

Abstract
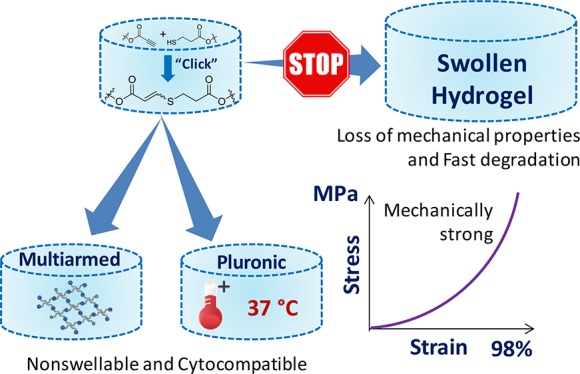
A key drawback of hydrogel materials for tissue engineering applications is their characteristic swelling response, which leads to a diminished mechanical performance. However, if a solution can be found to overcome such limitations, there is a wider application for these materials. Herein, we describe a simple and effective way to control the swelling and degradation rate of nucleophilic thiol–yne poly(ethylene glycol) (PEG) hydrogel networks using two straightforward routes: (1) using multiarm alkyne and thiol terminated PEG precursors or (2) introducing a thermoresponsive unit into the PEG network while maintaining their robust mechanical properties. In situ hydrogel materials were formed in under 10 min in PBS solution at pH 7.4 without the need for an external catalyst by using easily accessible precursors. Both pathways resulted in strong tunable hydrogel materials (compressive strength values up to 2.4 MPa) which could effectively encapsulate cells, thus highlighting their potential as soft tissue scaffolds.
Introduction
Hydrogels have become a powerful platform as tissue engineering scaffolds as a consequence of their unique features (high water content, suitable porosity, synthetic versatility, and biocompatibility); however, they suffer from swelling-induced behavior which can limit their applications in this field.1−3 Furthermore, these materials must mimic unique biological environments by displaying specific mechanical strength, robustness, and stability. Therefore, the design of hydrogels with finely tuned properties for a specific bioapplication is still a challenging task.4−6
To address this issue, biorthogonal click-hydrogels that form rapidly under physiological conditions by covalently bonding nontoxic polymeric chains through easily accessible functional groups, have been synthesized.7−9 These materials, which are envisaged as promising soft tissue scaffolds, can be prepared using a wide range of click reactions, for example, thiol–ene, oxime, inverse electron demand Diels–Alder, and strain promoted azide–alkyne cycloaddition (SPAAC), and copper-mediated azide– alkyne cycloaddition (CuAAC).7,8 Although these chemistries form efficient networks that are biocompatible, some of the functional groups can be difficult to synthesize onto polymer backbones (e.g. strained alkynes). Furthermore, some click reactions follow a UV-initiated radical pathway, using a photoinitiator to conduct the cross-linking reaction releasing free radicals during the cross-linking process. It has been demonstrated that the release of free radicals during the network formation can be cytotoxic to some cell lines, for example, human mesenchymal stem cells (hMSCs) and can cause more toxicity issues than the presence of the photoinitiator.10−12 Therefore, by using a radical pathway the cyctotoxcity of the cross-linking reaction can limit the number of different cell lines which can be encapsulated into the network. In contrast, the nucleophilic thiol–yne addition reaction13 is highly suitable for hydrogel synthesis as a consequence of its efficient and rapid nature, through the use of easily accessible functional end groups (activated alkyne and thiol functionalities). This allows for the design of unique hydrogels with predefined and robust features.14−17
In our previous work,16 we reported the synthesis of robust poly(ethylene glycol) (PEG) thiol–yne click-hydrogels with tunable properties. This work utilizes the nucleophilic pathway to form hydrogels using thiol–yne chemistry under slightly basic conditions, pH 7.4. To exploit the nucleophilic pathway, PEG precursors are easily functionalized with either an activated alkyne (carbonyl adjacent to the alkyne) or thiol end groups allowing the reaction to take place under physiological conditions (37 °C in PBS solution, pH 7.4) without the need of an external catalyst. Through the optimization of the molecular weight, architecture, and composition of the alkyne- and thiol-terminated PEG precursors, these hydrogels displayed a wide range of tunable compressive strengths (up to 2.4 MPa) and stiffness.
For a hydrogel material to meet the needs of a specific biological environment, the networks need to be designed accordingly. Usually, their mechanical properties are compared to soft tissue (i.e., soft tissue stiffness in the range between 0.25–2.2 kPa).18 In addition to this, and most importantly, the hydrogel’s network should be characteristic of an environment that resembles the in vivo setting (i.e., aqueous conditions). However, although some hydrogel systems remain unaltered when immersed in aqueous solutions,19,20 in general most swell in aqueous environments at 37 °C, which not only expands and deforms the polymeric network isotropically but also has a major impact on their performance. Swelling-induced effects include loss of mechanical performance,21 changes in hydrogel stiffness,22 enhanced hydrolytic degradation,1 or compromised patient health by excessive compression to the surrounding tissue when used in vivo, thus greatly limiting their biomedical application. Consequently, research into the mechanical response of hydrogels in the swollen state over time is an interesting study for these materials.23
Attempts to overcome this drawback rely on synthesizing nonswellable hydrogels, which are able to retain their high compressive modulus and strength despite being immersed in aqueous media.17,24−26 Specifically, the swelling induced by the hydrophilic segments of the hydrogel backbone is counterbalanced by either exploiting the thermoresponsive properties of polymers that collapse above a certain temperature, ideally body temperature,17,26,27 or by modulating the architecture of the polymeric precursors (i.e., multiarm precursors result in dense hydrogel networks with high cross-linking density and reduced hydrophilicity).25
Recently, the former approach was exemplified by Truong et al.17 who applied the commercially available triblock PEG–PPG–PEG (Pluronic) in the preparation of nucleophilic thiol–yne click hydrogels with reduced water uptake at a physiologically relevant temperature. The viscoelastic properties of the resulting hydrogels were characterized by rheology. Notably, however, the authors found that these systems were not suitable for 3D cell encapsulation and instead led to high levels of cell death, and the effect of swelling-suppression on the compressive strength of thiol–yne click-hydrogels was not assessed.
Herein, we aim to further improve the performance of thiol–yne PEG hydrogels,16 by rendering them nonswellable and thus controlling their degradation profiles and mechanical performance in aqueous environments. To that end, the thiol-functionalized PEG precursor was altered to effectively supress swelling by utilizing two different approaches. One route increased the hydrophobic nature of the PEG hydrogels by increasing the number of cross-linking sites in the network using alkyne and thiol-functionalized PEG precursors displaying a 3- or 4-arm architecture (previously 2-arm thiol or alkyne-functionalized PEG were used).16,17 This has been previously shown by Kamata and co-workers24 as a versatile way of controlling the swelling properties of PEG hydrogels. For the thiol–yne reaction, the reactive end groups are very small, and therefore there is little difference between the unfunctionalized and functionalized precursors, which allows for a straightforward control of the hydrophobicity of the hydrogels through the PEG architecture.
Second, we modified a 2-arm thermoresponsive segment (Pluronic L-64) as the thiol-moiety within the click-hydrogel network, which allowed us to finely tune their swelling properties by varying the hydrophilic/hydrophobic ratio of the hydrogel polymeric chains. During the characterization of the resulting click hydrogel, comparisons have been drawn between the two routes applied, especially when monitoring the evolution of their compressive strength with time after being immersed in PBS solution at 37 °C. Most importantly, their potential as soft tissue scaffolds has been assessed through encapsulated cell studies over a 72 h period.
Experimental Section
Materials
4-arm PEG-tetrahydroxyl (molar mass 2.0 kg mol–1) was purchased from JenKem Technology, U.S.A. All other reagents were purchased from Sigma-Aldrich or Fisher Scientific and used without purification.
Instrumental Methods
1H NMR spectra were recorded on a Bruker DPX-400 spectrometer at 293 K. Chemical shifts are reported as δ in parts per million (ppm) and referenced to the chemical shift of the residual solvent resonances (CDCl3, 1H δ = 7.26 ppm; (CD3)2CO, 1H δ = 2.05 ppm).
Size exclusion chromatography (SEC) was used to determine the molar masses and molar mass distributions (dispersities, ĐM) of the synthesized polymers. SEC conducted in chloroform (CHCl3) (0.5% NEt3) used a Varian PL-SEC 50 system equipped with 2× PLgel 5 μM MIXED-D columns in series and a differential refractive index (RI) detector at a flow rate 1.0 mL min–1. The system was calibrated against a Varian Polymer Laboratories Easi-Vial poly(styrene) (PS) standard and analyzed by the software package Cirrus v3.3.
SEC conducted in N,N-dimethylformamide (DMF) (5 mM NH4BF4) used a Varian PL-SEC 50 system equipped with 2× PLgel 5 μM MIXED-C+ guard columns in series and a differential RI detector at a flow rate of 1.0 mL min–1. The systems were calibrated against Varian Polymer Laboratories Easi-Vial linear poly(methyl methacrylate) (PMMA) standards and analyzed by the software package Cirrus v3.3.
Lower critical solution temperature (LCST) and cloud point measurements for Pluronic L-64 before and after modification were recorded using a PerkinElmer UV–vis Spectrometer (Lambda 35) equipped with a Peltier temperature control system, using a wavelength of 500 nm and a heating/cooling rate of 1 °C min–1. The polymer was dissolved in water at 1% wt concentration and submitted to three heating–cooling cycles. Transmittance curves were normalized for clarity, and the cloud point of each sample measured at 50% of normalized transmittance.
