

Abstract
The heterotrimeric cardiac troponin complex is a key regulator of contraction and plays an essential role in conferring Ca2+ sensitivity to the sarcomere. During ischemic injury, rapidly accumulating protons acidify the myoplasm, resulting in markedly reduced Ca2+ sensitivity of the sarcomere. Unlike the adult heart, sarcomeric Ca2+ sensitivity in fetal cardiac tissue is comparatively pH insensitive. Replacement of the adult cardiac troponin I (cTnI) isoform with the fetal troponin I (ssTnI) isoform renders adult cardiac contractile machinery relatively insensitive to acidification. Alignment and functional studies have determined histidine 132 of ssTnI to be the predominant source of this pH insensitivity. Substitution of histidine at the cognate position 164 in cTnI confers the same pH insensitivity to adult cardiac myocytes. An alanine at position 164 of cTnI is conserved in all mammals, with the exception of the platypus, which expresses a proline. Prolines are biophysically unique because of their innate conformational rigidity and helix-disrupting function. To provide deeper structure-function insight into the role of the TnC-TnI interface in determining contractility, we employed a live-cell approach alongside molecular dynamics simulations to ascertain the chemo-mechanical implications of the disrupted helix 4 of cTnI where position 164 exists. This important motif belongs to the critical switch region of cTnI. Substitution of a proline at position 164 of cTnI in adult rat cardiac myocytes causes increased contractility independent of alterations in the Ca2+ transient. Free-energy perturbation calculations of cTnC-Ca2+ binding indicate no difference in cTnC-Ca2+ affinity. Rather, we propose the enhanced contractility is derived from new salt bridge interactions between cTnI helix 4 and cTnC helix A, which are critical in determining pH sensitivity and contractility. Molecular dynamics simulations demonstrate that cTnI A164P structurally phenocopies ssTnI under baseline but not acidotic conditions. These findings highlight the evolutionarily directed role of the TnI-cTnC interface in determining cardiac contractility.
Introduction
Cardiac contractility requires tightly regulated and highly orchestrated structural rearrangements between multiple proteins within the sarcomere assembly (1). At the center of this chemo-mechanical regulation is the troponin complex, which ultimately directs the molecular motor of the sarcomere, myosin, to produce force and motion (2, 3). Genetic and environmental perturbations can markedly alter these intermolecular interactions, resulting in depressed cardiac myocyte function and declining heart pump function (4). Consequently, gaining a deeper understanding of the structure-function relationship in sarcomeric proteins is critical to developing strategies to effectively redress the ischemic and failing heart.
The cardiac troponin complex is a heterotrimeric protein assembly consisting of the tropomyosin-binding subunit cardiac troponin T, which anchors the complex to the actin thin filament via tropomyosin; the Ca2+-responsive subunit cardiac troponin C (cTnC); and the allosteric regulator of sarcomeric function cardiac troponin I (cTnI). In systole, Ca2+ is released from the sarcoplasmic reticulum, causing a rapid rise in intracellular concentration (5). Ca2+ then binds to the N-terminus of cTnC, initiating a series of concerted conformational changes in the troponin complex. As Ca2+ binds, conformational changes in cTnC reveal a patch of hydrophobic residues in cTnC, allowing the cTnI switch peptide (residues 147–163) to then bind to initiate activation of the sarcomere (6). Concurrent with the cTnI switch peptide binding the cTnC N-terminal hydrophobic pocket, the cTnI inhibitory peptide (residues 128–146) and the cTnI mobile domain (residues 172–210) shift away from the actin filament, thereby allowing tropomyosin to azimuthally rotate and expose myosin strong-binding sites (7, 8). In diastole, myoplasmic Ca2+ concentration drops precipitously as Ca2+ is re-sequestered in the sarcoplasmic reticulum or extruded across the sarcolemma. As a consequence, the dynamic TnC-TnI protein interactions in systole are reversed, and the cTnI inhibitory peptide binds to the actin filament to inhibit strong myosin cross-bridge formation. Collectively, these changes constitute the molecular switch function of cTnI (9).
The allosteric function of cTnI as both a sarcomere inhibitory ligand and regulator of cardiac contractility poises it to be a critical effector of contractility. Here, cTnI interacts with key regulatory proteins in the thin filament: cTnC, cardiac troponin T, actin, and tropomyosin, highlighting its crucial position in the beat-to-beat regulation of cardiac muscle (10, 11, 12). Because of its essential role in mediating thin filament regulation, it is also highly sensitive to disruptions in the local biochemical milieu and genetic mutations. The majority of human disease-causing mutations in cTnI are located in the C-terminal half of the protein (13). This demonstrates the critical role of the C-terminal half of cTnI in determining cardiac lusitropy and diastolic function of the heart (14).
During acute ischemia, as oxygen tension drops, protons rapidly accumulate in the myoplasm to acidify the sarcomere, resulting in markedly reduced Ca2+ sensitivity of the sarcomere (15). Unlike its adult counterpart, myofilament Ca2+ sensitivity of fetal cardiac tissue is comparatively pH insensitive. Targeted replacement of the adult cTnI isoform with the fetal troponin I (ssTnI) isoform has been shown to confer myofilament-based pH insensitivity to adult cardiac tissue (16). Troponin I chimeric and structure-function studies have revealed the origin of insensitivity to be histidine 132 in ssTnI (17). Substitution of histidine at the cognate position 164 in cTnI confers marked pH insensitivity to adult tissue (18, 19, 20, 21).
Interestingly, the presence of an alanine at position 164 of cTnI is conserved across mammals, with a single notable exception: Ornithorhynchus anatinus (platypus), which instead harbors a proline at this position (13). Phylogenetically, the egg-laying mammals branched off from their viviparous counterparts close to 200 million years ago to give rise to the modern order Monotremata (22). The only five extant monotremes are the platypus and four species of echidna. Consequently, the platypus represents an interesting divergence in mammalian evolution in terms of troponin molecular switch structure-function (23, 24). We seek to examine this intriguing evolutionary substitution in troponin to further elucidate cTnI structure-function and its role as a determinant of cardiac contractility.
The high level of conservation in the cTnI switch peptide, and especially helix 4, is indicative of its critical role as a determinant of contractility. Accordingly, the introduction of a biophysically unique amino acid in this region merits further investigation. Proline substitutions are of special interest because of their structural implications. The distinctive cyclic structure of proline underlies its role as a “helix breaker” because of its innate ability to disrupt the α-helical protein motif (25). The principal goal of this study was to assess the biophysical consequences of the cTnI-A164P evolutionary stepping stone in the context of isolated adult cardiac myocytes by acute gene transfer. In complement, atomistic molecular dynamics (MD) simulations were constructed to better understand how cardiac troponin proteins have been modified throughout evolution to accommodate environmental and physiological requirements of cardiac muscle. The new—to our knowledge—findings detailed here enhance our understanding of the TnC-TnI interface as a central regulator of cardiac contractility.
