

Abstract
The uncontrolled growth of tumors provides metabolic dependencies that can be harnessed for therapeutic benefit. Although tumor cells exhibit these increased metabolic demands due to their rapid proliferation, these metabolic processes are general to all cells, and furthermore, targeted therapeutic intervention can provoke compensatory adaptation that alters tumors’ characteristics. As an example, a subset of melanomas depends on the transcriptional coactivator PGC1α function to sustain their mitochondrial energy-dependent survival. However, selective outgrowth of resistant PGC1α-independent tumor cells becomes endowed with an augmented metastatic phenotype. To find PGC1α−dependent components that would not affect metastasis in melanomas, an unbiased proteomic analyses was performed and uncovered the orphan nuclear receptor ERRα, which supports PGC1α’s control of mitochondrial energetic metabolism, but does not affect the anti-oxidant nor anti-metastatic regulatory roles. Specifically, genetic or pharmacologic inhibition of ERRα reduces the inherent bioenergetic capacity and decreases melanoma cell growth, but without altering the invasive characteristics. Thus within this particularly aggressive subset of melanomas, which is characterized by heighted expression of PGC1α, ERRα specifically mediates pro-survival functions and represents a tangible therapeutic target.
Implications
ERRα, a druggable protein, mediates the bioenergetic effects in melanomas defined by high PGC1α expression, suggesting a rational means for therapeutic targeting of this particularly aggressive melanoma subtype.
Keywords: ERRα, oxidative metabolism, bioenergetics, PGC1α, melanoma
INTRODUCTION
Tumor cells generally use aerobic glycolysis (Warburg effect) to fuel their growth, yet many also rely on mitochondrial oxidative phosphorylation to support their anabolic needs (1). To this end, we recently identified that a significant fraction of human melanomas (8-10%) have heightened expression levels of PGC1α (gene name: PPARGC1A) – a transcriptional coactivator that promote mitochondrial biogenesis and respiration, wherein genetic suppression leads to decreased energetic capacity, impaired antioxidant capacity and subsequent apoptosis (2,3). Rare melanoma cells that survive chronic depletion of PGC1α shift their metabolism towards increased aerobic glycolysis (4), and in addition, adapt invasive and metastatic traits, which is driven by transcriptional upregulation of integrin, TGFβ and WNT signaling components (5). PGC1α consequently have opposing functions in melanoma pathogenesis — favoring tumor growth and survival by promoting oxidative metabolism, but also suppressing a transcriptional network that controls invasion and metastatic spread.
While potentially amenable for therapeutic inhibition, the functional ramifications of inhibiting PGC1α in melanoma may consequently prove counterproductive. However, PGC1α is a transcriptional coactivator that is recruited to promoters of genes by certain transcription factors involved in cellular metabolic functions (6). Because our recent data indicated that PGC1α promotes mitochondrial-dependent survival distinct from the mechanism through which PGC1α suppress metastasis (5), it is likely that PGC1α employs alternate transcriptional cues to regulate these divergent melanoma growth phenotypes. To this end, successful identification of transcription factors that controls each of growth and metastasis may inform selective targets for therapeutic pursuit of PGC1α-positive melanomas.
The estrogen-related orphan nuclear receptors (ERRα, β and γ) are known PGC1α interacting proteins that mediate mitochondrial biogenesis in oxidative tissues (7–9). In addition to participate in normal metabolism, ERRs also play important functions in various malignancies (1,10), wherein heightened ERRα has been associated with worse prognosis in certain carcinomas, including breast, ovarian, uterine, prostate, and colorectal tumors (10). The ERRα/PGC1α complex has been mechanistically demonstrated to promote glutamine into de novo fatty acid biosynthesis, and thereby confer growth advantages to ERBB2-positive breast cancers (11). Increased glutamine metabolism and ROS detoxification by the ERRα/PGC1α axis in breast cancers have also been found to promote metabolic adaptation following receptor tyrosine kinase inhibition using lapatinib (12). Further, the ERRα/PGC1α complex suppresses one-carbon metabolism in response to AMPK stimulation, thus rendering breast cancer cells more vulnerable to anti-folate drugs such as methotrexate (13). On the other hand, in prostate cancer, ERRα has been found to promote catabolic metabolism that subsequently suppresses the metastatic ability (14). In melanoma, it is however not clear to what extent ERRα functionally contribute to tumor growth.
As a nuclear receptor ERRα is a drugable target, and multiple reverse agonists (antagonists) have been developed that effectively block its transcriptional activation both in vitro and in vivo (15). Most of these compounds were designed to block the interaction between ERRα and PGC1α, such as XCT790, Compound (Cpd) A and 29, and have been shown to exhibit growth-inhibitory therapeutic effects in certain cancers (16–18). For example, in combination with PI3K inhibition, Cpd29 significantly inhibits the progression of breast cancer in nude mice (18). Cpd29 has also been shown to suppress ERRα-mediated metabolic reprogramming and to overcome drug resistance in a mammary tumor model (12). Hence, ERRα antagonist have shown preclinical activity in certain tumor models, but their potential efficacy have not been characterized in melanoma, with particular emphasis on PGC1α-positive melanomas that are highly dependent on oxidative metabolism.
In the present study, we have employed a proteomic approach in human melanoma cells to identify functional PGC1α protein complexes. We characterized ERRα as a critical factor that mediates PGC1α’s growth and survival functions. Specifically, ERRα physically interacts with PGC1α, which complex promotes expression of genes involved in mitochondrial oxidative phosphorylation, but do not affect expression of the cellular antioxidant or invasive/metastatic programs. Like suppression of PGC1α, depletion of ERRα impairs mitochondrial bioenergetic capacity, in vitro cell proliferation and in vivo tumor growth in PGC1α-positive melanoma cells. However, in contrast to genetic manipulation of PGC1α levels (5), depletion of ERRα did not promote melanoma cell invasion. Strikingly, pharmacological ERRα antagonism phenocopies genetic ERRα deletion, therefore ERRα might constitute an attractive and amenable therapeutic target in a subset of PGC1α-positive melanomas.