Rheological testing was carried out using an Anton Parr MCR 302 rheometer equipped with parallel plate configuration with a diameter of 50 mm. A Peltier system was used to maintain the temperature at 20 °C throughout the study. Data was analyzed using RheoCompass software.
Compression testing was carried out using single column universal materials testing machine M100-1CT Testometric with a load cell of 1 kN. Data was analyzed using Wintest analysis software.
Cryogenic scanning electron microscopy (cryo-SEM) was performed on ZEISS SUPRA 55-VP equipped with cold stage and sample preparation chamber.
Evaluation of cell viability during 3D cell encapsulation experiments was carried out using a Zeiss LSM 880 confocal fluorescence microscope. Staining for live cells (Calcein, Ex. = 495 nm, Em. = 515 nm) was excited with a 488 nm laser, while staining for dead cells (Ethidium homodimer, Ex = 528 nm, Em = 617 nm) was excited with a 514 nm laser. Z-stacks with an average thickness of 400 μm were collected from different zones of the samples and maximum intensity projections were obtained from each Z-stack. Images were processed using Zen (blue edition) and ImageJ software.
Synthesis of Alkyne and Thiol Precursors
Synthesis of 4-Arm Alkyne Functionalized PEG
To a suspension of 4-arm PEG2k-OH (molar mass 2.0 kg mol–1, 10 g, 5 mmol) in benzene (75 mL) and toluene (75 mL) was added 2 drops of concentrated H2SO4. The solution was heated to 80 °C with stirring to obtain a clear homogeneous solution. To this solution, propiolic acid (2.8 g, 40 mmol) was added and the solution was heated to reflux under Dean–Stark conditions. After no more water was collected in the condenser (about 20 h), the solution was allowed to cool to room temperature and the solvents were removed in vacuo. The resultant oil was dissolved in CH2Cl2 (100 mL) and washed with saturated NaHCO3 solution (20 mL) and brine (20 mL). The organic phase was dried (MgSO4) and stirred with charcoal (about 0.1 g) for 30 min at 40 °C. The solution was filtered through Celite 545 and the solvent was evaporated to collect product as a clear to light yellow oil (yield 6.1 g, 61%). 1H NMR ((CD3)2CO, 400 MHz): δ 4.32–4.35 (t, 3JHH = 8 Hz, −CH2OCO-), 3.90 (s, −CH≡CC(O)O−), 3.72–3.74 (m, −OCH2CH2O−), 3.6 (s, CCH2O). 1H NMR spectroscopy indicated about 93% conversion of the hydroxyl group to propiolate group. SEC (DMF): Mn = 5.9 kg mol–1 (ĐM = 1.08).
Synthesis of 3-Arm Alkyne Functionalized PEG
In a typical esterification, as stated in the above procedure, glycerol ethoxylate (molar mass 1.0 kg mol–1, 10 g, 10 mmol) was esterified with propiolic acid (4.2 g, 60 mmol) to collect the product as a light yellow oil (yield 8.1 g, 70%). 1H NMR ((CD3)2CO, 400 MHz): δ 4.32–4.34 (t, 3JHH = 8 Hz, −CH2OCO−), 3.89 (s, −CH≡CC(O)O−), 3.71–3.74 (m, −OCH2CH2O−), 3.58 (s, −CCH2O). 1H NMR spectroscopy indicated about 92% conversion of the hydroxyl group to propiolate group. SEC (DMF): Mn = 3.3 kg mol–1(ĐM = 1.04).
Synthesis of 2-Arm Thiol Functionalized PEG (1, 2, or 3 kg mol–1)
In a typical esterification, as stated in the above procedure, 2-arm PEG-OH (10 g, 10 mmol) was esterified with 3-mercaptopropionic acid (2 equiv per arm/0.5 kg mol–1) to collect the product as a white solid.
PEG1k(SH)2 (Yield 9.4 g, 80%)
1H NMR (CDCl3, 400 MHz): δ 4.25–4.27 (t, 3JHH = 8, −CH2OCO−), 3.65 (m, −OCH2CH2O−), 2.75–2.78 (q, 3JHH = 12, −OCCH2CH2SH), 2.66–2.69 (t, 3JHH = 12, −OCCH2CH2SH), 1.66–1.68 (t, 3JHH = 8, −SH), 1H NMR spectroscopy indicated about 96% conversion of the hydroxyl group to mercaptopropionate group. SEC (CHCl3): Mn = 1.1 kg mol–1 (ĐM = 1.24).
PEG2k(SH)2 (Yield 9.2 g, 85%)
1H NMR (CDCl3, 400 MHz): δ 4.26–4.28 (t, 3JHH = 8, −CH2OCO−), 3.65 (m, −OCH2CH2O−), 2.75–2.80 (q, 3JHH = 12, −OCCH2CH2SH), 2.67–2.70 (t, 3JHH = 12, −OCCH2CH2SH), 1.67–1.69 (t, 3JHH = 8, −SH), 1H NMR spectroscopy indicated about > 99% conversion of the hydroxyl group to mercaptopropionate group. SEC (CHCl3): Mn = 2.7 kg mol–1 (ĐM = 1.26).
PEG3k(SH)2 (Yield 9.3 g, 88%)
1H NMR (CDCl3, 400 MHz): δ 4.26–4.28 (t, 3JHH = 8, −CH2OCO−), 3.65 (m, −OCH2CH2O−), 2.71–2.78 (q, 3JHH = 12, −OCCH2CH2SH), 2.63–2.67 (t, 3JHH = 12, −OCCH2CH2SH), 1.62–1.68 (t, 3JHH = 8, −SH), 1H NMR spectroscopy indicated about 84% conversion of the hydroxyl group to mercaptopropionate group. SEC (DMF): Mn = 8.7 kg mol–1 (ĐM = 1.1).
Synthesis of 2-Arm Thiol-Functionalized Pluronic L-64 (PEG–PPG–PEG 2.9 kg mol–1)
In a typical esterification, as stated in the above procedure, PEG–PPG–PEG (10 g, 3.5 mmol) was esterified with 3-mercaptopropionic acid (1.46 g, 13.8 mmol) to collect the product as a white solid (yield 8.8 g, 78%). 1H NMR (CDCl3, 400 MHz): δ 4.20–4.23 (t, 3JHH = 8, −CH2OCO−), 3.65 (m, −OCH2CH2O−), 3.49 (m, −OCH2CH(CH3)O−), 2.69–2.76 (q, 3JHH = 12, −OCCH2CH2SH), 2.61–2.65 (t, 3JHH = 12, −OCCH2CH2SH), 1.61–1.66 (t, 3JHH = 8, -SH), 1.08–1.09 (d, 3JHH = 4, −OCH2CH(CH3)O−), 1H NMR spectroscopy indicated about 93% conversion of the hydroxyl group to mercaptopropionate group. SEC (DMF): Mn = 5.8 kg mol–1 (ĐM = 1.2).
Synthesis of 3-Arm Thiol Functionalized PEG (1 kg mol–1)
In a typical esterification as stated in the above procedure, glycerol ethoxylate (molar mass 1.0 kg mol–1, 10 g, 10 mmol) was esterified using 3-mercaptopropionic acid (4.2 g, 60 mmol) (yield 9.4 g, 74%). 1H NMR (CDCl3, 400 MHz): δ 4.20–4.22 (t, 3JHH = 8, −CH2OCO−), 3.58 (m, −OCH2CH2O−), 2.69–2.74 (q, 3JHH = 12, −OCCH2CH2SH), 2.61–2.64 (t, 3JHH = 12, −OCCH2CH2SH), 1.61–1.66 (t, 3JHH = 20, −SH), 1H NMR spectroscopy indicated about >99% conversion of the hydroxyl group to mercaptopropionate group. SEC (DMF): Mn = 10.6 kg mol–1 (ĐM = 1.08).
Synthesis of 4-Arm Thiol Functionalized PEG (2 kg mol–1)
In a typical esterification, as stated in the above procedure, 4-arm PEG2k–OH (molar mass = 2.0 kg mol–1, 10 g, 5 mmol) was esterified using 3-mercaptopropionic acid (4.2 g, 40 mmol) (yield 9.1 g, 77%). 1H NMR (CDCl3, 400 MHz): δ 4.21–4.24 (t, 3JHH = 12, −CH2OCO−), 3.58 (m, −OCH2CH2O−), 2.71–2.76 (q, 3JHH = 20, −OCCH2CH2SH), 2.63–2.66 (t, 3JHH = 12, −OCCH2CH2SH), 1.63–1.67 (t, 3JHH = 16, −SH), 1H NMR spectroscopy indicated about > 99% conversion of the hydroxyl group to mercaptopropionate group. SEC (DMF): Mn= 4.7 kg mol–1 (ĐM = 1.06).
Hydrogel Fabrication
A 1:1 molar ratio of alkyne group to thiol group was used for all gelations by thiol–yne chemistry, and the precursor content was kept at 10 wt %. In a typical procedure for making a thiol–yne PEG gel, PEG1k-(SH)2 (22.9 mg, 1.95 × 10–3 mmol) was dissolved in 200 μL of PBS pH 7.4 solution. A separate solution of PEG2k-(C≡CH)4 (21.5 mg, 9.74 × 10–3 mmol) in 200 μL of PBS pH 7.4 solution. The two solutions were mixed together on a vortex mixer for 5 s. The mixture was then injected into a mold suitable for the chosen analysis. All the preparation steps for thermoresponsive hydrogels were performed below 10 °C by cooling the solutions in an ice bath. For the blended hydrogels, a 1:1 molar ratio of alkyne group to thiol group was maintained. Different molar ratio of each thiol was used to tune the properties of the resulting hydrogels. The precursors were diluted with PBS solution and the thiol precursors were mixed together before being added to the diluted alkyne precursor.