Materials and Methods
Adenovirus generation and purification
We used a pGEM3Z vector containing cTnI-Flag and the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) to generate site-directed A164P-Flag mutants according to the manufacturer’s protocol. Mutated DNA was sequenced before subcloning of mutant cTnI complementary DNA into an pDC316 adenovirus shuttle vector. All DNA sequences were verified by overlapping sequence runs. Recombinant serotype-5 adenoviruses were generated by cotransfection of an adenovirus genome cosmid into HEK293 cells. A164P cTnI-Flag adenovirus plaques were isolated by a noble agar overlay then isolated, amplified, and purified by a cesium chloride gradient. Adenoviruses for cTnI-Flag and A164H cTnI-Flag were generated previously and used as controls here (21).
Immunoblot detection
Adult cardiac myocytes on day 3 after transduction were scraped off glass coverslips and placed in a Laemmli sample buffer with a protease inhibitor cocktail (#S8220; Sigma-Alrdich, St. Louis, MO). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane for immunodetection. After blocking in 5% milk in Tris-buffered saline, membranes were probed with a cTnI-specific antibody (4C2, 1:4000; AbCam, Cambridge, MA). Indirect immunodetection was carried out using a fluorescently labeled anti-mouse secondary antibody (Infrared Dye 680nm-conjugated, 1:10,000; Li-Cor, Lincoln, NE). Western blot analysis was accomplished using the infrared imaging system Odyssey (Li-Cor), and images were analyzed using Odyssey software v3.0.
Ventricular myocyte isolation and gene transfer
Adult rat ventricular myocyte isolation was performed as previously described (14, 21, 26). Adult female Sprague-Dawley rats were anesthetized by inhalation of isoflurane followed by intraperitoneal injection of heparin (1500 U/kg) and Fatal-Plus (150 U/kg) (Vortech Pharmaceuticals, Dearborn, MI). After anesthesia, rats were euthanized by exsanguination and the heart was rapidly excised, cannulated via the aorta, and perfused retrogradely with collagenase in Ca2+-free Krebs buffer. After enzymatic digestion, the cardiac ventricles were mechanically triturated, and viable myocytes were fractionated and pooled. Cardiac myocytes were plated on laminin-coated glass coverslips (2 × 104 myocytes per coverslip) and cultured in M199 media (Sigma-Aldrich) supplemented with 26.2 mmol/L sodium bicarbonate, 0.02% bovine serum albumin, and 50 U/mL penicillin-streptomycin, with the pH adjusted to 7.4 by NaOH from the initially more acidic media. Additionally, insulin (5 μg/mL), transferrin (5 μg/mL), and selenite (5 ng/mL) (I1884; Sigma-Aldrich) were added. One hour after plating, nonadherent cells were aspirated, and recombinant adenoviruses expressing cTnI-Flag isoforms were applied to the remaining cells as previously described (14, 21, 26). Cells were cultured for 72 h to provide sufficient time for stoichiometric replacement of the troponin I isoforms. All animal experiments were performed in compliance with University of Minnesota Institutional Animal Care and Use Committee animal protocols.
Sarcomere and calcium dynamics
Sarcomere length measurements were performed using an IonOptix calcium and contractility system (IonOptix, Westwood, MA), wherein laminin-coated coverslips plated with isolated adult rat ventricular cardiac myocytes were bathed in M199 media (#M3274; Sigma-Aldrich) at 37°C. Myocytes were stimulated at 25 volts by field stimulation using platinum electrodes at a pacing frequency of 1 Hz. Sarcomere length and Ca2+ transients were measured in real time and averaged over 10–20 contractile cycles. Ca2+ transients were measured simultaneously using a fluorescent Ca2+ indicator, Fura-2-acetoxymethyl ester (#F1221; Thermo Fisher Scientific, Waltham, MA), loaded at 1 μM for 10 min and then de-esterified in M199 before measurement as previously described (14, 21, 26).
For acidosis experiments, electrical field stimulation was paused, and M199 basal media on cells were aspirated and replaced with M199 media with a pH adjusted to 6.2 by NaOH from the initially more acidic media. After media were replaced, cells were allowed to come to equilibrium over a period of 10 min before field stimulation was resumed. Cells were then measured again with a pacing frequency of 1 Hz over 10–20 contractile cycles, and sarcomere dynamics were averaged.
MD simulations and interatomic distance calculations
The MD simulations were conducted as previously described (21, 27). Briefly, the starting structures were obtained from the Protein Data Bank (PDB): 1J1E chains D and F for cTnC (residues 1–90) and cTnI (residues 148–174), respectively. For ssTnI (residues 115–140) PDB: 1YTZ was used, and ssTnI was modeled onto cTnC using Schrödinger Maestro (28). All mutations (A164H, A164P) and missing amino acid side chains of G50 and Q51 of cTnC were performed via standard amino acid substitution utilizing the Schrodinger’s Prime module that takes into account a rigorous conformation search of side-chain rotamers only and local structure optimization. The approach does not take into account backbone rotamers that can potentially lead to repacking of the hydrophobic core. Missing hydrogen atoms were added to the final model, and energy was minimized using the optimized potentials for liquid simulations 2005 force field (29).
The general continuum electrostatics method, which is commonly implemented to calculate the energetics of proton transfer and the corresponding pKa within a thermodynamic cycle, is described in greater detail elsewhere (30, 31). In this study, calculations of the theoretical pKa were carried out using the web-based H++ server that implements the linearized Poisson-Boltzmann implicit solvent model (32). For these calculations, the protein was treated as a low-dielectric medium in which εin = 40, and the surrounding solvent was assigned a high-dielectric constant of εout = 80. The choice of a low-protein dielectric constant for pKa calculation was previously discussed (33). The Debye-Hückel screening parameter κ was used to set the electrostatic screening effects of the salt concentration corresponding to physiological conditions of 0.145 M. The pKa values shown in Fig. S2 were derived by calculating the intrinsic pKa, which corresponds to the pKa of the group of interest wherein all of the other titratable amino acids are maintained in the neutral state (30).
The imidazole groups of all histidines in the structure were ionized according to the calculated pKas. All simulations were carried out using NAMD version 2.6 with a CHARMM27 protein force field and a transferable intermolecular potential and a three point (TIP3P) water model (34). Each protein complex was solvated in a rectangular box with a 15 Å water buffer from the protein. Na+ and Cl− counterions were added at 5 Å from the box boundary to neutralize the total charge of the system. Each system was initialized by a 5000-step conjugate gradient energy minimization, with protein heavy atoms restrained at 50 kcal/(mol · Å2). The restraint system was gradually heated from 25 to 300 K at 25 K increments at every 10 ps interval for 100 ps, followed by a 100 ps equilibration with gradual removal of the heavy atoms’ restraint at every 10 ps interval under a number volume temperature condition. The final unrestrained equilibration was carried out for 100 ps, followed by 40 ns of production simulation under 1 atm and 300 K NPT condition. The simulations were carried out under a periodic boundary condition using particle mesh Ewald (35). The SHAKE method was employed to restrain all hydrogen bonds’ lengths (36). A 2 fs time step was used with coordinates saved at 1 ps time intervals, resulting in a total of 40,000 configurations for each simulation. Three to five 40 ns simulations were carried out for each of the modeled complexes by random initialization of the starting velocities to improve sampling of conformational states. All calculated distances were evaluated in angstrom unit over the entire 40 ns of the simulation. For all complexes, the distance was evaluated between cTnI R171 (NE2) or ssTnI R139 (NE2) and cTnC E15 (OE2). Cα Root mean-square deviation was used to determine the relative changes of the protein structure over the course of the simulation (Fig. S3). These simulations were conducted using computational resources from the University of Minnesota Supercomputing Institute.