MATERIALS & METHODS
Tissue Culture
All melanoma cell lines were obtained from ATCC and their authentication was confirmed by either DNA fingerprinting with small tandem repeat profiling or in-house PCR testing of melanoma marker genes and BRAF mutation status. Mycoplasma contamination was tested in house with the PCR Mycoplasma Detection Kit (Lonza). Cells were maintained in DMEM (Sigma-Aldrich) with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin. All cells were cultured in humidified incubator at 37 °C with 5% CO2. All cells were used for less than 10 passages upon thawing from liquid nitrogen.
The Cancer Genome Atlas (TCGA) melanoma tumor analyses
Publicly available RNAseq-based gene expression, mutation, AJCC-stage of sample retrieval, and patient outcome data for melanoma tumors was obtained from the TCGA portal (tcga-data.nci.nih.gov). Using data from the 366 samples obtained at AJCC stage III (local metastasis) and IV (distant metastasis), within-cohort normalized expression levels were calculated for PPARGC1A (PGC1α) and ESRRA (ERRα) to compare their correlative expression (based on Pearson r), relationship between PGC1α rank-based expression and ERRα levels (un-paired, two-sided t-test), and mutual (normalized average) expression rank (MER) with patient outcome (survival based on Mantel-Cox log-rank test).
Reagents and Antibodies
Compound 29 (4-{4-[(2,4-dioxo-1,3-thiazolidin-5-ylidene)methyl]-2-methoxyphenoxy}-3-(trifluoromethyl)benzonitrile, 95%) (17) was synthesized by MolPort. Oligomycin, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), antimycin A and rotenone were all purchased from Sigma-Aldrich. The following antibodies were used: PGC1α (H300, Santa Cruz; ST1202, EMD Millipore), ERRα (N1, GeneTex), mtCOI (Abcam), actin (Cell Signaling), SOD2 (GeneTex), GPX1 (GeneTex), pFAK-Y397 (Cell Signaling), COX4 (Abcam), COX5A (Abcam), UQCRC2 (Abcam), tubulin (Abcam), histone H3 (Abcam), and Ki-67 (Thermo Fisher).
Lentiviral Generation and Transduction
Lentiviruses encoding shRNAs or sgRNAs (lentiCRISPRv2, Addgene #52961) were produced in HEK293T cells with packaging vectors (pMD2G and psPAX2) using Escort™ IV Transfection Reagent (Sigma-Aldrich)(19). Lentiviruses particles were collected 48 h after post-transfection and used to infect melanoma cells in the presence of 8 μg/ml polybrene, and infected cells were selected with 2 μg/ml of puromycin, or 7 μg/ml blasticidin for at least 4 days prior to experiments. The shERRα-#1 is TRCN0000330256 and #2 is TRCN0000022181 (http://portals.broadinstitute.org/gpp/public/). The sgRNA sequences for ERRα are: #1 AGGCTCGGTCTCTGTCTCCG, #2 GACAGAGACCGAGCCTCCTG, and #3 AGTGGGCTGGGGGCTCACCC.
Immunoprecipitation, Immunoblotting and Quantitative real time-PCR
For co-immunoprecipitation (co-IP), cells were first permeabilized by Swelling Buffer (25 mM HEPES pH 7.6, 1.5 mM MgCl2, 10 mM KCl, 0.5% NP40) on ice for 15 min, followed by centrifugation at 2000 rpm at 4 °C for 10 min to collect the intact nucleus. The pelleted nucleus was then lysed in RIPA buffer without SDS and sodium deoxycholate but supplemented with 10% glycerol, followed by sonication at low power for 30 seconds and rotation at 4 °C for one hour. Exogenously expressed Flag/HA-tagged PGC1α by adenoviruses in the nuclear lysate was subsequently subjected to immunoprecipitation with beads conjugated with anti-Flag M2 antibody (Sigma) and anti-HA antibody (Sigma) at 4 °C overnight, followed by wash for 3 times and elution with respective peptides. Elution was performed by 5 mg/mL Flag peptide (Sigma) or 5 mg/mL HA peptide (Sigma) in the same buffer at 37 °C with gentle shaking at 100 rpm for one hour. Total elution was subjected to mass spec analysis. For endogenous PGC1α co-IP, 5 mg nuclear lysate with 2 μL of PGC1α antibody (ST1202) was incubated overnight at 4 °C, followed by precipitation using DynaBead A/G (Invitrogen) for 2 hours at 4 °C.
For immunoblotting, cells were lysed in RIPA buffer and quantified by DC protein concentration assay kit (Pierce) before subjecting to SDS-PAGE gel. Total RNA was isolated with Trizol (Invitrogen) by Direct-zol RNA MiniPrep kit (Zymo Research), and 2 μg of total RNA was used for cDNA synthesis using high capacity cDNA reverse transcription kit (Applied Biosystems). qPCR was carried out using SYBR Green PCR Master Mix (Bio-Rad). Experimental Ct values were normalized to 36B4, and relative mRNA expression was calculated using the ΔΔCt method. Sequences for all primers are available upon request.
Seahorse Respirometry
Respirometry was performed using the XFe24 platform (Seahorse Biosciences). Briefly, 4~8×104 cells with indicated genetic manipulations were seeded in the Seahorse plates and allowed overnight for the cells to attach to the plates. Cells were then washed once with Seahorse buffer (unbuffered DMEM medium without FBS). All the chemicals loaded in the Seahorse cartridge ports were diluted in Seahorse buffer, and the pH was adjusted to 7.4. Following measurement of basal respiration, 2 μM Oligomycin (Complex V/ATP synthase inhibitor) was used to measure ATP production; uncoupled respiration by adding 4 μM FCCP was measured to determine the maximal respiration of the cells; non-mitochondrial respiration was recorded by the addition of antimycin A (4 μM) and rotenone (2 μM). Oxygen consumption values were normalized to cell number(2,20).
ATP production assay
Cells were lifted by trypsin, counted and suspended in PBS. 1×105 cells in 50 μL were used for ATP assay using CellTiter-Glo Luminescent Kit (Promega), following the manufacturer’s instruction.