Hydrogel Characterization
Gel Fraction (GF) and Equilibrium Water Content (EWC)
Hydrogels were fabricated as stated in the above procedure. To determine the gel fraction (GF) the hydrogels were lyophilized and their weights (Wg) recorded. The hydrogels were then allowed to swell in deionized water for 3 days with frequent changes in water to extract unreacted polymers. The hydrogels were then lyophilized and their weights (Wr) were recorded again. All measurements were repeated in triplicate. The gel fraction was expressed as
| 1 |
To determine the equilibrium water content (EWC) the prepared hydrogels were allowed to swell in PBS solution for 1 day so that swelling could reach equilibrium. The surface water was then removed with soft tissue paper and the weights recorded (Ws). The hydrogels were then lyophilized and the weights recorded (Wd). All measurements were repeated in triplicate. The equilibrium water content was expressed as
| 2 |
Mesh Size Calculations
Flory–Rehner calculations were used to determine each thiol–yne PEG hydrogel mesh size.28,29 All measurements were repeated in triplicate.
| 3 |
Rheological Testing
All rheology was performed on an Anton Parr MCR 302 rheometer fitted with a parallel plate configuration (diameter of 50 mm) at 20 °C. In a typical rheological test for gelation kinetics, PEG1k(SH)2 (56.8 mg, 4.83 × 10–2 mmol) and PEG2k(C≡CH)4 (53.3 mg, 2.41 × 10–2 mmol) were dissolved in separate solutions of 500 μL PBS pH 7.4. The two solutions were drawn up in a 1 mL syringe and injected on to the lower plate, at 20 °C. The upper plate was immediately lowered to a plate separation of 0.5 mm and the measurement was started. A frequency of 5 Hz and a strain of 5% was applied to minimize interference with the gelation process and to keep the measurement within the linear viscoelastic region. The normal force was also kept constant at 0 N. The gelation kinetics was characterized by the evolution of storage moduli (G′) and loss moduli (G″) as a function of time. The gel point was determined by the crossover between the G′ and G″. A point was recorded each second until the G′ and G″ plateaued. The amplitude sweeps were carried out on the gel formed from this experiment. The amplitude sweep applied a constant frequency of 10 rad/s and the strain was ramped logarithmically from 0.01% to 100%. The normal force was kept constant at 0 N and 6 points were recorded for each decade. All measurements were repeated in triplicate and representative charts are shown.
Uniaxial Compressive Tests
All uniaxial compressive testing was performed on a M100-1CT Testometric fitted with a load cell of 1 kN. Hydrogel samples were prepared with a 2 mL syringe to give a cylindrical shape with a diameter of 9 mm and length of 4 mm. Samples were left to cure for 1 h after forming, to ensure the cross-linking reaction was complete before being tested. A preload force of 0.1 N was set and each test was carried out at a compression velocity of 5 mm min–1. Each gel was subject to 98% strain in order to determine the ultimate compressive stress and strain. All compression tests were repeated 10 times and an average of the data was taken to find the ultimate compressive stress and strain. Data was analyzed using Wintest analysis software. Young’s modulus was calculated from the initial 10% of the stress/strain curve and is defined as
| 4 |
Cryogenic Scanning Electron Microscopy
Cryogenic scanning electron microscopy (cryo-SEM) was performed on ZEISS SUPRA 55-VP equipped with cold stage and sample preparation chamber. In a typical procedure, a hydrogel sample was formed in situ on a stub and the top layer was sliced off. The stub was placed in a prefrozen stub adaptor and frozen in liquid nitrogen (−195 °C) which was obtained by placing the sample in liquid nitrogen under vacuum. The stub was then transferred to the cold stage (at −125 °C) of the preparation chamber connecting to the SEM chamber. The frozen sample was surface fractured and sublimated at −95 °C for 15 min to reveal the cross-sectional surface. The temperature was then brought down to −125 °C and the sample was sputter coated with platinum before being transferred under vacuum into the SEM chamber, which was kept at −186 °C for imaging. The accelerating voltage was set at 2 kV to avoid burning the sample.
Swelling and Degradation Studies
Hydrogels were fabricated as stated in the previous procedure and left to cure for 1 h at room temperature. The prepared hydrogels were then placed in PBS solution pH 7.4 and incubated at 37 °C in an orbital shaker-incubator (Model ES-20, Grant Instruments (Cambridge) Ltd.) with a shaking speed of 80 rpm. The PBS solution was replaced regularly to remove unreacted PEG precursors and to prevent the buildup of solute concentration. At preset time intervals, the hydrogels were removed, gently blotted dry and the weight was recorded. The swelling factor (SF) and degradation was monitored by the percentage of weight of the hydrogel at each time point (t) compared to the weight before submersion which is defined as
| 5 |
where Wt is the weight measured at specific time point and W0 is the initial wet weight before immersed (after 1 h).
Biocompatibility Studies and 3D Cell Encapsulation
MC3T3-E1 were purchased from Public Health England and cultured as advised by supplier in MEM-α medium with addition of 10% FBS and 1% penicillin/streptomycin. For the biocompatibility studies, hydrogels were prepared in culture medium in 24 wells cell culture inserts with a 0.4 μm pore PET membrane, as described in the previous section, in a total volume of 200 μL. After 30 min incubation at 37 °C, 100,000 cells suspended in 20 μL of medium were seeded on top of the hydrogel, and the material was incubated for 2 h before addition of fresh medium on top and around the hydrogel. Bioviability was assessed using a PrestoBlue viability assay. Fluorescence was measured using a BioTek plate reader (λEx. = 530 nm, λEm. = 590 nm). For 3D encapsulation studies, 500,000 cells per hydrogel were suspended with the alkyne precursor, followed by addition of the thiol precursor prior mixing to obtain a 200 μL hydrogel. Cells were incubated up to 72 h, after which they were stained with calcein for live cells (λEx. = 495, λEm. = 515) and ethidium homodimer for dead cells (λEx. = 528, λEm. = 617) and imaged using a confocal fluorescence microscope. Finally, cell viability was also evaluated in the presence of degradation products from hydrogels incubated in complete culture medium (MEM-α with 10% FBS and 1% pen/strep). Hence, three hydrogels for each system were prepared as previously described and then immersed in culture medium (5 mL) for 6 days at 37 °C under slight shaking (80 rpm). The solutions were then sterile filtered (0.22 μm). MC3T3 cells were seeded at 2000 cells cm−2 in 24 well plates and incubated in culture medium for 24 h. Serial dilutions of each degraded gel were prepared (from 0.625 to 5 mg mL−1), and 1 mL of each dilution was added to each well and incubated under standard culture conditions (37 °C, 5% CO2). Cell viability was evaluated using the PrestoBlue (λEx. = 530 nm, λEm. = 590 nm) metabolic assay at three time points, 24 h, 72 h, and 7 days. Experiments were performed in triplicate.
Results and Discussion
Synthesis of Alkyne- and Thiol-Terminated PEG Precursors
The design of nonswellable hydrogels has been achieved by two accessible strategies that alter the swelling characteristics of our previous robust PEG-based hydrogels prepared through the nucleophilic thiol–yne addition reaction (Scheme 1).16 Overall, both routes use simple end group modifications which are highly efficient with conversion values higher than 84% (Figures S1–S9 and Table S1). The hydrogel nomenclature is dependent on the PEG precursors, each precursor is denoted considering the number of arms, molecular weight, and the incorporated functionality, S for thiol or A for alkyne. Hence, 31S refers to the 3-arm thiol-terminated PEG precursor (1 kg mol–1).
Scheme 1. Nucleophilic Base-Catalyzed Thiol–Yne Reaction.
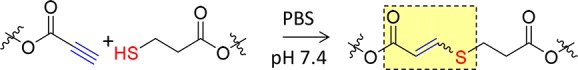
To increase the cross-linking density of the hydrogel network (route one), and thus reducing the hydrophilicity of the system, alkyne- and thiol-terminated polymeric precursors were prepared by modifying multiarm PEG-hydroxyl groups (Scheme 2a). Specifically, 3-arm (1 kg mol–1) and 4-arm (2 kg mol–1) PEG polymers were functionalized through Fischer esterification of the hydroxyl end-group with either propiolic acid or mercaptopropionic acid. The thermoresponsive nonswellable click hydrogels (route two) were synthesized using a thiol-terminated Pluronic PEG precursor, PEG–PPG–PEG (2900 g mol−1, Pluronic L-64, 40% of EG content) with mercaptopropionic acid, hereafter denoted 2P3S (Scheme 2b).17 After modification, the lower critical solution temperature (LCST) of the commercially available Pluronic decreased from 55−60 °C down to 25–35 °C (SI, Figure S10), which is below 37 °C, and thus highly suitable for our application. Consequently, when injected into the body, the 2P3S segments will induce the collapse of the click-hydrogel network preventing it from swelling.
Scheme 2. Nonswellable Click-Hydrogels.