Free-energy perturbation calculation
The relative binding free energy of the Ca2+ ion between the wild-type (WT) and its mutant ΔΔ was evaluated using the free-energy perturbation (FEP) method that has been used previously for the studies of charges in solution (37, 38) and proteins (39). Briefly, Molaris calculates the energy of changing the charge of a calcium ion by changing the charge in small steps from a fully charged +2 to 0 while allowing the coordinating atoms around the calcium ion to undergo reorganization. Thus, FEP is a measure of the free-energy change associated with the change in calcium ion discharging as a proxy for TnC-calcium binding affinity.
We calculated the binding free energy, ΔG, associated with ionizing a charge within its water or protein-water environment. This involves the adiabatic charging process by which the charge (Q) of the Ca2+ ion is gradually changed from Q = 0 → Q = Q0, where Q0 = +2 for the Ca2+ ion. The free energy is given by the following equation:
where εm is the mapping potential and λm is the mapping parameter. The complete charging process involved 11 mapping steps, with each mapping step performed at 300 K for 20 ps with a step size of 1 fs. Harmonic distance constraints of 200 kcal/(mol · Å2) were applied on the cTnC site II Ca2+ with its chelating residues D65, D67, D73, E76, and S69, and the charge of the Ca2+ ion was systematically changed from 0 to +2. The use of distance restraint between the Ca2+ ion and the chelating residues prevents dissociation of the Ca2+ ion from the binding site when the ionic charge is perturbed to zero. The binding free energy was averaged over the multiple simulation snapshots for the given isoform. Four independent frames from 10 to 40 ns were analyzed for each simulation on the three to five simulations run per variant. The relative association binding free energy (kilocalories per mole) was then evaluated by the difference between the WT and its mutant. The FEP calculations were conducted within the Molaris 9.09 software modeling package (40).
Results
cTnI replacement and incorporation
To study the structure-function relationship of the interaction between the cTnI switch peptide and the N-terminus of cTnC as it relates to isolated myocyte contractility, we used an ex vivo cellular model. Recombinant adenoviruses were generated to express single-amino-acid substitutions at position 164 of cTnI in the switch peptide domain. The three amino acid substitutions highlighted in this study represent distinct switch peptide alterations during chordate evolution: cTnI alanine/histidine/proline.
To study the effects of these cTnI substitutions, adult rat cardiac myocytes were transduced with recombinant adenoviruses encoding WT cTnI, cTnI A164H, or cTnI A164P substitutions in the context of the adult cTnI isoform. Adenovirus transduction of adult myocytes is well established as a nearly 100% efficient system that results in stoichiometric replacement of the endogenous cTnI (14, 16, 41, 42). All cTnI vectors were Flag-tagged to allow tracking compared to the endogenous cTnI. We have previously demonstrated that this epitope tagging method does not alter myocyte functionality (14, 27, 43). At 72 h after viral transduction, adult cardiac myocytes were harvested and replacement was quantified by Western blot densitometry (Fig. 1). Here, cTnI gene transfer showed stoichiometric replacement of the endogenous protein with the Flag-tagged cTnI substitutions (Fig. 1 A). All of the cTnI substitutions had 45–60% replacement, and the extent of sarcomeric replacement was not significantly different between groups (Fig. 1 B). Previous studies have shown that this experimental approach is targeted to TnI replacement and does not affect the content or isoform profile of other key sarcomeric proteins (14, 21, 41, 44, 45).
Figure 1.

Replacement and incorporation of the adenovirus-transduced adult cardiac myocytes. (A) Shown is a representative Western blot of adult rat ventricular myocytes on day 3 after transduction, probed with a TnI antibody. Expected sizes are denoted on the left; all adenovirus-transduced cTnI isoforms are Flag tagged. All protein samples are from the same blot with the intervening lanes removed. (B) Shown is the densitometry of cTnI replacement based on a Western blot comparing exogenous (cTnI Flag band) to total cTnI (n = 3–5 for each group from two to three independent experiments). Mean ± standard error (SE) are presented.
Cardiac myocyte contractility
Sarcomere length dynamics in membrane-intact single adult rat cardiac myocytes transduced with cTnI substitutions were examined to determine the functional consequence of these single-amino-acid substitutions under baseline physiological conditions (pH 7.4) and during acidosis (pH 6.2). All measurements were made 72 h after gene transfer. Myocyte contractility is described here in terms of fractional shortening percentage—i.e., the percent change in sarcomere length from baseline that occurs during the average contractile cycle (Fig. 2, A and B). Under baseline physiological conditions, the cTnI A164P substitution resulted in increased contractile amplitude (Fig. 2, A and B). Substitution of cTnI A164P and cTnI A164H had no significant effect on the kinetics of contractility (Fig. S1).
Figure 2.

Sarcomere length and cytosolic calcium dynamics at physiologic baseline. (A and B) Shown are representative traces of sarcomere length dynamics, with normalized baseline sarcomere lengths shown as a percent change in length from baseline; a group summary is also given. n = 37–55 myocytes from four independent experiments for each group. ∗p < 0.05 from a one-way analysis of variance. (C) Shown is a representative Fura-2-acetoxymethyl ester fluorescence trace of a cTnI Flag adenovirus-transduced myocyte (solid line) and a cTnI A164P adenovirus-transduced myocyte (dotted line). (D) Peak amplitude of the calcium transient reveals no changes in the amplitude of the calcium transient; n = 6–12 myocytes from two different experiments for each group. Mean ± SE are presented, and results were not significant by Student’s t-test.
Under acidosis, we have previously demonstrated that A164H substitution significantly preserves contractility (17, 21, 27, 46, 47). Thus, to test whether the proline substitution at position 164 would lead to a similar pH insensitivity, myocytes were exposed to media at pH 6.2 and studied for sarcomere dynamics. Consistent with previous reports, substitution of cTnI A164H preserved contractility under acidosis (Fig. 3, B, D, and E). However, introduction of proline at position 164 did not protect myocytes from depressed contractility (Fig. 3, C–E). These contrasting contractile properties under acidosis suggest that the mechanisms of increased contractility between A164H and A164P are different.
Figure 3.

Sarcomere length dynamics at physiologic baseline and in acidosis. (A–C) Shown are representative traces of sarcomere length dynamics, with normalized baseline sarcomere lengths shown as a percent change in length from baseline (solid trace = pH 7.4, dotted track = pH 6.2). (D) pH 6.2 sarcomere length dynamics showing the fractional shortening summary are given. (E) Shown is the change in fractional shortening from pH 6.2 to pH 7.4. n = 37–55 myocytes from four independent experiments for each group. Mean ± SE are presented. ∗p < 0.05 from a one-way analysis of variance.