In vitro migration assay
Transwell chambers were purchased from Corning Life Science. Generally, A375P (1×105), G361 (5×104) or A2058 (1×104) cells in 0.1 mL of FBS-free medium were seeded into the upper chamber and incubated overnight. Cells that had migrated were then fixed and stained with Crystal Violet. The membrane attached with migrated cells was placed on a glass slide; total cells from three images taken from three random fields under 10X magnifications with a Nikon 80i Upright microscope were quantified.
Cellular ROS measurement by DCF-DA
DCF-DA-based cellular ROS detection was performed according to manufacturer’s instruction (Abcam). Briefly, cells were lifted by trypsin and stained with 20 μM DCF-DA at 37°C for 30 min, followed by FACS analysis(2,21). Cells with 1 mM H2O2 treatment were used as positive controls.
Proliferation Assay and Clonogenic Assay
Melanoma cells with indicated manipulation were seeded in either 6-well or 12-well plates at a density of 1×104/well in triplicate. Cell number was counted at indicated time points. For compound treatment experiments, the next day after seeding, cells were exposed to DMSO or indicated concentration of Cpd29, followed by cell counting. For clonogenic assay, cells were seeded at the density of 1×104/well in 6-well plates in triplicate and cultured for 10 to 14 days, followed by fixation with 100% ethanol and staining with Crystal Violet. Culture medium was refreshed every other day for all these experiments.
Animal experiments
All animal experiments were designed and conducted following the protocol approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee. Eight- to 10-week-old male NCr nude mice (for G361 cell line, purchased from Taconic Biosciences) and female NCI Ath/Nu mice (for MeWo cell line, purchased from Charles River) were used for this study. Mice were housed in a controlled environment of 12:12 hour dark and light cycle and provided food and water ad libitum. All mice from a single experiment were at the same age. 2×106 tumor cells were injected s.c. into the flanks of mice, and the tumor development was monitored three time per week. Tumor volume was calculated based on the equation V=(width (in mm)2Xlength (in mm))/2(22). Tumor-bearing mice were given 30 mg/kg Cpd29 (in 10% NMP/90% PEG300)(18) or vehicle via oral administration six times a week.
Histology
Tissue samples were fixed in 10% buffered formalin overnight and stored in 70% ethanol prior to paraffin embedding, sectioning and hematoxylin/eosin (H&E) staining (by the Rodent Histopathology Core, Harvard Medical School)(23). Immunohistochemistry was performed with Ki-67 (SP6, Thermo Fisher) antibody with protocol described previously(24). Negative control was done by replacing the primary antibody with species-matched total IgG.
Statistics
All statistics are described in figure legends and were performed with GraphPad Prism. In general, for two experimental comparisons, a two-tailed unpaired Student’s t-test was used. For multiple comparisons, one-way ANOVA was applied. When cells were used for experiments, three replicates per treatment were chosen as an initial sample size. All n values defined in the legends refer to biological replicates unless otherwise indicated. Statistical significance is represented by asterisks corresponding to *P < 0.05, **P < 0.01, ***P < 0.001, if not otherwise indicated.
RESULTS
ERRα is an interacting partner of PGC1α in a subset of human melanoma cells
To identify proteins that could selectively mediate PGC1α–dependent mitochondrial bioenergetics, survival, or metastatic suppression, we set out to analyze PGC1α proteome complexes in melanoma cells. To accommodate this goal, we ectopically expressed double-FLAG/HA-tagged PGC1α in the high PGC1α-expressing melanoma cell line A375P, and performed mass spectrometric analyses of associated proteins after sequential immunoprecipitation (Fig. S1). Among the top ten most abundant peptides that were in close stoichiometry with PGC1α, we identified the estrogen related receptor alpha (ERRα; gene name ESRRA) as the only transcription factor (Fig. 1A). To validate the presence of PGC1α-ERRα complexes in cells, we confirmed the endogenous interaction between these two proteins using immunoprecipitation analysis from different PGC1α-positive melanoma cell extracts (Fig. 1B).
Figure 1. ERRα is associated with PGC1α in melanoma cells.
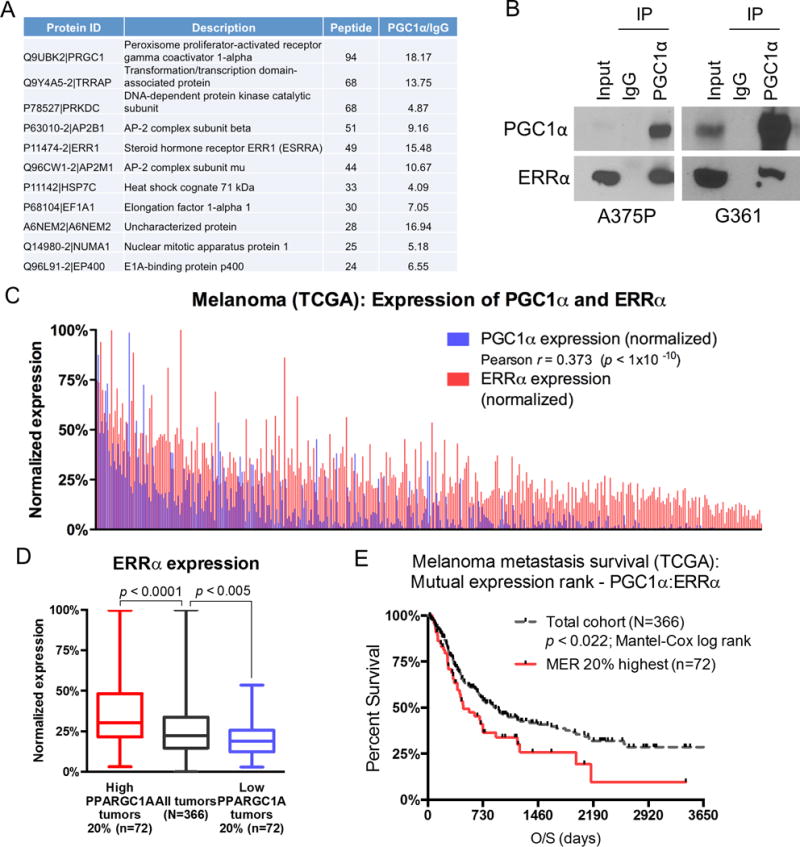
A. A list of the most abundant proteins co-immunoprecipitated with Flag/HA-tagged PGC1α in A375P melanoma cells as identified by mass spectrometry. B. Endogenous PGC1α is interacting with ERRα in PGC1α-positive melanoma cell lines A375P and G361. Immunoprecipition of PGC1α was followed by immunoblotting using the indicated antibodies. C. Pearson-based correlation between PGC1α and ERRα expression levels across metastatic melanoma samples within TCGA (N=366). D. Based on 2-sample, 2-sided t-test statistics, the 20 percentile highest and lowest PGC1a expression levels associate with highest and lowest ERRα expression levels, respectively. E. Based on Mantel-Cox log rank test, the top 20 percentile mutual expression rank (MER) of PGC1α:ERRα segregates poorer overall survival relative to the metastatic melanoma cohort average (p < 0.022).
