

Abstract
Multiple system atrophy (MSA) is a fatal neurodegenerative disorder characterized by the abnormal accumulation of toxic forms of the synaptic protein alpha-synuclein (α-syn) within oligodendrocytes and neurons. The presence of α-syn within oligodendrocytes in the form of glial cytoplasmic inclusions is the diagnostic hallmark of MSA. However, it has been postulated that α-syn is produced in neurons and propagates to oligodendrocytes, where unknown mechanisms lead to its accumulation. The presence of α-syn within neurons in MSA has not been so extensively studied, but it may shed light into neuropathological mechanisms leading to oligodendroglial accumulation. Here we summarize the principal neuropathological events of MSA, and discuss how a deeper knowledge of these mechanisms may help develop effective therapies targeting α-syn accumulation and spreading.
Multiple system atrophy (MSA) is a rapidly progressing, sporadic and fatal neurodegenerative disorder that belongs to the synucleino-pathy spectrum (Farrer et al., 1999; Spillantini, 1999; Takeda et al., 1998; Wakabayashi et al., 1998a). Clinically, MSA is characterized by parkinsonian features and cerebellar, autonomic and urogenital dysfunction, which are a reflection of striatonigral degeneration and oli-vopontocerebellar atrophy (Gilman et al., 2008). There are two major subtypes of MSA, distinguished by their symptoms at the time of diagnosis (Gilman et al., 2008): the parkinsonian subtype (MSA-P), where parkinsonism is predominant, including bradykinesia, muscle rigidity, tremors, and postural instability; and the cerebellar subtype (MSA-C), characterized by cerebellar ataxia. The prevalence of MSA is between 3.4 and 4.9 cases per 100,000 people, and the mean incidence is 0.6–0.7 cases per 100,000 people and year (Fanciulli and Wenning, 2015; Stefanova et al., 2009), making MSA an orphan disease (Lavandeira, 2002). In Western countries, MSA-P predominates, occurring in 66–82% of MSA patients (Wenning et al., 2013). However, MSA-C is more common in Eastern countries, occurring in 67% of MSA patients (Yabe et al., 2006). The rapid progression, its orphan disease status, and its neuropathological features make MSA an ideal candidate for accelerated drug development.
1. The neuropathology of MSA
The principal neuropathological characteristic of MSA is the presence of aggregates containing the synaptic protein alpha-synuclein (α-syn) within brain cells (Spillantini et al., 1998). Specifically, the presence of α-syn-positive inclusions in oligodendroglial cells in the form of glial cytoplasmic inclusions (GCIs) is the diagnostic hallmark of MSA (Dickson et al., 1999; Papp et al., 1989; Spillantini, 1999; Wakabayashi et al., 1998b). Interestingly, α-syn aggregates can also be observed as glial nuclear inclusions, neuronal cytoplasmic inclusions (NCIs), neuronal nuclear inclusions (NNIs) and dystrophic neurites, however these lesions all appear at lower frequencies than the GCIs (Papp and Lantos, 1992). The cellular distribution of α-syn aggregates in MSA has been the cause of intense research, as α-syn is considered a neuronal protein (Fortin et al., 2005; George et al., 1995) that abnormally accumulates within glial cells (oligodendrocytes). Although several groups have found no evidence of increased SNCA expression in MSA oligoden-drocytes (Jin et al., 2008; Miller et al., 2005; Ozawa et al., 2001), a more recent study reported that there is a 3-fold increase in SNCA mRNA levels in postmortem MSA oligodendrocytes (Asi et al., 2014). It is unknown if this increase would be enough to induce a significant accumulation of α-syn in oligodendrocytes, a cell type that does not express high basal levels of α-syn; or if the increased expression is a consequence of α-syn accumulation, rather than its cause.
Achieving a deeper understanding of the neuropathology of MSA has been one of the primary goals in the field. In this sense, a major unanswered question is why α-syn tends to accumulate to a greater extent in oligodendrocytes than in neurons. One possibility is that α-syn is produced by oligodendroglial cells which in turn over-express or fail to intrinsically clear out α-syn (Fig. 1); the other is that α-syn that propagates from neurons and cannot be cleared out by oligoden-drocytes due to defective clearance mechanisms (Fig. 1). In any case, the source of α-syn in oligodendroglial cells in MSA is still unclear. Given the high levels and widespread distribution of α-syn aggregates in MSA, it is possible that both propagation and oligodendroglial α-syn expression might be occurring simultaneously. Supporting the possibility of propagation, several studies have shown that α-syn aggregates can transmit from neuron to neuron (Desplats et al., 2009; Lee et al., 2012b), neuron to astroglial and oligodendroglial cells (Lee et al., 2010; Reyes et al., 2014), and oligodendroglial to astroglial cells (Valera et al., 2014), leading to neuronal dysfunction, apoptosis and neuroin-flammation (Desplats et al., 2009; Klucken et al., 2012; Lee et al., 2010; Valera et al., 2014; Volpicelli-Daley et al., 2011). Moreover, recent studies have shown that injection of homogenates from MSA brains propagate α-syn pathology in a prion-like fashion in the murine brain (Prusiner et al., 2015; Watts et al., 2013). Neuronal cells (donors) release α-syn aggregates into the extracellular environment by exocytosis and in clear vesicles and exosomes (Danzer et al., 2012; Lee et al., 2005), and α-syn is taken up by other neurons, oligodendrocytes and astrocytes (acceptors) via endocytosis (Lee et al., 2008a) (Fig. 1). This scenario could explain the presence of NCIs and NNIs in MSA neurons, however whether neurons showing α-syn accumulation are the source of extracellular α-syn in MSA has not been investigated.