To elucidate the mechanism of the enhanced contractility caused by cTnI A164P substitution, we monitored intracellular Ca2+ cycling with the Ca2+-sensitive fluorescent dye Fura-2 in isolated rat myocytes transduced with either a cTnI A164P adenovirus or a cTnI WT adenovirus. Here, cTnI A164P caused no significant change in the peak Ca2+ transient amplitude (Fig. 2, C and D). The similar Ca2+ amplitudes are evidence that the positive inotropy caused by the cTnI A164P substitution at baseline is not due to a change in Ca2+ cycling or sarcoplasmic reticulum Ca2+ storage; instead this implicates alterations in sarcomere activation as a driving force behind the increased myocyte contractility.
MD simulations
To further probe the mechanism of the enhanced baseline contractility observed in myocytes transduced by cTnI A164P, we used MD simulations to gain atomistic-level structural insights into how cTnI A164P introduction could alter cTnI-cTnC interactions. Simulations were performed with the Ca2+-saturated cTnC with the WT cTnI, cTnI A164P, cTnI A164H, and ssTnI switch peptides under both histidine deprotonated (HSD) and histidine protonated (HSP) conditions, in which all histidines are either deprotonated or protonated, respectively. In this model system, we are able to probe the biophysical consequences of acidosis by altering the protonation state of only the histidine residues since they are the only amino acids with a pKa in the physiological operating range (pKa ≈ 6.0). Starting structures were truncated cTnC residues 1–90; fast skeletal Troponin I 115–140 from the Ca2+-bound chicken fast skeletal troponin crystal structure (PDB: 1YTZ), mutated computationally to generate ssTnI; and cTnI residues 148–174 from the Ca2+-bound human cTn crystal structure (PDB: 1J1E, chains D and F), mutated computationally to generate the cTnI A164P and A164H structures. We carried out three simulations with each of the three starting structures at random starting velocities to probe the possible three-dimensional conformational spaces the troponin structure could assume. Here, each simulation was run for 40 ns or until the simulation reached a stable conformation. Representative final stable structures of each TnI computational mutant showed the N-terminus (i.e., switch peptide) of the TnI peptide to be in generally the same conformation (Fig. 4). In comparison, the structures differed significantly at their C-termini (i.e., helix 4) and its interactions with the N-terminal lobe of cTnC (Fig. 4). In particular, the angle between the switch peptide and helix 4 differed considerably; in ssTnI HSD and HSP, helix 4 was disordered and followed in parallel with the A helix of cTnC. This is in contrast with cTnI and cTnI A164H HSD and HSP, in which the helix 4 of cTnI was at ∼90–100° relative to the cTnC A helix. The most striking finding from the MD simulations with cTnI A164P HSD is that the final stable structure is qualitatively similar to the ssTnI structure, in which helix 4 and the cTnC A helix are parallel. By contrast, in the cTnI A164P HSP stable structure, cTnI A164P helix 4 remained parallel with the cTnC A helix but kinked away from the N-terminus of cTnC and extended toward the cTnC B helix. Collectively, these findings indicate that the cTnI-cTnC interaction is significantly altered by the introduction of a proline at position 164 of cTnI and reveals a structural intermediate between ssTnI and cTnI, wherein helix 4 is highly ordered but maintains close proximity to the cTnC A helix.
Figure 4.

Atomistic ribbon structure representations of the protein structures at the end of the 40 ns simulation. Red is cTnC 1–90, blue is cTnI 148–174 or ssTnI 115–140, and yellow spheres represent the calcium ion bound to cTnC site II. (A) Shown are cTnI-cTnC starting structures from PDB: 1J1E with key motifs labeled; amino acid position 164 is highlighted in yellow for reference. (B) Shown are HSP (histidine protonated) and HSD (histidine deprotonated) final 40 ns ribbon structures for each of the cTnI structures. In the lower right of each ribbon structure is a simplified stick figure representation of the orientation between the cTnC A helix (red) and the TnI switch peptide and helix 4 (blue). To see this figure in color, go online.
Interatomic distances and the A helix–helix 4 interaction
In ssTnI-cTnC HSD and HSP simulations, helix 4 (residues 130–142) of ssTnI was found to be in an unstructured region of the peptide that interacts at multiple points with the cTnC A helix (residues 14–28). Notably ssTnI-cTnC HSD has a salt bridge between R139 of cTnI and E15 of cTnC, a cognate interaction that has been previously implicated as important in maintenance of contractility under acidosis (Fig. 5, A and C) (21). When the histidine at position 132 was protonated in the HSP simulation, the simulation showed the R139-E15 salt bridge was gradually replaced in favor of H132-E19 interaction (data not shown). Despite the loss of the R139-E15 salt bridge, helix 4 of ssTnI remained in close proximity with the A helix of cTnC (Fig. 4 B).
Figure 5.

Atomistic ribbon structures of TnI helix 4 (blue) interactions with cTnC (red) (A) A representative 40 ns frame of ssTnI cTnI HSD showing R139 of ssTnI interaction with cTnC E15 is given. The inset shows distances between E15 and R139. (B) A representative 40 ns frame of cTnI A164P HSD showing R171 of cTnI interaction with cTnC E15 is given. The inset shows distances between E15 and R171. (C) Shown is the calculated distance measurement from TnI R139/171 for ssTnI and cTnI A164P, respectively, to cTnC E15 during the 40 ns simulation. (D) The mean distance between ssTnI R139 or cTnI A164P R171 to cTnC E15 over the entire stable 40 ns of each simulation is given. Error bars are ±SE, and ∗p < 0.05 from a one-way analysis of variance. To see this figure in color, go online.
In cTnI A164P, the cognate amino acid to ssTnI R139 is R171. In the A164P-cTnC HSD simulation, cTnI A164P helix 4 formed multiple contact points with the cTnC A helix (Fig. 4 B), including a unique salt bridge between A164P R171 and both cTnC E15 and E19. These two cTnC glutamates were previously demonstrated as structurally significant determinants of contractility (Fig. 5, B and C) (21). This juxtaposes the A164P-cTnC HSP simulation wherein helix 4 was kinked away from the A helix of cTnC and as a result the mean distance was twofold greater between R171 and E15 under HSP conditions throughout the course of the simulation (Fig. 5 D).
Free energy of cTnC Ca2+ binding
To determine if the increased contractility caused by introduction of cTnI A164P was related to an alteration in Ca2+ binding affinity, we evaluated the relative binding free energy of Ca2+ ion binding to cTnC. These calculations were based on representative sample frames from 10 to 40 ns for WT, A164P, and A164H cTnI isoforms using the FEP method. The relative free energy of Ca2+ binding is shown in Fig. 6.
Figure 6.