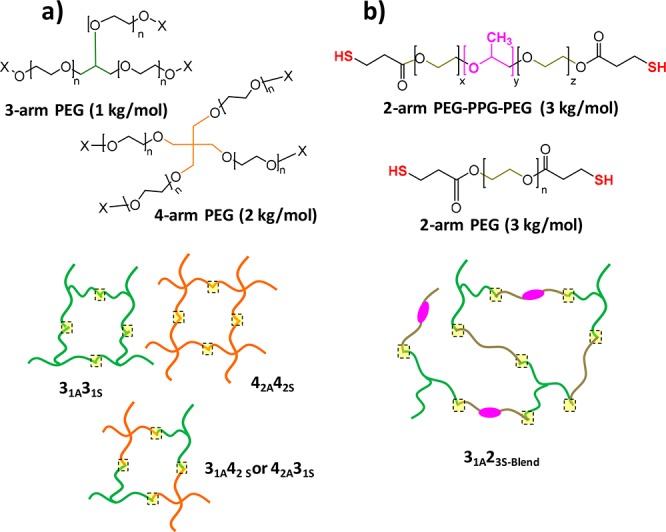
Based on (a) multi-arm and (b) thermoresponsive PEG precursors.
Once the modified PEG precursors were synthesized, click-hydrogels with 10% w/v polymer content were subsequently prepared by mixing solutions containing a 1:1 molar ratio of alkyne to thiol end groups in PBS solution at pH 7.4.
Characterization of Swellable PEG-Containing Click-Hydrogels
In general, most hydrogels swell when placed in aqueous solution; consequently, their volume increases, and severe alterations of their mechanical properties occur. In order to characterize the swelling behavior of the previously reported related system, thiol–yne PEG-based click-hydrogels (31A21S, 31A22S, and 31A23S) were prepared using 2-arm thiol-terminated PEG precursors to provide control measurements. As expected, these systems undergo swelling up to 179.9% ± 29.4%, 443.0% ± 7.4%, and 408.0% ± 9.0%, respectively, before degrading completely in the aqueous solution within 3 and 4 days (Figure 1a). For thiol–yne click-hydrogels, bulk hydrolytic degradation occurs, which results from the combination of the diffusion of water into a polymer network and the subsequent random cleavage of hydrolyzable bonds.30−32 In addition, carboxylic acid groups, from the ester cleavage, display an autocatalytic effect. The polymeric nature of these degradation products, many of which are likely to remain attached to the network, accelerate degradation and contribute to the loss of structural and mechanical stability. Ultimately, as we observe, the hydrogel network swells and then dissolves into the PBS solution.33
Figure 1.
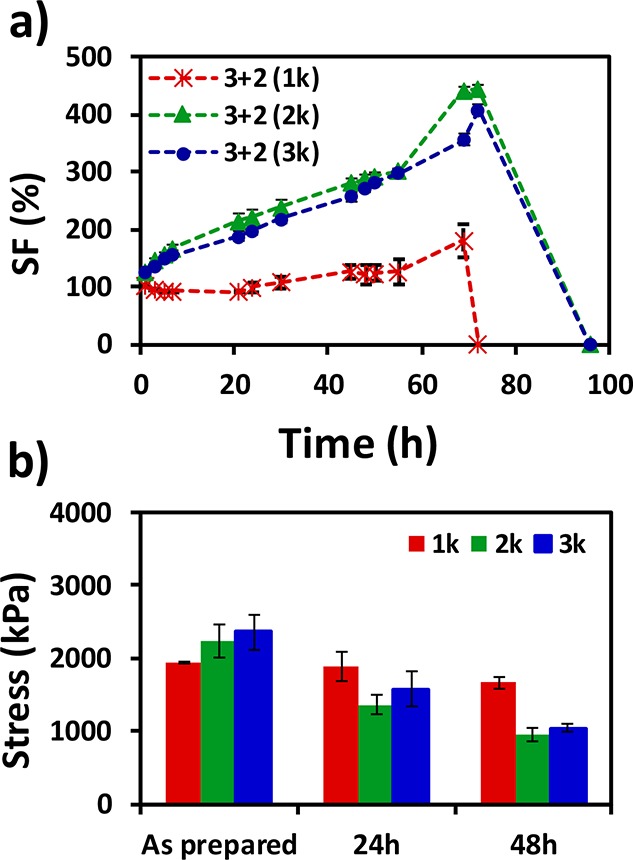
Swellable hydrogels characterization (31A21S, red; 31A22S, green; 31A23S, blue). (a) Swelling factor (SF) (%) as a function of time in PBS solution at 37 °C and (b) evolution of stress at breaking with swelling time. Compressive modulus data can be found in the SI (Table S3 and Figure S11).
Additionally, swelling experiments also reveal information on the equilibrium water content (EWC) and mesh size of the hydrogel networks. Both features are key factors for tissue engineering scaffolds: cells require high water content, as well as an adequate diffusion of nutrients, oxygen, and growth factors in order to grow and proliferate. High EWC percentages (94–95%, Table 1) confirmed that the swellable click-hydrogels contained porous structures with the ability to hold large amounts of water. The average mesh size of these thiol–yne networks, calculated using the Flory–Rehner equation,29 ranged between 5.8 and 6.4 nm (Table 1). Although the pore size of the hydrogels determined by Cryo-SEM observation differs from the previous values (i.e., pore size in the range of μm), a porous structure was observed for the three systems (data not shown).
Table 1. Gelation Time (GT), Gelation Fraction (GF), and Swelling Kinetics (Equilibrium Water Content, EWC) of Hydrogels at 10 w/v% in PBS Solution (1:1 Molar Ratio of Alkyne to Thiol End Groups)a.
| GTb (s) | GF (%) | EWC (%) | Mesh Sizec (nm) | |
|---|---|---|---|---|
| 31A21S | 210 ± 7 | 74 ± 2.6 | 95 ± 0.9 | 5.8 ± 0.49 |
| 31A22S | 290 ± 10 | 80 ± 1.9 | 94 ± 0.8 | 6.4 ± 0.16 |
| 31A23S | 438 ± 5 | 78 ± 4.9 | 95 ± 0.1 | 5.9 ± 0.03 |
| 31A31S | 26 ± 3 | 78 ± 1.2 | 90 ± 0.2 | 4.2 ± 0.07 |
| 31A42S | 25 ± 9 | 75 ± 1.3 | 90 ± 0.5 | 4.7 ± 0.14 |
| 42A31S | 15 ± 5 | 92 ± 0.7 | 87 ± 1.7 | 4.4 ± 0.47 |
| 42A42s | 11 ± 2 | 89 ± 1.8 | 86 ± 0.9 | 4.4 ± 0.24 |
| 31A2P3S | 10 min | 79 ± 1.3 | 58 ± 9.8 | 1.2 ± 0.48 |
| 31A23S-6040 | 8–10 min | 84 ± 1.2 | 86 ± 0.2 | 3.7 ± 0.04 |
The hydrogel naming (XZAYZS) denotes its structure, where X = number of arms of the alkyne precursor, Y = number of arms of the thiol precursor, and Z = molecular weight of the PEG precursor.
Measured via the vial tilt methods.
Calculated from the Flory–Rehner equation (see SI).
As expected, the swelling response of the control click-hydrogels (31A21S, 31A22S, and 31A23S) had a significant negative effect on their mechanical performance (Figure 1). The compressive strength and modulus values decrease with immersion time (Figure 1b and Table S2), which becomes more evident with increasing molecular weight of the thiol-terminated PEG precursor. For instance, for the 31A23S system, the compressive modulus before and after 48 h of swelling was 33.6 ± 2.6 kPa and 5.6 ± 1.1 kPa, respectively; whereas the compressive strength decreased from 2.4 ± 0.3 MPa to 1.0 ± 0.05 MPa during the same swelling time.
All the hydrogels broke when 98% strain was reached therefore demonstrating a hydrogel with longer, more flexible PEG chains can withstand large amounts of strain. However, these PEG chains also display a hydrophilic character, thus attracting more water molecules into the network, which in turn accelerates degradation rates and impairs the mechanical performance. Hence, to counteract this effect, we targeted specific modifications of the PEG precursors to render the click-hydrogels nonswellable (Scheme 2). The resulting nonswellable hydrogels were immersed in PBS solution at 37 °C for 30 days or until degradation, and their swelling response and mechanical properties in the swollen state were assessed periodically.
Characterization of Nonswellable PEG-Containing Click-Hydrogels
The water uptake of thiol–yne click-hydrogels immersed in PBS solution at 37 °C was effectively regulated by applying two straightforward approaches that are based on different principles. The first route relies on rendering the polymeric network more hydrophobic by increasing the number of cross-linking sites and also reducing the molecular weight of the hydrophilic polymer backbone.
Hence, in our multiarm thiol–yne hydrogel systems, the 2-arm thiol-functionalized PEG precursor was replaced with either a 3- or 4-arm precursor (Scheme 2a). Four multiarm PEG precursors were synthesized (31S,31A,42S,42A), and accordingly four different click-hydrogel systems were prepared as a result of different combinations: 31A31S, 31A42S, 42A31S, and 42A42S.
In the second strategy, the polymeric network 31A2P3S (Scheme 2b) was designed to contain thermoresponsive segments (2P3S) that collapse above a certain temperature (i.e., cloud point 25–35 °C). Gel fraction (GF) values (Table 1), which indicate the efficiency of the cross-linking process, were higher than 74% for all the systems tested regardless of the followed route, thus highlighting the suitability of the nucleophilic thiol–yne chemistry for hydrogel synthesis.