Because of the tight physical interaction between these two proteins, coupled to our recent data indicating that higher PGC1α expression associates with poor metastatic melanoma survival (2,3), we examined whether the intersection of high ERRα and PGC1α levels correlates across melanoma tumors. Using RNAseq data from the 366 metastatic melanoma lesions within the Cancer Genome Atlas (TCGA), we found a significant association between PGC1α and ERRα expression levels (Fig. 1C: Pearson r = 0.373; p < 1×10−10). In addition, each of the highest and lowest 20 percentile PGC1α expressing metastatic tumors have higher versus lower ERRα expression compared to the overall average, respectively (Fig. 1D). Consistent with PGC1α levels as a means to stratify metastatic-disease survival, there is a strong correlation between poor overall survival and the 20 percentile highest PGC1α and ERRα mutual expression rank, as compared to the cohort average, indicating an improved precision to stratify these aggressive tumors (Fig. 1E; p < 0.022 using Mantel-Cox log rank test). Together, these biochemical and clinical correlative data suggest that the association between PGC1α and ERRα may be heightened functional importance, and therefore, may inform therapeutic opportunities in this particularly aggressive subclass of melanoma tumors.
ERRα promotes mitochondrial oxidative metabolism in melanoma cells
ERRα has been shown to promote mitochondrial biogenesis through promoting transcription of genes involved in oxidative phosphorylation and other metabolic pathways (9). Because of the stoichiometric interaction between ERRα and PGC1α in melanoma cells, we analyzed the transcriptional gene expression programs specifically controlled by this complex. Using targeted CRISPR/Cas9 genome editing, we therefore manipulated ERRα expression in a cohort of melanoma cells with heighted PGC1α levels, including A375P, G361 and SK-MEL-30. Using this approach, we found that depletion of ERRα significantly decreased the expression of mitochondrial genes including components of the electron transport chain (COX5A, COX4, CYTC, mtCO1, NDUFB3, NDUFA4 and UQCRC2) and the tricarboxylic acid cycle (IDH3A and FH) (Fig. 2A-C, S2A). Similar gene expression pattern was also observed in PGC1α-positive melanoma cell lines with PGC1α suppression (Fig. S2B) (2), or with shRNA-mediated ERRα knockdown (Fig. S2C, D). In addition, ERRα -depleted cells functionally displayed impaired mitochondrial respiration (Fig. 2D, S2E, F) and lower ATP levels (Fig. 2E).
Figure 2. ERRα is required to support mitochondrial oxidative metabolism.
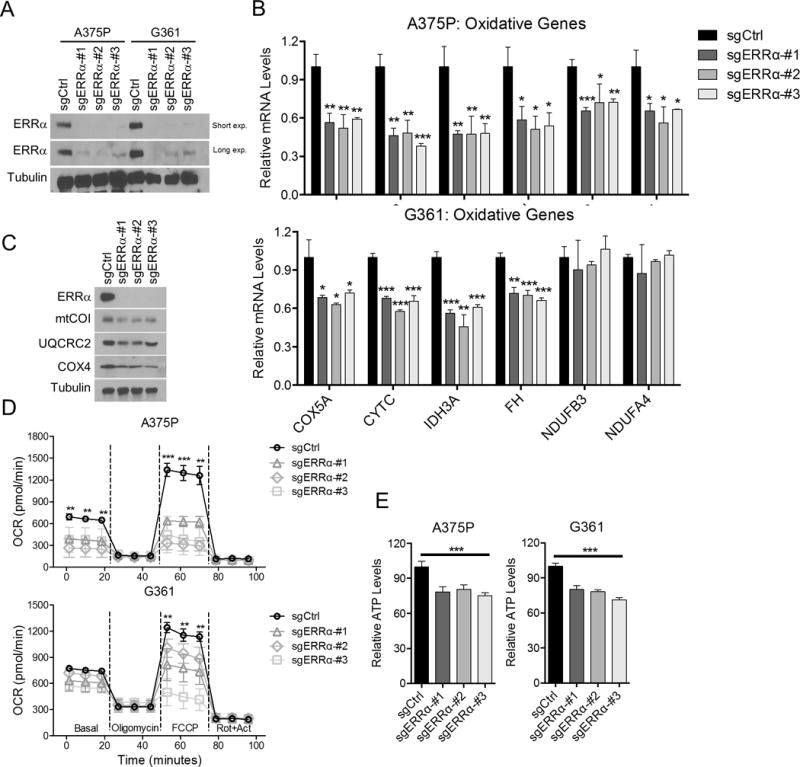
A. Immunoblotting of melanoma cells upon ERRα depletion by CRISPR. Small guide control vector: sgCtrl; small guide RNA for ERRα: sgERRα. B. Expression of mitochondrial related genes at the mRNA level in melanoma cells upon ERRα depletion. C. Immunoblotting of mitochondrial related genes in ERRα-depleted melanoma cells. D. Mitochondrial activity of melanoma cells with ERRα depletion presented as oxygen consumption rate (OCR) measured by seahorse flux assay. E. Intracellular ATP levels in cells with ERRα depletion. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by t-test (B) or one-way ANOVA (D, E).