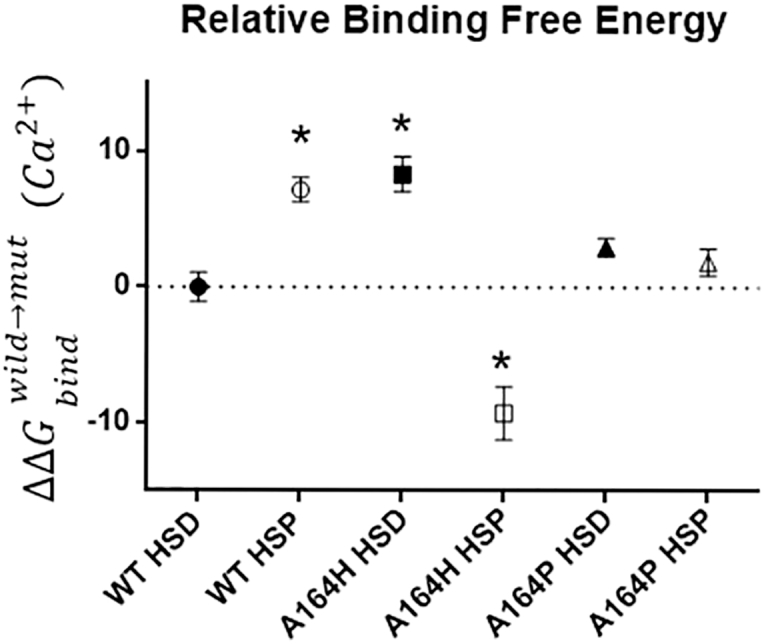
Relative Ca2+ binding free energy. The change in Ca2+ binding free energy is normalized to the average of WT cTnI, plotted as ΔΔ . Four representative frames from all simulations for a given isoform were calculated and averaged; error bars are ±SE. Mean ± SE are presented. ∗p < 0.05 from a Student’s t-test to the WT HSD value.
FEP calculations reveal increased Ca2+ binding free energy for cTnI HSP compared to cTnI HSD, indicative of decreased cTnC Ca2+ binding affinity and consistent with the decreased isolated myocyte contractility observed under acidosis (Fig. 2 F). Moreover, the Ca2+ binding free energies calculated from the cTnI A164P HSD and HSP simulations were not statistically different from their WT HSD or HSP counterparts, indicating that cTnC Ca2+ binding affinity does not underlie the increased contractility of myocytes transduced by cTnI A164P. We further compared FEP-calculated Ca2+ binding free energy of the previously published cTnI A164H HSD and HSP (21, 27). Here, cTnI A164H HSD indicates increased Ca2+ binding free energy (decreased cTnC Ca2+ binding affinity), whereas cTnI A164H HSP had decreased Ca2+ binding free energy (increased cTnC Ca2+ binding affinity). Although the observation of decreased Ca2+ binding free energy of cTnI A164H HSP is consistent with the increased contractility observed in isolated cells under acidosis (Fig. 2 F), the increased Ca2+ binding free energy of cTnI A164H HSD suggested that a different mechanism than altered Ca2+ affinity was responsible for the increased baseline contractility of cTnI A164P and A164H (Fig. 2, B, C, and F).
Discussion
We used a live-cell structure-function approach in complement with MD simulations to gain new—to our knowledge—mechanistic insights into the TnC-TnI interface during cardiac contraction. We introduced a proline residue into position 164 in helix 4 of cTnI to probe the biophysical consequences of this key transitional modification that arose during evolution. By monitoring the Ca2+ and contractile dynamics of live cardiac myocytes substituted with cTnI A164P, along with state-of-the-art MD simulations, this study contributes to a growing model wherein the effective concentration of the troponin I switch peptide at cTnC is a primary determinant of contractile function (48, 49, 50, 51, 52). A key new—to our knowledge—finding of this study is that cTnI A164P substitution in isolated adult rat cardiac myocytes causes an increase in contraction at baseline independent of alterations in the Ca2+ transient. These data indicate that increased fractional shortening mediated by cTnI A164P is not due to altered TnC-Ca2+ binding, as suggested by MD-guided FEP analysis. Finally, using MD simulations, we observe a novel TnC-TnI interface conformation wherein cTnI A164P enhances interactions with cTnC that are similar to those found at baseline with ssTnI. This positive cellular inotropy caused by cTnI A164P is distinct from previously described positively charged residue substitutions at this position, which provide an increase in contractility compared to WT cTnI during acidosis (27). That is to say that whereas cTnI A164P enhances contractility at normal pH, cTnI A164P functions identically to WT cTnI with similarly depressed live-cell contractility during acidosis.
To provide further mechanistic insight, we probed whether increased cTnC-Ca2+ binding affinity underlies the cTnI A164P baseline sensitization, and FEP calculations of stable cTnI-cTnC conformations were performed using MD simulations. Consistent with the contractility experiments in isolated myocytes, in silico acidosis by histidine protonation has a profound effect on the Ca2+ binding affinity, as previously established in the literature (21, 53). However, not only was the relative Ca2+ binding affinity for A164H HSD decreased compared to cTnI WT HSD, the Ca2+ binding affinity for A164P HSD and HSP was also unchanged. Based on these data, we posit that the cTnI A164P heightened calcium sensitivity derived from the intact myocyte shortening experiments can be explained by a dramatically different conformation of the cTnI A164P peptide with respect to cTnC. As such, this finding provides evidence that the TnC-TnI interface, and not TnC-Ca2+ binding affinity, is the central determinant of cardiac myocyte contractility under these conditions.
The cTnI A164P MD simulations provide further support for a model in which the cTnI switch peptide effective concentration at the N-terminus of cTnC is a critical determinant of contractility (48). At baseline physiological pH, ssTnI and cTnI A164P indicate increased contractility compared to cTnI WT. In acidosis, ssTnI and cTnI A164H maintain normal contractility. The atomistic structure of the TnC-TnI interaction reveals a distinct bend in the TnI switch peptide between helix 3 and helix 4 that occurs in both the ssTnI and A164P structures, but not in WT cTnI. An analogous but less distinct bend in this region has been previously reported in the cTnI A164H structure, but only under conditions of acidosis (21, 27). We posit that this bend in the cTnI switch peptide enhances TnC-TnI interactions by increasing the effective concentration of cTnI at the cTnC A helix interface. This is supported by quantifying the distance between the end of helix 4 (TnI R139/171) and the transition from the N helix to the A helix in cTnC (E15). Although noncovalent interactions between TnI helix 4 and TnC helix A are independent and relatively weak, the data suggest summation of multiple interactions results in a stable protein-protein interaction. We don’t consider this to be an exhaustive list of all possible interactions between TnC and TnI; instead, this residue has been highlighted to show a significant alteration in the orientation of cTnI helix 4 relative to cTnC helix A between these conditions. The TnC-TnI interface findings of this study are summarized in Fig. 7.
Figure 7.

Summary of isoform-specific substitutions and pH as molecular determinants of TnI function. ↔ indicates no change in contractility compared to cTnI at pH 7.4, ↑ indicates increased contractility compared to cTnI at pH 7.4, and ↓ indicates decreased contractility with acidosis relative to the same isoform at baseline. Atomistic stick figures show the orientation of cTnC helix A (red) in relation to cTnI switch peptide and helix 4 (blue) from MD simulations. + indicates the presence of a stable salt bridge interaction between helix 4 of cTnI and the A helix of cTnC, and − indicates the absence of any observed stable salt bridges. To see this figure in color, go online.