Gelation Time
All the multiarm click-hydrogels formed almost immediately after mixing the alkyne- and thiol-containing solutions. Gelation times for these systems, which were first determined by the vial tilt method, range between 11 and 26 s (Table 1). The high rate of reaction for the nucleophilic thiol–yne addition was attributed to the elevated number of cross-linking points that are now spatially closer, thus resulting in this rapid gelation. This effect is more pronounced for the 42A42S hydrogel, which is formed with the 4-arm PEG precursors, which displays the highest number of reactive groups per molecule. In contrast, all the control systems, which are formed with a 2-arm thiol PEG precursors, displayed higher gelation times that increase with molecular weight.
The gelation time for the thermoresponsive hydrogel (31A2P3S) was slower (10 min) as a consequence of the low temperature during preparation, which affected the click-reaction rate. Lower temperatures were required as a consequence of the PEG precursors cloud point (cloud point 25–35 °C).17 The solution of the thiol-terminated Pluronic (2P3S) at 10 wt % was cloudy at room temperature. Therefore, all the preparation steps for hydrogels containing 2P3S as the thiol moiety were performed below 10 °C by cooling the solutions in an ice bath. In doing so, we assured that the thermoresponsive polymer was completely soluble in the aqueous buffer for an effective cross-linking. After mixing the PEG-containing precursors, gelation proceeded at room temperature (23 °C), and the resulting hydrogels were completely transparent.
Among the different theories applied to the gelation of polymers, the Flory–Stockmayer theory states that gelation occurs when systems of high functionality produce an infinite molecular weight network through cross-linking provided that the reaction is carried far enough, thus shifting the system from the liquid to the gel phase.34−36 This definition is derived from the probability of finding a cross-linking point during gelation. Hence, the gelation point, which is defined at a certain extent of reaction, is reached when a critical number of intermolecular linkages have been exceeded. More specifically, this theory states that the theoretical critical amount of cross-links needed to form a gel, pc, can be calculated by using the following equation 6
| 6 |
where fA is the functionality of the alkyne PEG precursors, fB is the functionality of the thiol PEG precursors, and r is the ratio between the total number of alkyne and thiol groups (i.e., r = 1 in our case because we are working under stoichiometric conditions).37
For our systems with a trifunctional alkyne precursor and a bifunctional thiol precursor (i.e., 31A21S, 31A22S, 31A23S, 31A2P3S, 31A23S-6040), the value that the Flory–Stockmayer theory predicts for αc is 0.71, which means that gelation takes place when the amount of alkyne PEG precursors that have reacted is higher than 71%. In the case of the multiarmed hydrogel systems with either a tri- or tetrafunctional PEG precursor, the value obtained for αc is 0.50, 0.41, and 0.33 for 31A31S,31A42S or 42A31S, and 42A42S, respectively, thus indicating that these systems reach the gelation point with fewer intermolecular linkages. Hence, although other factors such as temperature or molecular weight of the PEG precursors influence gelation, the gelation time values observed by the vial tilt method are in good agreement with the values predicted from the Flory–Stockmayer theory.
Swelling Profile
Both strategies were able to suppress swelling and thus control the increase in volume; however, the resulting click-hydrogels exhibited distinct swelling behaviors (Figure 2). In the multiarm click-hydrogels, the rate at which each hydrogel expelled and attracted water was dependent on the architecture of the thiol- and alkyne-terminated PEG precursors. All four systems rapidly shrunk after the initial 24 h immersed in PBS solution at 37 °C (Figure 3a,b). We postulate that the increased density of cross-linked sites promotes the hydrophobic nature of the hydrogel network, thus repelling water out of system. Although the hydrogels remained unchanged for 5 days with swelling factor (SF) values in the range between 46% ± 0.1% and 71% ± 3.1%, they started to swell back slowly to 100%. After 21 days, signs of hydrogel degradation appeared which allowed an increased amount of water into the network, thus accelerating the swelling rate rapidly and in turn the degradation rate.
Figure 2.
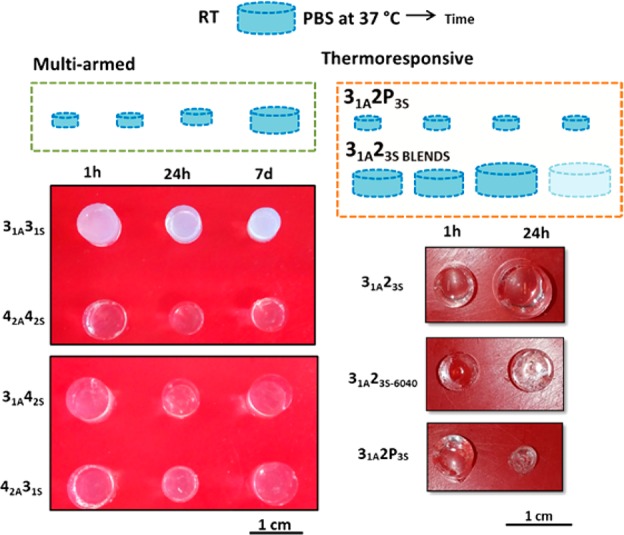
Thiol–yne click-hydrogels response when immersed in PBS solution at 37 °C.
Figure 3.
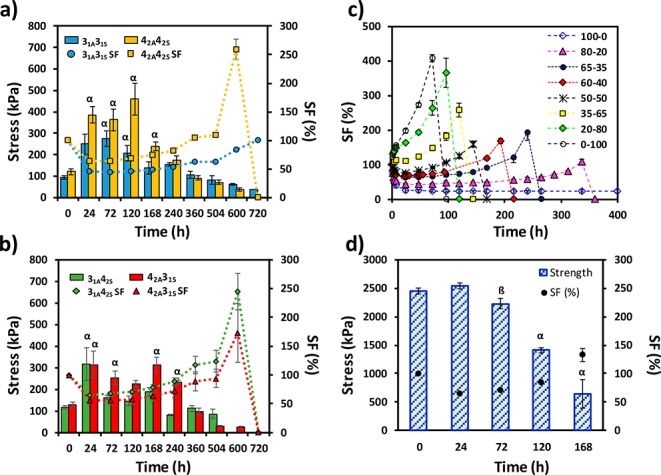
(a,b) Evolution of the compressive mechanical properties (bar data; left axis) with the SF (symbols, right axis) for the nonswellable nucleophilic thiol–yne PEG hydrogels displaying multiarm architecture: (a) 31A31S (blue circle) and 42A42S (yellow square); (b) 31A42S (green diamond) and 42A31S (red triangle). Greek letters on the bars refer to significant differences (p-value <0.05): α vs 1 h. (c) SF (%) as a function of time in PBS solution at 37 °C for 31A23S-blends; (d) compressive strength values with immersion time at 37 °C for 31A23S-6040 hydrogels. Greek letters on the bars refer to significant differences (p-value <0.05): α vs all, β vs 24, 120, and 168 h.
Moreover, despite the shrinkage of the polymeric network, multiarm PEG hydrogels also display high EWC values (between 86% and 90%, Table 1) in comparison to control swellable hydrogels, which evidence that changes in the architecture of the PEG precursor have little effect on the network’s ability to hold water. In contrast, they display a reduced average mesh size (4.2–4.7 nm) as a consequence of the increased number of cross-linking sites and shorter polymeric chains.
The 42A42S, 42A31S, and 31A42S behaved similarly; these systems shrunk to 64% after 24 h and then recovered, swelling up to 250% after 25 days in PBS solution before degrading completely. In contrast, the 31A31S hydrogel displayed a SF of 45% after 24 h, which is maintained for 7 days before the system starts to slowly swell to 100%, with no signs of degradation after 30 days. Therefore, we propose that the ratio between cross-linked sites and the length of the PEG arm controls the swelling profile of multiarm click-hydrogels. Indeed, the 31A31S system, which displays the shortest average arm length between each cross-linking site (i.e., 0.33 kg mol–1), results in a very dense system with enhanced hydrophobic nature and reduced ability to attract water. Consequently, the rate of ester hydrolysis is low in this system, which reduces the degradation rate of the polymeric network.
In contrast, the 31S2P3S click-hydrogels started to shrink almost immediately after immersing in PBS solution. The SF decreased to 27.1% ± 5.4% after 21 h, which indicates that water is quickly being expelled from the polymeric network (Figure S16a). However, at this point, the SF value remained constant for 30 days with a mean value of 24.5% ± 1.5%. Accordingly, the EWC value for 31A2P3S click-hydrogels is 58% ± 9.8%. The low water content for this system is responsible for slowing down the hydrolysis of the ester bonds present in the polymeric network, and thus 31S2P3S hydrogels did not undergo any degradation process within the observed period of time, as opposed to multiarm click-hydrogels, which started to degrade after 21 days of swelling.
Nevertheless, 31A2P3S click-hydrogels can hardly be considered suitable materials for tissue scaffolds. Although their mechanical properties are adequate (vide infra), we suspected that their low water content might endanger the viability of cells encapsulated within. Hence, we decided to exert a more direct control over the swelling response of the system by blending 2P3S and 23S as thiol-terminated PEG precursors. Accordingly, hydrogels with varying 2P3S:23S molar ratio displayed an intermediate swelling response between that shown by 31A23S and 31A2P3S (Figure 3c). Blended hydrogels with a content of 2P3S higher than 50% reached a stable SF below 100% within 20 h, whereas hydrogels containing a higher proportion of the PEG precursor 23S underwent considerable swelling and degraded in less than 5 days. Thus, by varying the thermoresponsive content, not only does this approach allow for good control of the water uptake but also affects the degradation rate of the hydrogels, which are stable for a period of time that ranges between 5 and 15 days, and at least 30 days in the case of 100% Pluronic content (Figure S16a). Besides, all the thermoresponsive click-hydrogels retain their original transparency at 37 °C (Figure 2), which suggests that their network structure is homogeneous regardless the content of Pluronic content.