In addition to increasing mitochondrial bioenergetic metabolism, PGC1α also regulates ROS detoxification capacity in melanoma cells (2,4). In contrast to PGC1α knockdown, however, depletion of ERRα did not affect the levels of PGC1α -controlled antioxidant response (Fig. S3A, C, D), suggesting that ERRα is not involved in the maintenance of cellular ROS scavenging. Importantly, targeted deletion of ERRα did not alter expression of the genes promoting metastatic spread of melanoma cells which are controlled by PGC1α (Fig. S3B), and we could not detect any significant differences in the resulting migratory ability (Fig. S3E). Although PGC1α overexpression was able to block the migration of invasive melanoma cells A2085, depletion of ERRα in these cells did not cause significant difference (Fig. S3F), further supporting the notion that ERRα is not involved in the regulation of metastasis by PGC1α. Collectively, our data indicate that ERRα preferentially mediates the mitochondrial functions of PGC1α in melanoma cells, but not contribute to ROS detoxification or affect the invasive/metastatic functions. Consequently, and similar to PGC1α, ERRα supports mitochondrial oxidative metabolism in melanoma cells, but PGC1α has broader functions.
Depletion of ERRα selectively inhibits the growth of PGC1α -positive but not -negative melanomas
We have previously found that PGC1α -positive melanoma cells depend on mitochondrial oxidative metabolism to maintain their growth and survival (2,3). Because ERRα like PGC1α also participates in control of mitochondrial bioenergetics, we determined whether ERRα supports melanoma proliferation. Targeted suppression of ERRα function using CRISPR/Cas9, or alternatively shRNA, significantly dampened in vitro proliferation of PGC1α -positive melanoma cells, including A375P, G361, SK-MEL-30, MeWo and KO29A (Fig. 3A and S4A, B). In addition, when implanted subcutaneously in athymic (nude) mice, PGC1α-positive melanoma cells with engineered ERRα deficiency displayed significantly slower tumor growth (Fig. 3B), consistent with their reduced Ki67 staining (Fig. S4C). Importantly, these growth suppressive effects of ERRα depletion were specific to melanoma cells with heightened PGC1α expression because melanoma cells with low expression, such as A375, A2058 and M14, were essentially unaffected by targeted deletion of ERRα (Fig. 3C-E and S4D). Similarly, A375P cells selected to grow with shRNA against PGC1α were essentially insensitive to ERRα deletion (Fig. S4E), indicating that the proliferative defects following ERRα depletion are dependent on PGC1α. Collectively, ERRα supports growth of melanomas with elevated PGC1α expression, but is dispensable in those with low PGC1α. In contrast to PGC1α depletion, however, ERRα deficiency did not induce accumulation of cellular ROS, which has been demonstrated to cause stabilization of the master glycolytic regulator HIF1α (1), suggesting that targeting ERRα is less likely to trigger compensatory metabolic reprogramming.
Figure 3. Depletion of ERRα compromises the growth of human melanomas.
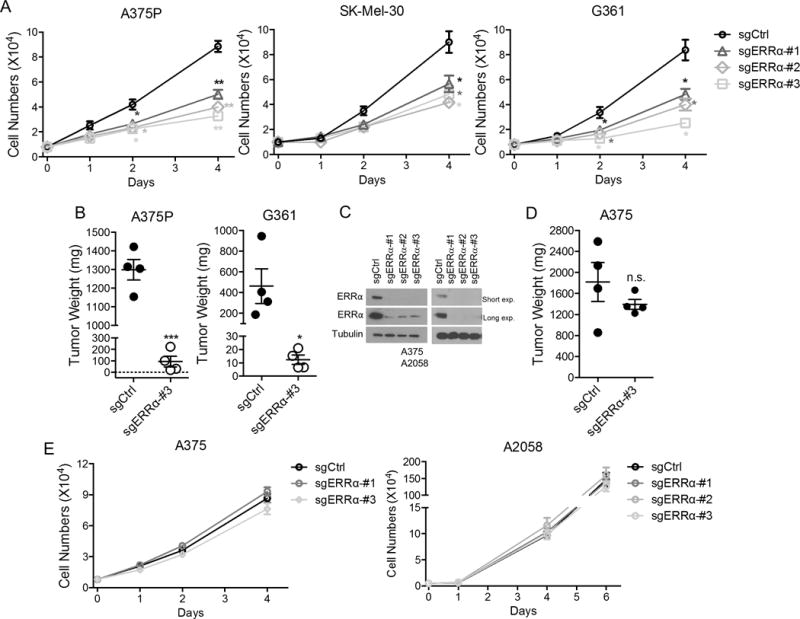
A. Growth curve of various PGC1α -positive melanoma cell lines with ERRα depletion. B. End-point tumor weight of PGC1α-positive melanoma cells with ERRα depletion after inoculated into nude mice. Data were presented as mean with SD. C. Immunoblotting of PGC1α-negative melanoma cells upon ERRα depletion by CRISPR. D. Growth curve of PGC1α -negative melanoma cell lines with ERRα depletion. E. End-point tumor weight of PGC1α-melanoma cell line A375 with ERRα depletion in nude mice. *P < 0.05, **P < 0.01, ***P < 0.001 and n.s. (not significant) as determined by t-test if not otherwise indicated.
Suppression of ERRα activity phenocopies ERRα depletion in melanoma cells
The ligand binding domain of ERRα is amenable for therapeutic intervention. To this end, Compound 29 (Cpd29) acts as an ERRα reverse agonists (antagonist), which interferes with the interaction between ERRα and PGC1s to block transcriptional activation (17). Considering that melanoma cells with high PGC1α expression were particularly susceptible to genetic depletion of ERRα, we examined whether ERRα could be pharmacologically targeted using the Cpd29 compound. Similar to ERRα depletion, inhibition of ERRα activity by Cpd29 caused a dose-dependent downregulation of ERRα/PGC1α target genes involved in mitochondrial oxidative metabolism (Fig. 4A, B), leaving the PGC1α-regulated antioxidant and metastasis promoting programs gene sets largely unaffected (Fig. S5A, B). Specifically, depletion of ERRα in the PGC1α-positive A375P cells rendered these cells insensitive to the inhibitory effects of Cpd29 (Fig. S5C) In addition, Cpd29 treatment affected oxygen consumption activity (Fig. 4C), lowered levels of intracellular ATP (Fig. 4D), and importantly, decreased cell proliferation (Fig. 4E and Fig. S5D). Taken together, pharmacological interference with ERRα function in vitro largely recapitulates ERRα depletion in PGC1α-expressing melanoma cells.