Proline has several unique biochemical characteristics. Under normal physiological conditions it is a nonpolar and aliphatic amino acid. Proline is the only amino acid whose alpha amino group is attached directly to the side chain, giving rise to a distinctive cyclic structure with exceptional conformational rigidity (54). When present in protein secondary structures, proline may act as a structural disrupter because of its locked φ dihedral angle (54). In complement with the restricted dihedral angle, the ring structure side chain is connected directly to the β-carbon of the peptide backbone, allowing for fewer degrees of rotation. In the context of cTnI, position 164 is at the transition from helix 3 to helix 4. Accordingly, introduction of a proline at this position disrupts the transition between cTnI helices 3 and 4, causing a dramatic conformational change, as observed in MD simulations (Fig. 7). Here, an approximation of the TnI switch peptide is shown relative to cTnC as well as whether or not there is evidence of an interaction between TnI helix 4 and cTnC helix A.
Taken together, these data support a model wherein an increase in interactions between TnI helix 4 and cTnC helix A results in positive inotropy. Maintenance of this interaction in acidosis is critical for maintaining the ability to contract under these disease-mimetic conditions. The introduction of proline at position 164 of cTnI disrupts the regularity of the α-helical backbone. This proline acts as a “helix breaker” and results in a kink in helix 4 at ∼90° that qualitatively mimics the structural signature of ssTnI under baseline and acidotic conditions, and cTnI A164H during acidosis (Fig. 7). In the cTnI A164P and WT cTnI MD simulations, the only histidine present is H173, which is not thought to directly interact with cTnC. Interestingly, protonation of H173 seems to have significant effects on the structure of the cTnI switch peptide, likely because of intramolecular interactions with the protonated cTnI H173. By contrast, ssTnI contains a polar uncharged asparagine at the cognate position (N141). However, investigation of the role of H173 was not the primary goal of this study and would require further examination before drawing broader conclusions about its role in determining contractility.
An alternate explanation, which we could not explore given computational limitations, is that the bend in helix 4 of TnI introduced by A164P substitution has an effect on the TnI interaction with actin and tropomyosin in the relaxed state. In addition to the inhibitory peptide (residues 128–146) of cTnI, there is a second putative actin biding domain at the C-terminus of cTnI, the mobile domain (residues 172–210) (55). The significant conformational change introduced by cTnI A164P may cause the mobile domain to position itself in a manner that limits its actin and/or tropomyosin interactions and therefore favors the active state. However interesting this hypothesis would be to investigate, computational limitations as well a lack of atomistic resolution of the cTnI mobile domain complicate this type of analysis at this time.
Numerous fish, amphibian, and bird species express unique troponin primary amino acid sequences to accommodate environmental pressures such as frequent cold and hypoxia. This allows for a wider range of Ca2+ sensitivities than the typically narrow operating range exhibited under the physiological constraints of mammals (13, 56, 57, 58, 59, 60, 61, 62). As such, it is intriguing that in the critical switch peptide region of cTnI, position 164 is 100% conserved in mammals. This position is always a nonpolar hydrophobic residue, whereas in nonmammalian chordates, the cognate position is exclusively a polar histidine (with the exception of Taeniopygia guttata, which expresses a proline) (13). In contrast with all other described mammals that express an alanine at position 164 of cTnI, the platypus (O. anatinus) expresses a unique proline substitution. The platypus represents a rather unique branch on the evolutionary tree as one of the few extant members of the mammalian order Monotremata (63). This animal occupies an evolutionary position that may provide insights into the biophysical transformation of the TnC-TnI interface and how this proline substitution may represent an evolutionary stepping stone. Interestingly, the nucleotide codon sequence for a histidine requires only a single-base transition to exchange a histidine for a proline; similarly, only one additional single-base transition is required to exchange a proline for an alanine. The naturally occurring nucleotide transition rate in mammals reveals proline as the most commonly occurring amino acid exchange from histidine. Moreover, an alanine is the most commonly exchanged amino acid from a proline (64). This probabilistic scheme underscores the platypus’s persistent expression of a proline at position 164, in stark contrast to an alanine or a histidine as observed in all other mammals or nonmammalian chordates, respectively.
Several allometric differences have been noted in the basal physiology across the extant mammalian subclasses (Prototheria, Marsupilia, and Euthera), which can be juxtaposed with the basal physiology of nonmammalian chordates. Of particular note is the difference in core body temperature—the platypus and other monotremes operate at ∼31°C compared to the average 35°C of marsupials and 37°C of eutherians. One prevailing hypothesis in the literature is that heightened calcium sensitivity is a compensatory mechanism required to maintain cardiac pump function at lower temperatures (57, 58, 59, 60, 61, 62, 65, 66). Additionally, anoxia creates a condition of depressed cardiac pump function, and this too may be ameliorated by calcium-sensitizing mutations, as seen in certain freshwater turtles and crucian carp. During anoxia, these animals have been observed to alter their cardiac physiology to maintain contractility, including increasing cardiac output. The platypus is a semifossorial animal that digs its burrows at depths of up to 30 m deep and spends considerable time diving underwater for hunting (67, 68). It has been established that monotremes have an exceptionally low ventilatory response when challenged with CO2 (64), which equips the platypus to endure the prolonged bouts of hypoxia that occur during these activities. Taken together with our study, this suggests a model in which the platypus has several physiological adaptations to environmental conditions, whereas cardiac performance would be blunted in other species not similarly adapted to these harsh conditions.
In conclusion, this study combines live-cell structure-function with in silico MD simulations to provide, to our knowledge, new mechanistic insights into the critical role of the TnI-TnC interface in modulating cardiac performance. We propose that enhancing the interaction between TnI helix 4 and the A helix of TnC increases contractility and that disruption of this interaction during acidosis is one of the key factors contributing to decreased contractility of the heart during ischemia. Furthermore, this work enhances our knowledge of how disease-causing mutations in the TnI switch peptide may be affecting contractility, establishing a foundation for designing new therapeutics that target the TnC-TnI interface.
Author Contributions
The experimental design of this manuscript was conducted by A.D.V., E.M.H., B.R.T., Y.Y.S., and J.M.M. The experiments contained within were performed by A.D.V., E.M.H., J.J.S., and B.R.T. The data was analyzed by A.D.V., E.M.H., B.R.T., and J.M.M. The final manuscript was written by A.D.V. and J.M.M.
Acknowledgments
The authors acknowledge the Minnesota Supercomputing Institute at the University of Minnesota for providing resources that contributed to the research results reported within this article. We also thank Professor Arieh Warshel from University of Southern California for providing the Molaris software package for the FEP calculation.
This work was supported by grants from the American Heart Association (to A.D.V.) and National Institutes of Health (to J.M.M.)
Editor: David Warshaw.
Footnotes
Three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(18)30218-2.