It is important to note a few considerations: the degree of swelling expected for an injectable hydrogel should be close to 100%, which means just enough swelling for an “expansion fit” of the hydrogel within the void space. A considerable increase in volume could potentially damage the nearby tissue, causing pain and triggering an inflammatory response, while a sudden shrinkage not only implies low water content, but also results in a smaller hydrogel which will not fill the vacant space. Moreover, hydrogels as tissue engineering scaffolds are required to be mechanically stable and robust for as long as possible, allowing cells to adhere, grow, and eventually develop new tissue while the hydrogel slowly degrades.
Taking these considerations into account, we evaluated all the Pluronic thiol–yne hydrogels compositions and calculated (Figure S12a) that a click-hydrogel with a 2P3S:23S molar ratio of 60:40 (hereafter denoted 31A23S-6040) met the required criteria for further characterization. Indeed, this system, which displays a gelation time of 8–10 min and a GF value of 84% ± 0.2% (Table 1), exhibits a mean SF of 71.0% ± 4.4% for an overall period of 7 days, when it finally starts to swell and degrades (day 10) (Figure 3c). The polymeric network of the blended click-hydrogel 31A23S-6040 does not shrink as abruptly as in the case of 31A2P3S, thus the hydrogel still retains a high amount of water at 37 °C (EWC is 86.0 ± 0.2%). However, despite the presence of thermoresponsive units, the immersion temperature has a severe effect on the hydrogel’s degradation rate (Figure S12b); 31A23S-6040 hydrogels immersed in PBS solution at RT and 4 °C display longer stability (22 and at least 37 days, respectively).
Morphology
As a consequence of the general collapse of the thermoresponsive click-hydrogel network, the mesh pore size derived from swelling kinetics was calculated to be 1.2 ± 0.48 nm and 3.7 ± 0.04 nm for 31A2P3S and 31A23S-6040 hydrogels, respectively. Both values are clearly smaller than those displayed by multiarm PEG hydrogels (4.2–4.7 nm) showing a dependence on the mesh size with swelling. These mesh sizes could affect how cells interact with the network as it has been reported that smaller mesh size and stiffer matrices restrict 3D cell spreading,38 as well as influence cell morphology.39−41
Cryo-SEM characterization was also conducted to understand the morphology of the networks and the correlation between ice crystal growth and the different PEG precursors. For all systems, the structure revealed is porous (Figure 4 and S13) with a range of different sizes that ice crystals can grow to during the freezing process. The difference sizes ranged between 1.64 ± 0.36 μm and 4.20 ± 2.74 μm, which corresponded to 31A23S-6040 and 42A42S, respectively (Figure S14). Multiarm click-hydrogels prepared with 42A exhibited a much broader distribution of ice crystal sizes, while using 31A resulted in click hydrogels with a more homogeneous structure. Additionally, the ice crystal structure for the 31A42S is significantly smaller in comparison to the other three multiarm hydrogel systems. Similarly, thermoresponsive hydrogels, which are prepared with 31A, also reveal a narrower and more homogeneous distribution of ice crystals. Consequently, using 31A as alkyne-modified PEG precursor results in hydrogel networks which are more densely packed, which could influence cell behavior.
Figure 4.

Cryo-SEM images of nonswellable thiol–yne PEG hydrogels: (a) 31A23S, (b) 31A31S, (c) 31A2P3S, and (d) 31A23S-6040 (Scale bar = 2 μm).
Rheological Characterization
Rheological tests were carried out to fully evaluate the viscoelastic properties of the nonswellable thiol–yne click-hydrogels and determine any possible drawback derived from either of the approaches followed to suppress the swelling response. For comparison purposes, the control hydrogel 31A23S was also characterized.
First, by monitoring the evolution of both the storage (G′) and loss (G″) moduli of a sample with time, the gelation time, which is defined as the crossover point between G′ and G″, can be determined. The gelation time for the four multiarm systems was within 30 s (Figure 5a), which is in good agreement with the results from the vial tilt method, thus indicating that the hydrogels are fully cross-linked into stable and robust networks despite the quick gelation process. As previously observed, increasing the concentration of reactive end groups in solution reduces the gelation time. The click hydrogels with thermoresponsive units showed a gelation time of about 10 min (Figure 5c) and also present a stable structure after 25 min.
Figure 5.
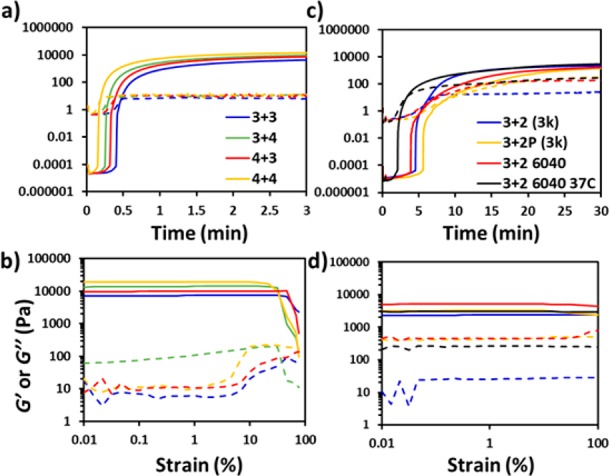
Representative curves of storage (G′; solid line) and loss (G″; dashed line) modulus as a function of (a,c) time, and (b,d) strain for nonswellable PEG click-hydrogels with multiarm architecture (left column) and thermoresponsive units (right column).
The viscoelastic properties were determined by monitoring the storage (G′) and loss (G″) moduli as the strain on the hydrogel increased (Figure 5b). All the multiarm hydrogels showed high G′, which is indicative of highly viscoelastic materials. Moreover, G′ increased with the number of functional groups per molecule, thus suggesting that a more rigid structure is formed as a consequence of the higher density of cross-linking points. Consequently, the stiffer materials were unable to withstand high levels of strain. For instance, G′ decreases at 32% of strain for 42A42A and 31A42S, whereas click-hydrogels with a more elastic network (42A31S and 31A31S) start to yield at higher strain (47%). In contrast, the thermoresponsive click-hydrogels exhibited G′ values which are similar to the control hydrogel 31A23S and comparable to those displayed by multiarm click-hydrogels (Figure 5d and Table S3) regardless of their composition.
Moreover, the high G′ values remained stable during the whole amplitude sweep range, even at high strain, which reveals a more elastic nature of the polymeric network as a consequence of using 2-arm instead of 3- or 4-arm thiol-functionalized PEG precursors. Therefore, although the architecture of the PEG precursors affects the viscoelastic properties of the resulting click-hydrogels, it can be exploited to finely tune these features, which can be potentially useful for biological applications, such as tuning the scaffold stiffness to influence cell morphology and spreading.38−40
Furthermore, temperature had no significant effect on the rheological properties of the blended click-hydrogel 31A23S-6040; both G′ and G″ were retained despite the partial shrinkage of the hydrogel at 37 °C, thus further confirming the suitability of this system for an in vivo bioapplication.
Mechanical Characterization in the Swollen State
Uniaxial compression testing was performed over a 30-day swelling period to evaluate the mechanical performance of nonswellable PEG-containing click-hydrogels in the swollen state. Specifically, hydrogels were prepared and left to cure for 1 h to allow for the hydrogels to be fully cross-linked, before immersing them in PBS solution at 37 °C. After a set period of time, their mechanical properties were characterized.
The thiol–yne multiarm click-hydrogels presented a range of compressive strengths that was related to both their swelling factor (SF) and immersion time. After the initial 24 h in PBS solution at 37 °C, their compressive strength values increased (Figure S15). Specifically, for the 42A42S system, which displayed the greatest change, the compressive strength increased from 120 ± 13 kPa to 385 ± 37.5 kPa in 24 h (Figure 3a,b, Tables S2 and S3). This response is in good agreement with the decrease of the SF. As water molecules are expelled from the hydrogel network, the polymer content increases and the mechanical properties of the multiarm hydrogels improve. Moreover, as long as the SF remained constant, no changes in the mechanical performance were observed. For instance, the 31A42S system reached its maximum strength after 24 h, while the 42A42S system displayed its highest stress values after 5 days, which also reflects the unique properties of each one of the click-hydrogels based on the molecular weight of the PEG precursors, as well as their swelling response.
However, when the swelling factor increased past 100% (after 15 days), the mechanical properties of the multiarm click-hydrogels were severely compromised. Once swollen, the accelerated rate of ester hydrolysis promotes the degradation process and thus weakens the hydrogel structure, which is reflected by a reduction in the compressive strength after 25 days of immersion. Finally, when SF values higher than 150% were reached, the click-hydrogels degraded very rapidly and dissolved into the PBS solution.
It is important to note that the compressive strength values exhibited by multiarm click-hydrogels are not as high as those displayed by the control swellable systems, mainly as a consequence of the increased stiffness of the system. The polymeric chains are highly cross-linked and unable to adapt and withstand an external stress; the multiarm hydrogels break at lower strain values (48–66% for the multiarm series versus 95–96% for the thermoresponsive series Table S2). Nevertheless, despite this fact, they are able to retain their mechanical performance over time in the swollen state for longer (Figures 3a,b and S17), and thus can be effectively applied as biodegradable scaffolds for soft tissue regeneration in a biological context where extremely robust hydrogels are not required.
