulate G protein-dependent activation of phosphatidylinositol-specific phospholipase C by tumor necrosis factor-alpha in MC3T3 E1 osteoblasts. . J Cell Biochem. 1997;65:198–208. doi: 10.1002/(SICI)1097-4644(199705)65:2<198::AID-JCB6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Wilkinson MF, Earle ML, Triggle CR, Barnes S. Interleukin-1β, tumor necrosis factor-α and LPS enhance calcium channel current in isolated vascular smooth muscle cells of rat tail artery. FASEB J. 1996;10:785–796. doi: 10.1096/fasebj.10.7.8635696. [DOI] [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Garcia G, Arias-Diaz J, Balibrea JL, Vara E. Modifications in calcium uptake may contribute to the effects of tumor necrosis factor on fetal islets. Transplant Proc. 1994;26:3496–3498. [PubMed] [Google Scholar]
- Krown KA, Yasui K, Brooker MJ, Dubin AE, Nguyen C, Harris GL, McDonough PM, Glembotski CC, Palade PT, Sabbadini RA. TNF alpha receptor expression in rat cardiac myocytes: TNF alpha inhibition of L-type Ca2+current and Ca2+ transients. FEBS Lett . 1995;376:24–30. doi: 10.1016/0014-5793(95)01238-5. [DOI] [PubMed] [Google Scholar]
- Renzetti LM, Paciorek PM, Tannu SA, Rinaldi NC, Tocker JE, Wasser man MA, Gater PR. Pharmacological evidence for tumor necrosis factor as a mediator of allergic inflammation in the airways. J Pharmacol Exptl Ther. 1996;278:847–853. [PubMed] [Google Scholar]
- Amrani Y, Martinet N, Bronner C. Potentiation by tumour necrosis factor-α of calcium signals induced by bradykinin and carbachol in human tracheal smooth muscle cells. Br J Pharmacol . 1995;114:4–5. doi: 10.1111/j.1476-5381.1995.tb14896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani Y, Panettieri RA, Jr, Frossard N, Bronner C. Activation of the TNFα-p55 receptor induces myocyte proliferation and modulates agonist-evoked calcium transients in cultured human tracheal smooth muscle cells. Am J Respir Cell Mol Biol. 1996;15:55–63. doi: 10.1165/ajrcmb.15.1.8679222. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Krymskaya V, Maki C, Panettieri RA., Jr Mechanisms underlying TNFa effects on agonist-mediated calcium homeostasis in human airway smooth muscle cells. Am J Physiol (Lung Cell Mol Physiol) . 1997;273/17:L1020–L1028. doi: 10.1152/ajplung.1997.273.5.L1020. [DOI] [PubMed] [Google Scholar]
- Hotta K, Emala CW, Hirshman CA. TNF-a upregulates Gi a and Gq a protein expression and function in human airway smooth muscle cells. Am J Physiol (Lung Cell Mol Physiol) 1999;276/20:L405–L411. doi: 10.1152/ajplung.1999.276.3.L405. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Nishimura Y, Nishiumua T, Maeda H, Yokoyama M. Tumor necrosis factor-a augments contraction and cytosolic Ca2+ through phospholipase A2 in bovine tracheal smooth muscle. . Eur J Pharmacol. 2000;392:175–182. doi: 10.1016/S0014-2999(00)00087-X. [DOI] [PubMed] [Google Scholar]
- Parris JRM, Cobban HJ, Littlejohn AF, MacEwan DJ, Nixon GF. Tumor necrosis factor-α activates a calcium sensitization pathway in guinea-pig bronchial smooth muscle. J Physiol (Lond) 1999;518:561–569. doi: 10.1111/j.1469-7793.1999.0561p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Lazaar AL, Hoffman R, Amin K, Ousmer S, Panettieri RA., Jr Activation of the p55 TNFR1 coupled to TRAF2 stimulates ICAM-1 expression by modulating a thapsigargin-sensitive pathway in human tracheal smooth muscle cells. Mol Pharmacol. 2000;58:237–245. doi: 10.1124/mol.58.1.237. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV. The TNF receptor-1 associated protein TRADD signals cell death and NF-κB activation. . Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu SB, Pan MG, Baichwal V, Goeddel DV. TRADD-TRAF-2 and TRAD-FADD interactions define two distinct TNF receptor-1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Lazaar AL, Panettieri RA., Jr Up-regulation of ICAM-1 by cytokines in human tracheal smooth muscle cells involves an NF-κB-dependent signaling pathway that is only partially sensitive to dexamethasone. J Immunol. 1999;163:2128–2134. [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: Inhibition of NF-κB activity through induction of IκB synthesis. . Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- Thastrup O. Role of Ca2+-ATPases in regulation of cellular Ca2+ signalling, as studied with the selective microsomal Ca2+-ATPase inhibitor, thapsigargin. . Agents Actions. 1990;29:8–15. doi: 10.1007/BF01964706. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Magnier C, Wuytack F, Enouf J, Bronner C. Ca2+increase and Ca2+ influx in human tracheal smooth muscle cells: role of Ca2+ pools controlled by sarco-endoplasmic reticulum Ca2+-ATPase 2 isoforms. . Br J Pharmacol. 1995;115:1204–1210. doi: 10.1111/j.1476-5381.1995.tb15026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly BA, Brostrom MA, Brostrom CO. Regulation of protein synthesis in ventricular myocytes by vasopressin. J Biol Chem . 1998;273:3747–3755. doi: 10.1074/jbc.273.6.3747. [DOI] [PubMed] [Google Scholar]
- Aktas H, Fluckiger R, Acosta JA, Savage JM, Palakurthi SS, Halperin JA. Depletion of intracellular Ca2+ stores, phosphorylation of eIF2a, and sustained inhibition of translation initiation mediate the anti-cancer effects of clotrimazole. Proc Natl Acad Sci USA. 1998;95:8280–8285. doi: 10.1073/pnas.95.14.8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL, Baeuerle PA. The ER-overload response: activation of NF-κB. Trends Biochem Sci. 1997;22:63–67. doi: 10.1016/S0968-0004(96)10073-6. [DOI] [PubMed] [Google Scholar]
- Adebanjo OA, Anandatheerthavarada HK, Koval AP, Moonga BS, Biswas G, Sun L, Sodam BR, Bevis PJ, Huang CL, Epstein S, Lai FA, Avadhani NG, Zaidi M. A new function for CD38/ADP ribosyl cyclase in nuclear Ca2+homeostasis. Nature Cell Biol. 1999;1:409–414. doi: 10.1038/15640. [DOI] [PubMed] [Google Scholar]