Supporting Material
References
- 1.Huxley H., Hanson J. Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature. 1954;173:973–976. doi: 10.1038/173973a0. [DOI] [PubMed] [Google Scholar]
- 2.Rayment I., Rypniewski W.R., Holden H.M. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 3.Granzier H.L., Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ. Res. 2004;94:284–295. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- 4.Thompson B.R., Asp M.L., Metzger J.M. Molecular mechanism of sarcomeric cardiomyopathies. In: Garry D., Wilson R., Vlodaver Z., editors. Congestive Heart Failure and Cardiac Transplantation. Springer International Publishing; 2017. pp. 151–160. [Google Scholar]
- 5.Farah C.S., Reinach F.C. The troponin complex and regulation of muscle contraction. FASEB J. 1995;9:755–767. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- 6.Pineda-Sanabria S.E., Robertson I.M., Sykes B.D. Interaction between the regulatory domain of cardiac troponin C and the acidosis-resistant cardiac troponin I A162H. Cardiovasc. Res. 2013;97:481–489. doi: 10.1093/cvr/cvs348. [DOI] [PubMed] [Google Scholar]
- 7.Robinson J.M., Dong W.J., Cheung H.C. Switching of troponin I: Ca(2+) and myosin-induced activation of heart muscle. J. Mol. Biol. 2004;340:295–305. doi: 10.1016/j.jmb.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 8.Dong W.J., Xing J., Cheung H.C. Structural kinetics of cardiac troponin C mutants linked to familial hypertrophic and dilated cardiomyopathy in troponin complexes. J. Biol. Chem. 2008;283:3424–3432. doi: 10.1074/jbc.M703822200. [DOI] [PubMed] [Google Scholar]
- 9.Takeda S., Yamashita A., Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 10.Perry S.V. Troponin I: inhibitor or facilitator. Mol. Cell. Biochem. 1999;190:9–32. [PubMed] [Google Scholar]
- 11.Lehrer S.S., Morris E.P. Dual effects of tropomyosin and troponin-tropomyosin on actomyosin subfragment 1 ATPase. J. Biol. Chem. 1982;257:8073–8080. [PubMed] [Google Scholar]
- 12.Westfall M.V., Borton A.R., Metzger J.M. Myofilament calcium sensitivity and cardiac disease: insights from troponin I isoforms and mutants. Circ. Res. 2002;91:525–531. doi: 10.1161/01.res.0000034710.46739.c0. [DOI] [PubMed] [Google Scholar]
- 13.Palpant N.J., Houang E.M., Metzger J.M. Pathogenic peptide deviations support a model of adaptive evolution of chordate cardiac performance by troponin mutations. Physiol. Genomics. 2010;42:287–299. doi: 10.1152/physiolgenomics.00033.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis J., Wen H., Metzger J.M. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ. Res. 2007;100:1494–1502. doi: 10.1161/01.RES.0000268412.34364.50. [DOI] [PubMed] [Google Scholar]
- 15.Lee J.A., Allen D.G. Mechanisms of acute ischemic contractile failure of the heart. Role of intracellular calcium. J. Clin. Invest. 1991;88:361–367. doi: 10.1172/JCI115311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westfall M.V., Rust E.M., Metzger J.M. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc. Natl. Acad. Sci. USA. 1997;94:5444–5449. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Day S.M., Westfall M.V., Metzger J.M. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat. Med. 2006;12:181–189. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 18.Wolska B.M., Arteaga G.M., Solaro R.J. Expression of slow skeletal troponin I in hearts of phospholamban knockout mice alters the relaxant effect of beta-adrenergic stimulation. Circ. Res. 2002;90:882–888. doi: 10.1161/01.res.0000016962.36404.04. [DOI] [PubMed] [Google Scholar]
- 19.Fentzke R.C., Buck S.H., Leiden J.M. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J. Physiol. 1999;517:143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westfall M.V., Albayya F.P., Metzger J.M. Chimera analysis of troponin I domains that influence Ca(2+)-activated myofilament tension in adult cardiac myocytes. Circ. Res. 2000;86:470–477. doi: 10.1161/01.res.86.4.470. [DOI] [PubMed] [Google Scholar]
- 21.Thompson B.R., Houang E.M., Metzger J.M. Molecular determinants of cardiac myocyte performance as conferred by isoform-specific TnI residues. Biophys. J. 2014;106:2105–2114. doi: 10.1016/j.bpj.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musser A.M. Review of the monotreme fossil record and comparison of palaeontological and molecular data. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2003;136:927–942. doi: 10.1016/s1095-6433(03)00275-7. [DOI] [PubMed] [Google Scholar]
- 23.Warren W.C., Hillier L.W., Wilson R.K. Genome analysis of the platypus reveals unique signatures of evolution. Nature. 2008;453:175–183. doi: 10.1038/nature06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rowe T., Rich T.H., Woodburne M.O. The oldest platypus and its bearing on divergence timing of the platypus and echidna clades. Proc. Natl. Acad. Sci. USA. 2008;105:1238–1242. doi: 10.1073/pnas.0706385105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williamson M.P. The structure and function of proline-rich regions in proteins. Biochem. J. 1994;297:249–260. doi: 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herron T.J., Vandenboom R., Metzger J.M. Calcium-independent negative inotropy by beta-myosin heavy chain gene transfer in cardiac myocytes. Circ. Res. 2007;100:1182–1190. doi: 10.1161/01.RES.0000264102.00706.4e. [DOI] [PubMed] [Google Scholar]
- 27.Palpant N.J., Houang E.M., Metzger J.M. pH-responsive titratable inotropic performance of histidine-modified cardiac troponin I. Biophys. J. 2012;102:1570–1579. doi: 10.1016/j.bpj.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shrödinger . Schrödinger; LLC: 2016. Maestro-Desmond Interoperability Tools. [Google Scholar]
- 29.Jorgensen W.L., Maxwell D.S., Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 30.Sham Y.Y., Chu Z.T., Warshel A. Consistent calculations of pKa’s of ionizable residues in proteins: semi-microscopic and microscopic approaches. J. Phys. Chem. B. 1997;101:4458–4472. [Google Scholar]
- 31.Bashford D., Karplus M. pKa’s of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 32.Gordon J.C., Myers J.B., Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33(Suppl 2):W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schutz C.N., Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 34.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 35.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;1995:8577–8593. [Google Scholar]
- 36.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 37.Sham Y.Y., Warshel A. The surface constraint all atom model provides size independent results in calculations of hydration free energies. J. Chem. Phys. 1998;109:7940–7944. [Google Scholar]
- 38.Warshel A. Dynamics of reactions in polar solvents. Semiclassical trajectory studies of electron-transfer and proton-transfer reactions. J. Phys. Chem. 1982;86:2218–2224. [Google Scholar]