Similar to the response displayed by the multiarm nonswellable hydrogels, the mechanical performance of 31A2P3S click-hydrogels was altered after being immersed in PBS solution at 37 °C (Figure S16, Tables S2 and S3). However, in contrast to the multiarm system, the high compressive strengths observed in the related hydrogels that we have previously reported16 were maintained. Specifically, after only 24 h of immersion time, both the compressive strength and Young’s Modulus of the system increased from 2.5 ± 0.2 MPa and 18 ± 2.2 kPa (i.e., values comparable to those displayed by control swellable hydrogels) to 5.5 ± 0.2 MPa and 83.7 ± 11.3 kPa, respectively, a fact that is attributed also to the low water content in the hydrogel, which makes it extremely strong; hydrogels were rubbery and did not break after reaching 98% compression during testing. Since the SF of 31A2P3S click-hydrogels does not change significantly with time after a constant SF value of about 25% is reached after 24 h (i.e., no degradation process was observed as opposed to multiarm click-hydrogels), they retain this excellent mechanical performance even after 14 days of immersion (Figure S16).
Finally, we confirmed that, as long as the blended click-hydrogel 31A23S-6040 is not highly swollen, the mechanical performance is preserved (Figure 3d and S17). The high compressive strength of the blended system, which is 2.5 ± 0.06 MPa as-prepared, is significantly lower after 5 and 7 days of swelling (1.4 ± 0.04 MPa and 0.64 ± 0.25 MPa, respectively). Although there is indeed a decrease in the mechanical performance, the compressive strength values shown by the swollen blended click-hydrogels 31A23S-6040 are still high. Furthermore, the Young’s modulus experiences a similar decreasing trend, from 23.7 ± 1.9 kPa to 8.1 ± 1.7 kPa and 5.8 ± 2.2 kPa after 5 and 7 days of swelling, respectively.
Overall, the major factor influencing the thiol–yne click hydrogels mechanical response is the swelling degree, and thus the condition of the polymeric matrix.25 On the one hand, the architecture of the PEG precursors has a fundamental influence on the swelling response of the resultant multiarm thiol–yne click-hydrogels, which in turn affects their compressive strength. Although the multiarm click-hydrogels display lower compressive strength values in comparison to the thermoresponsive systems (but within the range of soft load bearing tissue), their enhanced stability allowed them to retain their mechanical performance for at least 15 days. On the other hand, fine tuning of the swelling response as well as the degradation rate of robust thermoresponsive thiol–yne click-hydrogels is obtained by controlling the molar ratio of thiol-terminated PEG precursors (2P3S and 23S).
Assessing the Biocompatibility of Nonswellable Click-Hydrogels
Finally, to evaluate the suitability of thiol–yne nonswellable click-hydrogels in a biological setting, their biocompatibility was assessed. A representative hydrogel of each route was chosen, 31A42S as multiarm click-hydrogel and 31A23S-6040 as the thermoresponsive hydrogel. Also, 31A2P3S (100% Pluronic content) and 31A23S (2-arm thiol-terminated PEG-3k) hydrogels were used as control systems.
First, cell viability tests on MC3T3-E1 (murine preosteoblasts) cells were conducted by seeding them on top of the hydrogels, which had been prepared in situ directly in the wells of the TCPS plates, thus no washing steps were applied to the hydrogels. After 72 h the cell’s metabolic activity was measured using PrestoBlue metabolic assay (Figure S18). All hydrogels demonstrated high cell viability which indicated that the click-hydrogel networks are biocompatible once formed. Most importantly, the extended time points reveal that within that incubation time, their degradation products are not cytotoxic, which further reinforces their potential as tissue engineering scaffolds able to support cell growth.
Following the promising initial results, 3D cell encapsulation experiments were also undertaken, and cell viability was assessed after 24 and 72 h of incubation by live–dead fluorescent staining (Figure 6). Since the preparation of the thermoresponsive hydrogels is performed at low temperature, another control system that consisted in preparing 31A23S at low temperature was also included. After 24 h of incubation, all the hydrogels showed high cell viability with >95% live cells, which indicates that the materials are nontoxic at this stage (Figure S19). Moreover, we confirmed that the low preparation temperature had no effect on cell viability. In contrast, after a longer incubation time (72 h), a high percentage of cell death was observed only for the thermoresponsive hydrogel 31A2P3S, which contains 100% of Pluronic as thiol-terminated precursor (Figure S18). Both the blended PEG/Pluronic click-hydrogels and the multiarm PEG hydrogels displayed high percentage of live cells, and hence we attribute the low cell viability of encapsulated cells within 31A2P3S hydrogel to several factors: the severe collapse of the polymeric network (SF = 24.5% ± 1.5%), the small mesh size (1.2 ± 0.48 nm), the low water content of the hydrogel (58% ± 9.8%), as well as the cytotoxic effect of 2P3S (Figure S21). Interestingly, the stiffer nature of the multiarm 31A42S click-hydrogel did not affect the viability of 3D encapsulated cells.
Figure 6.
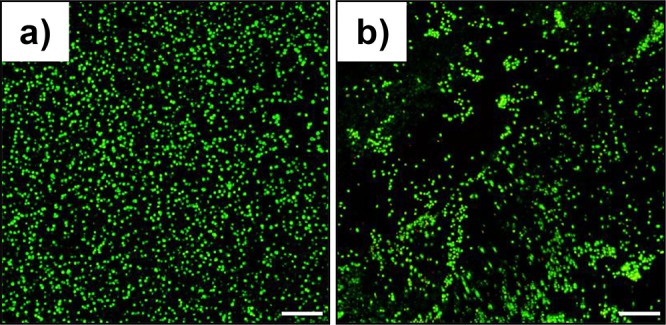
Biocompatibility of nonswellable thiol–yne PEG click-hydrogels. Images from 3D encapsulated cells after 72 h of incubation for (a) 31A42S and (b) 31A23S-6040. Scale bar is 200 μm.
Conclusions
The design of a biomaterial device that perfectly matches all the requirements for a specific biological context is challenging. Here, we have presented two approaches to overcome the negative effect of water uptake on the mechanical properties of robust click-hydrogels cross-linked by thiol–yne nucleophilic addition. Through the judicious choice of the alkyne- and thiol-terminated poly(ethylene glycol)-based precursors, either multiarm or thermoresponsive, the nonswellable click-hydrogels preserve their mechanical performance in the swollen state over time, as well as displaying good viscoelastic properties and finely tunable degradation rates. Most importantly, both approaches yielded nonswellable click-hydrogels that are highly biocompatible. Not only were cells able to adhere and grow on the top surface of the hydrogels, but they were also viable when encapsulated within the polymeric network as long as the water content and mesh size allow for adequate diffusion of oxygen and nutrients. Therefore, this work highlights the suitability of nonswellable click-hydrogels prepared by efficient thiol–yne chemistry as potential candidates to address the major issues of hydrogels as biomaterials.
Acknowledgments
Prof. Matthew Gibson is thanked for giving access to his cell lab facilities. The University of Warwick Advanced BioImaging Research Technology Platform, BBSRC ALERT14 award BB/M01228X/1 are thanked for confocal fluorescence microscopy analysis. M.M.P.-M. acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 703415. EPSRC are thanked for the award of a DTP studentship to L. J. M. and ERC are acknowledged for support to M. C. A and A. P. D. (grant number: 681559)
Glossary
ABBREVIATIONS
- PEG
poly(ethylene glycol)
- PBS
phosphate buffer saline
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.7b01204.