Figure 4. Inhibition of ERRα activity phenocopies ERRα depletion in melanoma cells.
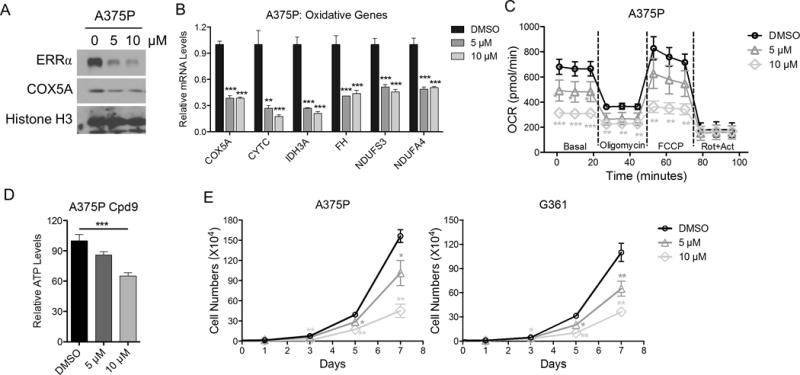
A. Immunoblotting of melanoma cells upon treatment with ERRα antagonist Cpd29. B. Expression of oxidative genes at the mRNA level in melanoma cells upon ERRα antagonist Cpd29. C. Mitochondrial activity of melanoma cells treated with ERRα antagonist Cpd29 as measured by seahorse flux assay. D. Intracellular ATP levels in cells treated with Cpd29. E. Growth curve of various melanoma cell lines treated with Cpd29. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by t-test (B, E) or one-way ANOVA (C, D).
Pharmacologic inhibition of ERRα activity compromises melanoma growth in vivo
Based on the results of pharmacologic inhibition of ERRα on melanoma proliferation in vitro, we next assessed whether the antagonist Cpd29 could affect tumor growth in vivo. To this end, we established subcutaneous xenografts from the PGC1α expressing melanoma cells G361 and MeWo, and determined their sensitivity to Cpd29 treatment. Following clearly palpable subcutaneous tumor growth (approx. 100mm) in athymic (nude) mice, Cpd29 or vehicle was administered by oral gavage six times a week. Consistent with the in vitro results, Cpd29 administration significantly blocked the tumors growth in vivo (Fig. 5A, B) without affecting the body weight (Fig. S6A). In agreement with reduced tumor growth, the Ki67 staining was also lower (Fig. S6B), underscoring the efficacy of ERRα inhibition to suppress melanoma growth. These results suggest that ERRα might constitute a valuable therapeutic target to treat melanoma tumors with elevated PGC1α expression.
Figure 5. Inhibition of ERRα activity suppresses melanoma growth in vivo.
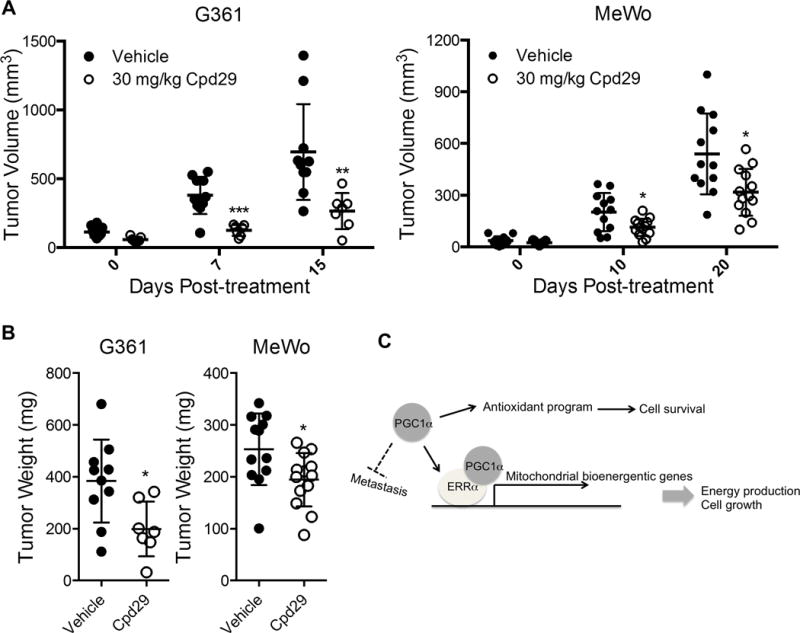
A. Growth curve of melanomas in nude mice upon treatment with 30 mg/kg ERRα antagonist Cpd29 (G361: n=10 for vehicle, n=7 for Cpd29; MeWo: n=12 for vehicle, n=13 for Cpd29). Data were presented as mean with SD. B. End-point tumor weight of melanoma xenografts upon Cpd29 treatment. Data were presented as mean with SD. C. A schematic model depicting the functional roles of ERRα in the mediating of PGC1α effects in melanoma cells. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by t-test if not otherwise indicated.