- 39.Warshel A., Sussman F., King G. Free energy of charges in solvated proteins: microscopic calculations using a reversible charging process. Biochemistry. 1986;25:8368–8372. doi: 10.1021/bi00374a006. [DOI] [PubMed] [Google Scholar]
- 40.Lee F.S., Chu Z.T., Warshel A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J. Comput. Chem. 1993;14:161–185. [Google Scholar]
- 41.Rust E.M., Westfall M.V., Metzger J.M. Stability of the contractile assembly and Ca2+-activated tension in adenovirus infected adult cardiac myocytes. Mol. Cell. Biochem. 1998;181:143–155. doi: 10.1023/a:1006802719136. [DOI] [PubMed] [Google Scholar]
- 42.Davis J., Wen H., Metzger J.M. Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants. J. Mol. Cell. Cardiol. 2008;44:891–904. doi: 10.1016/j.yjmcc.2008.02.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westfall M.V., Metzger J.M. Single amino acid substitutions define isoform-specific effects of troponin I on myofilament Ca2+ and pH sensitivity. J. Mol. Cell. Cardiol. 2007;43:107–118. doi: 10.1016/j.yjmcc.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lang S.E., Robinson D.A., Westfall M.V. Myofilament incorporation and contractile function after gene transfer of cardiac troponin I Ser43/45Ala. Arch. Biochem. Biophys. 2013;535:49–55. doi: 10.1016/j.abb.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Day S.M., Westfall M.V., Metzger J.M. Tuning cardiac performance in ischemic heart disease and failure by modulating myofilament function. J. Mol. Med. (Berl.) 2007;85:911–921. doi: 10.1007/s00109-007-0181-6. [DOI] [PubMed] [Google Scholar]
- 46.Palpant N.J., Day S.M., Metzger J.M. Single histidine-substituted cardiac troponin I confers protection from age-related systolic and diastolic dysfunction. Cardiovasc. Res. 2008;80:209–218. doi: 10.1093/cvr/cvn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westfall M.V., Metzger J.M. Troponin I isoforms and chimeras: tuning the molecular switch of cardiac contraction. News Physiol. Sci. 2001;16:278–281. doi: 10.1152/physiologyonline.2001.16.6.278. [DOI] [PubMed] [Google Scholar]
- 48.Siddiqui J.K., Tikunova S.B., Davis J.P. Myofilament calcium sensitivity: consequences of the effective concentration of troponin I. Front. Physiol. 2016;7:632. doi: 10.3389/fphys.2016.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis J.P., Shettigar V., Walton S.D. Designing proteins to combat disease: cardiac troponin C as an example. Arch. Biochem. Biophys. 2016;601:4–10. doi: 10.1016/j.abb.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Valen D., Haataja M., Phillips R. Biochemistry on a leash: the roles of tether length and geometry in signal integration proteins. Biophys. J. 2009;96:1275–1292. doi: 10.1016/j.bpj.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiroli A.O., Tasic L., Ramos C.H. Mapping contacts between regulatory domains of skeletal muscle TnC and TnI by analyses of single-chain chimeras. FEBS J. 2005;272:779–790. doi: 10.1111/j.1742-4658.2004.04515.x. [DOI] [PubMed] [Google Scholar]
- 52.Pineda-Sanabria S.E., Julien O., Sykes B.D. Versatile cardiac troponin chimera for muscle protein structural biology and drug discovery. ACS Chem. Biol. 2014;9:2121–2130. doi: 10.1021/cb500249j. [DOI] [PubMed] [Google Scholar]
- 53.Metzger J.M., Parmacek M.S., Leiden J.M. Skeletal troponin C reduces contractile sensitivity to acidosis in cardiac myocytes from transgenic mice. Proc. Natl. Acad. Sci. USA. 1993;90:9036–9040. doi: 10.1073/pnas.90.19.9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morris A.L., MacArthur M.W., Thornton J.M. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 55.Murakami K., Yumoto F., Wakabayashi T. Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J. Mol. Biol. 2005;352:178–201. doi: 10.1016/j.jmb.2005.06.067. [DOI] [PubMed] [Google Scholar]
- 56.Hastings K.E., Bucher E.A., Emerson C.P., Jr. Generation of troponin T isoforms by alternative RNA splicing in avian skeletal muscle. Conserved and divergent features in birds and mammals. J. Biol. Chem. 1985;260:13699–13703. [PubMed] [Google Scholar]
- 57.Blumenschein T.M., Gillis T.E., Sykes B.D. Effect of temperature on the structure of trout troponin C. Biochemistry. 2004;43:4955–4963. doi: 10.1021/bi035504z. [DOI] [PubMed] [Google Scholar]
- 58.Gillis T.E., Liang B., Tibbits G.F. Increasing mammalian cardiomyocyte contractility with residues identified in trout troponin C. Physiol. Genomics. 2005;22:1–7. doi: 10.1152/physiolgenomics.00007.2005. [DOI] [PubMed] [Google Scholar]
- 59.Gillis T.E., Marshall C.R., Tibbits G.F. Ca(2+) binding to cardiac troponin C: effects of temperature and pH on mammalian and salmonid isoforms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;279:R1707–R1715. doi: 10.1152/ajpregu.2000.279.5.R1707. [DOI] [PubMed] [Google Scholar]
- 60.Gillis T.E., Moyes C.D., Tibbits G.F. Sequence mutations in teleost cardiac troponin C that are permissive of high Ca2+ affinity of site II. Am. J. Physiol. Cell Physiol. 2003;284:C1176–C1184. doi: 10.1152/ajpcell.00339.2002. [DOI] [PubMed] [Google Scholar]
- 61.Liang B., Chung F., Tibbits G.F. Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol. Genomics. 2008;33:257–266. doi: 10.1152/physiolgenomics.00154.2007. [DOI] [PubMed] [Google Scholar]
- 62.Lillywhite H.B., Zippel K.C., Farrell A.P. Resting and maximal heart rates in ectothermic vertebrates. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 1999;124:369–382. doi: 10.1016/s1095-6433(99)00129-4. [DOI] [PubMed] [Google Scholar]
- 63.Brown S. Top billing for platypus at end of evolution tree. Nature. 2008;453:138–139. doi: 10.1038/453138a. [DOI] [PubMed] [Google Scholar]
- 64.Frappell P.B. Ventilation and metabolic rate in the platypus: insights into the evolution of the mammalian breathing pattern. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2003;136:943–955. doi: 10.1016/s1095-6433(03)00273-3. [DOI] [PubMed] [Google Scholar]
- 65.Shiels H.A., Calaghan S.C., White E. The cellular basis for enhanced volume-modulated cardiac output in fish hearts. J. Gen. Physiol. 2006;128:37–44. doi: 10.1085/jgp.200609543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shiels H.A., White E. The Frank-Starling mechanism in vertebrate cardiac myocytes. J. Exp. Biol. 2008;211:2005–2013. doi: 10.1242/jeb.003145. [DOI] [PubMed] [Google Scholar]
- 67.Bethge P., Munks S., Nicol S. Diving behaviour, dive cycles and aerobic dive limit in the platypus Ornithorhynchus anatinus. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2003;136:799–809. doi: 10.1016/s1095-6433(03)00198-3. [DOI] [PubMed] [Google Scholar]
- 68.Maule L. Habits and economy of the Ornithorhynchus. Proc. Zool. Soc. London. 1832;1832:145–146. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.