NMR and SEC data of alkyne and thiol precursors, LCST data for Pluonic3k(SH)2(2P3S), additional hydrogel characterization data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author Contributions
‡ L.J.M. and M.M.P.-.M. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Kamata H.; Li X.; Chung U.-i.; Sakai T. Design of Hydrogels for Biomedical Applications. Adv. Healthcare Mater. 2015, 4 (16), 2360–2374. 10.1002/adhm.201500076. [DOI] [PubMed] [Google Scholar]
- Gyles D. A.; Castro L. D.; Silva J. O. C.; Ribeiro-Costa R. M. A review of the designs and prominent biomedical advances of natural and synthetic hydrogel formulations. Eur. Polym. J. 2017, 88, 373–392. 10.1016/j.eurpolymj.2017.01.027. [DOI] [Google Scholar]
- Tam R. Y.; Smith L. J.; Shoichet M. S. Engineering Cellular Microenvironments with Photo- and Enzymatically Responsive Hydrogels: Toward Biomimetic 3D Cell Culture Models. Acc. Chem. Res. 2017, 50 (4), 703–713. 10.1021/acs.accounts.6b00543. [DOI] [PubMed] [Google Scholar]
- Li Y.; Rodrigues J.; Tomas H. Injectable and biodegradable hydrogels: gelation, biodegradation and biomedical applications. Chem. Soc. Rev. 2012, 41 (6), 2193–2221. 10.1039/C1CS15203C. [DOI] [PubMed] [Google Scholar]
- Slaughter B. V.; Khurshid S. S.; Fisher O. Z.; Khademhosseini A.; Peppas N. A. Hydrogels in Regenerative Medicine. Adv. Mater. 2009, 21 (32–33), 3307–3329. 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q.; Zou Y.; Arno M. C.; Chen S.; Wang T.; Gao J.; Dove A. P.; Du J. Hydrogel scaffolds for differentiation of adipose-derived stem cells. Chem. Soc. Rev. 2017, 46, 6255. 10.1039/C6CS00052E. [DOI] [PubMed] [Google Scholar]
- Kharkar P. M.; Kiick K. L.; Kloxin A. M. Designing degradable hydrogels for orthogonal control of cell microenvironments. Chem. Soc. Rev. 2013, 42 (17), 7335–7372. 10.1039/C3CS60040H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azagarsamy M. A.; Anseth K. S. Bioorthogonal Click Chemistry: An Indispensable Tool to Create Multifaceted Cell Culture Scaffolds. ACS Macro Lett. 2013, 2 (1), 5–9. 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko D. Y.; Shinde U. P.; Yeon B.; Jeong B. Recent progress of in situ formed gels for biomedical applications. Prog. Polym. Sci. 2013, 38 (3), 672–701. 10.1016/j.progpolymsci.2012.08.002. [DOI] [Google Scholar]
- Xu L.; Sheybani N.; Yeudall W. A.; Yang H. The effect of photoinitiators on intracellular AKT signaling pathway in tissue engineering application. Biomater. Sci. 2015, 3 (2), 250–255. 10.1039/C4BM00245H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. G.; Malik A. N.; Kim T. K.; Manson P. N.; Elisseeff J. H. Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation. Biomaterials 2005, 26 (11), 1211–1218. 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- Macaya D.; Spector M. Injectable hydrogel materials for spinal cord regeneration: a review. Biomed. Mater. 2012, 7 (1), 012001. 10.1088/1748-6041/7/1/012001. [DOI] [PubMed] [Google Scholar]
- Truong V. X.; Dove A. P. Organocatalytic, Regioselective Nucleophilic “Click” Addition of Thiols to Propiolic Acid Esters for Polymer–Polymer Coupling. Angew. Chem., Int. Ed. 2013, 52 (15), 4132–4136. 10.1002/anie.201209239. [DOI] [PubMed] [Google Scholar]
- Truong V. X.; Ablett M. P.; Richardson S. M.; Hoyland J. A.; Dove A. P. Simultaneous Orthogonal Dual-Click Approach to Tough, in-Situ-Forming Hydrogels for Cell Encapsulation. J. Am. Chem. Soc. 2015, 137 (4), 1618–1622. 10.1021/ja511681s. [DOI] [PubMed] [Google Scholar]
- Cai X. Y.; Li J. Z.; Li N. N.; Chen J. C.; Kang E.-T.; Xu L. Q. PEG-based hydrogels prepared by catalyst-free thiol-yne addition and their post-antibacterial modification. Biomater. Sci. 2016, 4 (11), 1663–1672. 10.1039/C6BM00395H. [DOI] [PubMed] [Google Scholar]
- Macdougall L. J.; Truong V. X.; Dove A. P. Efficient In Situ Nucleophilic Thiol-yne Click Chemistry for the Synthesis of Strong Hydrogel Materials with Tunable Properties. ACS Macro Lett. 2017, 6 (2), 93–97. 10.1021/acsmacrolett.6b00857. [DOI] [PubMed] [Google Scholar]
- Truong V. X.; Tsang K. M.; Forsythe J. S. Nonswelling Click-Cross-Linked Gelatin and PEG Hydrogels with Tunable Properties Using Pluronic Linkers. Biomacromolecules 2017, 18 (3), 757–766. 10.1021/acs.biomac.6b01601. [DOI] [PubMed] [Google Scholar]
- Chung C.; Lampe K. J.; Heilshorn S. C. Tetrakis(hydroxymethyl) Phosphonium Chloride as a Covalent Cross-Linking Agent for Cell Encapsulation within Protein-Based Hydrogels. Biomacromolecules 2012, 13 (12), 3912–3916. 10.1021/bm3015279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X.; Zhang Y.; Gao L.; Bai T.; Wang W.; Cui Y.; Liu W. A Mechanically Strong, Highly Stable, Thermoplastic, and Self-Healable Supramolecular Polymer Hydrogel. Adv. Mater. 2015, 27 (23), 3566–3571. 10.1002/adma.201500534. [DOI] [PubMed] [Google Scholar]
- Luo F.; Sun T. L.; Nakajima T.; Kurokawa T.; Zhao Y.; Sato K.; Ihsan A. B.; Li X.; Guo H.; Gong J. P. Oppositely Charged Polyelectrolytes Form Tough, Self-Healing, and Rebuildable Hydrogels. Adv. Mater. 2015, 27 (17), 2722–2727. 10.1002/adma.201500140. [DOI] [PubMed] [Google Scholar]
- Itagaki H.; Kurokawa T.; Furukawa H.; Nakajima T.; Katsumoto Y.; Gong J. P. Water-Induced Brittle-Ductile Transition of Double Network Hydrogels. Macromolecules 2010, 43 (22), 9495–9500. 10.1021/ma101413j. [DOI] [Google Scholar]
- Wang T.; Liu D.; Lian C.; Zheng S.; Liu X.; Tong Z. Large deformation behavior and effective network chain density of swollen poly(N-isopropylacrylamide)-Laponite nanocomposite hydrogels. Soft Matter 2012, 8 (3), 774–783. 10.1039/C1SM06484C. [DOI] [Google Scholar]
- Chen H.; Yang F.; Hu R.; Zhang M.; Ren B.; Gong X.; Ma J.; Jiang B.; Chen Q.; Zheng J. A comparative study of the mechanical properties of hybrid double-network hydrogels in swollen and as-prepared states. J. Mater. Chem. B 2016, 4 (35), 5814–5824. 10.1039/C6TB01511E. [DOI] [PubMed] [Google Scholar]
- Kamata H.; Kushiro K.; Takai M.; Chung U.-i.; Sakai T. Non-Osmotic Hydrogels: A Rational Strategy for Safely Degradable Hydrogels. Angew. Chem. 2016, 128 (32), 9428–9432. 10.1002/ange.201602610. [DOI] [PubMed] [Google Scholar]
- Kamata H.; Akagi Y.; Kayasuga-Kariya Y.; Chung U.-i.; Sakai T. Nonswellable” Hydrogel Without Mechanical Hysteresis. Science 2014, 343 (6173), 873–875. 10.1126/science.1247811. [DOI] [PubMed] [Google Scholar]
- Klouda L. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2015, 97, 338–349. 10.1016/j.ejpb.2015.05.017. [DOI] [PubMed] [Google Scholar]
- Koetting M. C.; Peters J. T.; Steichen S. D.; Peppas N. A. Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng., R 2015, 93, 1–49. 10.1016/j.mser.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zustiak S. P.; Leach J. B. Hydrolytically Degradable Poly(Ethylene Glycol) Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties. Biomacromolecules 2010, 11 (5), 1348–1357. 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal T.; Peppas N. A. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J. Biomed. Mater. Res. 1989, 23 (10), 1183–1193. 10.1002/jbm.820231007. [DOI] [PubMed] [Google Scholar]
- Uhrich K. E.; Cannizzaro S. M.; Langer R. S.; Shakesheff K. M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999, 99 (11), 3181–3198. 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- Brannigan R. P.; Dove A. P. Synthesis, properties and biomedical applications of hydrolytically degradable materials based on aliphatic polyesters and polycarbonates. Biomater. Sci. 2017, 5 (1), 9–21. 10.1039/C6BM00584E. [DOI] [PubMed] [Google Scholar]
- Nair L. S.; Laurencin C. T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32 (8), 762–798. 10.1016/j.progpolymsci.2007.05.017. [DOI] [Google Scholar]
- Lyu S.; Untereker D. Degradability of Polymers for Implantable Biomedical Devices. Int. J. Mol. Sci. 2009, 10 (9), 4033. 10.3390/ijms10094033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory P. J. Molecular Size Distribution in Three Dimensional Polymers. I. Gelation1. J. Am. Chem. Soc. 1941, 63 (11), 3083–3090. 10.1021/ja01856a061. [DOI] [Google Scholar]
- Flory P. J. Constitution of Three-dimensional Polymers and the Theory of Gelation. J. Phys. Chem. 1942, 46 (1), 132–140. 10.1021/j150415a016. [DOI] [Google Scholar]
- Stockmayer W. H. Theory of molecular size distribution and gel formation in branched-chain polymers. J. Chem. Phys. 1943, 11 (2), 45–55. 10.1063/1.1723803. [DOI] [Google Scholar]
- Schultz K. M.; Anseth K. S. Monitoring degradation of matrix metalloproteinases-cleavable PEG hydrogels via multiple particle tracking microrheology. Soft Matter 2013, 9 (5), 1570–1579. 10.1039/C2SM27303A. [DOI] [Google Scholar]
- Sala A.; Hanseler P.; Ranga A.; Lutolf M. P.; Voros J.; Ehrbar M.; Weber F. E. Engineering 3D cell instructive microenvironments by rational assembly of artificial extracellular matrices and cell patterning. Integr. Biol. 2011, 3 (11), 1102–1111. 10.1039/c1ib00045d. [DOI] [PubMed] [Google Scholar]
- Engler A. J.; Sen S.; Sweeney H. L.; Discher D. E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126 (4), 677–689. 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Kyburz K. A.; Anseth K. S. Three-dimensional hMSC motility within peptide-functionalized PEG-based hydrogels of varying adhesivity and crosslinking density. Acta Biomater. 2013, 9 (5), 6381–6392. 10.1016/j.actbio.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher D. E.; Janmey P.; Wang Y.-l. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310 (5751), 1139–1143. 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.