DISCUSSION
Melanoma is recognized as the deadliest among the most common skin cancers because of its highly aggressive nature, as well as its virtual resistance to chemotherapy (25). Despite recent advances in clinical management of melanoma using oncogene-targeted and immune checkpoint therapies, the genetic heterogeneity of melanoma tumors enables treatment resistance that ultimately blunts long-term benefit for patients. To this end, inherent metabolic dependencies in cancer cells represent additional means for therapeutic exploit that may favorably combine with existing anti-cancer precision medicines to extend patient benefit (26,27). To this end, a subset of melanomas are defined by heightened expression of the transcriptional coactivator PGC1α and that is characterized by functional dependence on mitochondrial oxidative metabolism for their growth and survival (2,3). However, we and others recently documented that PGC1α also exerts effects on suppressing metastatic spread, and consequently, PGC1α does not represent an advantageous target for therapeutic intervention (5). Using an unbiased approach, we have identified ERRα as a key mediator of PGC1α-dependent mitochondrial oxidative phosphorylation and growth in melanoma cells. Although the amount of ERRα peptides was ranking fourth in the proteomic list, considering the relatively smaller size of ERRα protein (423 amino acids vs. 3,859 for TRRAP, 4,128 for DNA-PK, and 937 for AP2B1), ERRα becomes the most enriched one if normalized to molecular weight. Thus, in melanomas with heightened expression of PGC1α, ERRα offers a metabolic vulnerability that may be exploited as a therapeutic target.
PGC1α is a transcription coactivator that binds and facilitates the activation of multiple transcription factors and nuclear receptors, which in turn determine the PGC1α function in different cell types and conditions (6). ERRα is one of the integral transcription factors that binds to a defining set of nuclear-encoded mitochondrial biogenesis genes and utilizes PGC1α to increase their expression (9). In human melanoma cells, PGC1α activates mitochondrial bioenergetic metabolism, protects against oxidative stress and suppresses cellular mobility (2–5), but based on our data mainly employs ERRα to support mitochondrial bioenergetic programs. PGC1α depletion in melanomas not only causes acute energy deficit due to mitochondrial failure, but also cause an accumulation of cytotoxic ROS, which stabilizes HIF1α to attenuate mitochondrial utilization and increase glycolysis that are hallmarks of the Warburg metabolic shift (4). Given that suppression of ERRα does not significantly compromise the antioxidant systems, the compensatory glycolytic reprogramming associated with PGC1α depletion is likely to be prevented. Although PGC1α/ERRα complex in prostate cancers has been demonstrated to elicit a catabolic state that compromises metastatic spread (14), yet ERRα in melanoma cells seems to segregate these PGC1α functions with regard to metastatic regulation. Furthermore, while PGC1α triggers a metabolism-independent transcription profile that suppresses melanoma metastasis (5), our data indicates that ERRα-mediated control of genes promoting oxidative metabolism is not involved in this regulatory circuit; however, these ERRα regulated genes are required to support melanoma cell proliferation (Fig. 5C).
Considering that suppression of ERRα selectively compromises cellular bioenergetics but does not affect other PGC1α–dependent melanoma growth-promoting functions, ERRα represents a potential therapeutic target. A series of small-molecule antagonists have been developed to repress the activity of ERRα, and have been applied to both chronic metabolic disorders and cancers (17). As an example, the ERRα antagonist Cpd29 has recently been shown to be effective in some breast cancers either as a single agent or in combination with other chemotherapeutic drugs (12,18). In the case of melanoma, we observed that Cpd29 mimics the effects of ERRα genetic depletion: decreased mitochondrial gene expression, reduced mitochondrial respiration and lowered ATP production. Importantly, this decline in bioenergetic capacity translates into significant reduction in tumor progression in animal models, highlighting the efficiency of antagonizing ERRα activity in the therapy of melanomas.
There are subsets of multiple different tumor-types that largely depend on mitochondrial oxidative metabolism for their survival and progression (2,3,28–34). As an example, it has been shown that a small population of slow-cycling melanoma cells within the bulk tumor predominantly utilizes oxidative phosphorylation to fuel their intrinsic resistance to conventional chemotherapies such as cisplatin (35), suggesting that blocking mitochondrial respiratory through ERRα inhibition could enhance the efficacy of conventional melanoma chemotherapy. In addition, PGC1α -mediated mitochondrial biogenesis is involved in the compensatory adaptation to MAP kinase pathway suppression (3,31). Through improving mitochondrial catabolic programs generally in cells, PGC1α also plays an important role in promoting tumor-associated M2 macrophage polarization (36). Because most cytolytic lymphocytes, such as effector T cells and natural killer cells, preferably rely on glycolysis (37,38), suppression of ERRα might therefore modify the microenvironment through decreasing M2 macrophages, while leaving cytolytic lymphocytes unaffected. Accordingly, ERRα inhibition might consequently represent an approach to improve immune checkpoint treatments for cancer treatment, acting directly by inhibiting the growth of oxidative phosphorylation addicted tumors, such as melanomas with heightened PGC1α expression, and by altering the balance between tumor cytolytic lymphocytes and immune-suppressing M2 macrophages. Nonetheless, we have here focused our attention to the cell intrinsic effects on inhibiting melanoma tumor growth, and consequently, future studies will be needed to examine therapeutic effects in the genuine tumor microenvironment.
In summary, our results indicate that the orphan nuclear receptor ERRα selectively mediates the growth-supporting bioenergetic functions of PGC1α. Hence, ERRα constitutes an appealing therapeutic target used to treat melanomas that are addicted to mitochondrial oxidative phosphorylation.
Supplementary Material
Acknowledgments
We thank Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Rodent Histopathology Core, which provided histology-processing services. We also thank the Nikon Imaging Center at Harvard Medical School for the use of microscopy. We thank members of the Puigserver lab for important discussions about this project. Research was supported by funds from NIH R01CA181217 and DFCI Claudia Adams Barr Award (both to P.P.); and from Outrun the Sun, Inc. 2016 National Research Scholar Program (to C.L.).
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Luo C, Widlund HR, Puigserver P. PGC-1 coactivators: shepherding tumors’ mitochondrial biogenesis. Trends in Cancer. 2016;2:619–631. doi: 10.1016/j.trecan.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer cell. 2013;23(3):287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer cell. 2013;23(3):302–15. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim JH, Luo C, Vazquez F, Puigserver P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer research. 2014;74(13):3535–45. doi: 10.1158/0008-5472.CAN-13-2893-T. [DOI] [PubMed] [Google Scholar]
- 5.Luo C, Lim JH, Lee Y, Granter SR, Thomas A, Vazquez F, et al. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature. 2016 doi: 10.1038/nature19347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocrine reviews. 2003;24(1):78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 7.Audet-Walsh E, Giguere V. The multiple universes of estrogen-related receptor alpha and gamma in metabolic control and related diseases. Acta pharmacologica Sinica. 2015;36(1):51–61. doi: 10.1038/aps.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaillard S, Dwyer MA, McDonnell DP. Definition of the molecular basis for estrogen receptor-related receptor-alpha-cofactor interactions. Molecular endocrinology. 2007;21(1):62–76. doi: 10.1210/me.2006-0179. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, et al. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(17):6472–7. doi: 10.1073/pnas.0308686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deblois G, St-Pierre J, Giguere V. The PGC-1/ERR signaling axis in cancer. Oncogene. 2013;32(30):3483–90. doi: 10.1038/onc.2012.529. [DOI] [PubMed] [Google Scholar]
- 11.McGuirk S, Gravel SP, Deblois G, Papadopoli DJ, Faubert B, Wegner A, et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer & metabolism. 2013;1(1):22. doi: 10.1186/2049-3002-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deblois G, Smith HW, Tam IS, Gravel SP, Caron M, Savage P, et al. ERRalpha mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nature communications. 2016;7:12156. doi: 10.1038/ncomms12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Audet-Walsh E, Papadopoli DJ, Gravel SP, Yee T, Bridon G, Caron M, et al. The PGC-1alpha/ERRalpha Axis Represses One-Carbon Metabolism and Promotes Sensitivity to Anti-folate Therapy in Breast Cancer. Cell reports. 2016;14(4):920–31. doi: 10.1016/j.celrep.2015.12.086. [DOI] [PubMed] [Google Scholar]
- 14.Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nature cell biology. 2016;18(6):645–56. doi: 10.1038/ncb3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deblois G, Giguere V. Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nature reviews Cancer. 2012;13(1):27–36. doi: 10.1038/nrc3396. doi nrc3396 [pii]10.1038/nrc3396. [DOI] [PubMed] [Google Scholar]
- 16.Riggins RB, Mazzotta MM, Maniya OZ, Clarke R. Orphan nuclear receptors in breast cancer pathogenesis and therapeutic response. Endocrine-related cancer. 2010;17(3):R213–31. doi: 10.1677/ERC-10-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patch RJ, Searle LL, Kim AJ, De D, Zhu X, Askari HB, et al. Identification of diaryl ether-based ligands for estrogen-related receptor alpha as potential antidiabetic agents. Journal of medicinal chemistry. 2011;54(3):788–808. doi: 10.1021/jm101063h. [DOI] [PubMed] [Google Scholar]
- 18.Park S, Chang CY, Safi R, Liu X, Baldi R, Jasper JS, et al. ERRalpha-Regulated Lactate Metabolism Contributes to Resistance to Targeted Therapies in Breast Cancer. Cell reports. 2016;15(2):323–35. doi: 10.1016/j.celrep.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheng J, Luo C, Jiang Y, Hinds PW, Xu Z, Hu GF. Transcription of angiogenin and ribonuclease 4 is regulated by RNA polymerase III elements and a CCCTC binding factor (CTCF)-dependent intragenic chromatin loop. The Journal of biological chemistry. 2014;289(18):12520–34. doi: 10.1074/jbc.M114.551762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatting M, Rines AK, Luo C, Tabata M, Sharabi K, Hall JA, et al. Adipose Tissue CLK2 Promotes Energy Expenditure during High-Fat Diet Intermittent Fasting. Cell metabolism. 2017;25(2):428–37. doi: 10.1016/j.cmet.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jena N, Sheng J, Hu JK, Li W, Zhou W, Lee G, et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia. 2016;30(5):1033–43. doi: 10.1038/leu.2015.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foley CJ, Luo C, O’Callaghan K, Hinds PW, Covic L, Kuliopulos A. Matrix metalloprotease-1a promotes tumorigenesis and metastasis. The Journal of biological chemistry. 2012;287(29):24330–8. doi: 10.1074/jbc.M112.356303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo C, Pietruska JR, Sheng J, Bronson RT, Hu MG, Cui R, et al. Expression of oncogenic BRAFV600E in melanocytes induces Schwannian differentiation in vivo. Pigment cell & melanoma research. 2015;28(5):603–6. doi: 10.1111/pcmr.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo C, Sheng J, Hu MG, Haluska FG, Cui R, Xu Z, et al. Loss of ARF sensitizes transgenic BRAFV600E mice to UV-induced melanoma via suppression of XPC. Cancer research. 2013;73(14):4337–48. doi: 10.1158/0008-5472.CAN-12-4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annual review of pathology. 2009;4:551–79. doi: 10.1146/annurev.pathol.3.121806.151541. [DOI] [PubMed] [Google Scholar]
- 26.Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nature biotechnology. 2012;30(7):671–8. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475(7355):231–4. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell cycle. 2010;9(17):3506–14. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. The FEBS journal. 2007;274(6):1393–418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 30.Denise C, Paoli P, Calvani M, Taddei ML, Giannoni E, Kopetz S, et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget. 2015;6(39):41706–21. doi: 10.18632/oncotarget.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. The Journal of clinical investigation. 2016;126(5):1834–56. doi: 10.1172/JCI82661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mouron S, et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell reports. 2016;15(12):2705–18. doi: 10.1016/j.celrep.2016.05.052. [DOI] [PubMed] [Google Scholar]
- 33.Luo C, Puigserver P, Widlund HR. Breaking BRAF(V600E)-drug resistance by stressing mitochondria. Pigment cell & melanoma research. 2016;29(4):401–3. doi: 10.1111/pcmr.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo C, Puigserver P. Stem cells: Dietary fat promotes intestinal dysregulation. Nature. 2016;531(7592):42–3. doi: 10.1038/531042a. [DOI] [PubMed] [Google Scholar]
- 35.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer cell. 2013;23(6):811–25. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell metabolism. 2006;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends in immunology. 2015;36(2):71–80. doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finlay DK. Metabolic regulation of natural killer cells. Biochemical Society transactions. 2015;43(4):758–62. doi: 10.1042/BST20150116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.