Fig. 1.
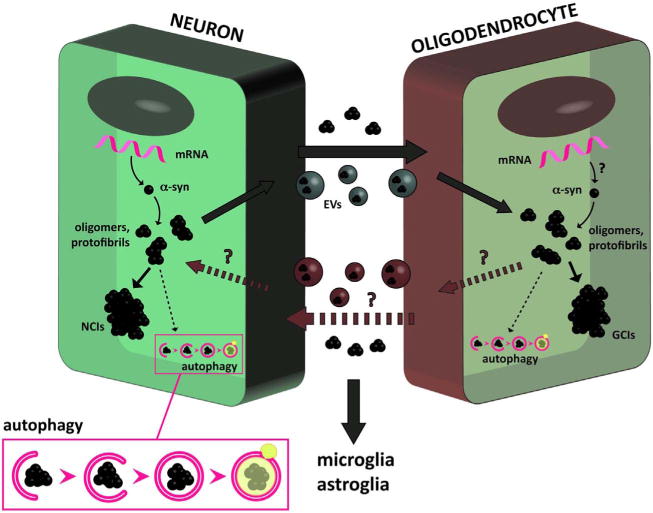
Neuropathology of MSA and cell-to-cell propagation of α-syn. It is believed that in MSA oligodendrocytes accumulate α-syn after a process of propagation from neurons or other oligodendroglial cells. Increased expression and/or reduced α-syn clearance in neurons may stimulate the accumulation of misfolded forms of the protein as NCIs, and their release and propagation to oligodendrocytes via exocytosis or within extracellular vesicles (EVs). Reduced α-syn clearance in oligodendrocytes may also enhance its accumulation in the form of GCIs, and induce its release to the extracellular environment. It is also possible that enhanced expression of the α-syn gene is present in oligodendrocytes. These neuropathological events represent potential targets for therapeutic intervention in MSA.
Whether its origin is intracellular or due to cell-to-cell propagation, recent evidence supports the notion that failure of intracellular protein clearance mechanisms (e.g. autophagy, unfolded protein response, proteolysis) might play a role in the process of α-syn aggregation, release and subsequent accumulation of α-syn pathological species in donor and acceptor cells (Klucken et al., 2012; Lee et al., 2013) (Fig. 1). Accumulation of toxic α-syn within MSA oligodendrocytes might be a direct consequence of impairments on those mechanisms. Free extracellular oligomeric α-syn is taken up by oligodendrocytes by clathrin-dependent endocytosis (Kisos et al., 2012; Konno et al., 2012), and endocytic vesicles containing α-syn are then directed to lysosomal degradation; however, cytosolic α-syn might also be degraded by other mechanisms such as UPR and proteolysis (Hoozemans et al., 2007; Xilouri et al., 2013). Impairments in clearance mechanisms such as autophagy have already been described in MSA and other synucleino-pathies (Lynch-Day et al., 2012; Schwarz et al., 2012).
1.1. Neuronal neuropathology in MSA
Histopathologically, the morphology and immunoreactivity of NCIs differ from that of the neuronal aggregates found in other synucleino-pathies (Spillantini et al., 1998), known as Lewy bodies. Interestingly, the immunohistochemical and ultrastructural features of NCIs seem to be virtually identical to those of GCIs (Yokoyama et al., 2001). NCIs are observed in the putamen and pons of all MSA cases, and they can also be observed in the cerebral cortex, medulla oblongata and spinal cord, with no NCIs present in the cerebellum and midbrain (Sugiura et al., 1995). NCI pathology follows a hierarchy of region-specific susceptibility, independent of the clinical phenotype, and the severity of the pathology is duration-dependent (Cykowski et al., 2015). Widespread NCIs have been identified not only in regions typically associated with the disease, but also within other areas such as anterior cingulate cortex, amygdala, entorhinal cortex, basal forebrain, hypothalamus, and in some cases cerebellar roof nuclei (Cykowski et al., 2015). These findings suggest that the neuronal pathology plays an important role in the developmental and progression of MSA. Interestingly, NCIs are heterogeneous, and in uncommon cases they may include Pick body-like inclusions that are strongly associated with neuronal loss in the hippocampus and amygdala (Aoki et al., 2015), potentially representing a novel subtype of frontotemporal lobar degeneration associated with α-syn.
In contrast, NNIs appear as a loosely woven network or irregularly arranged fibrils beneath the nuclear membrane (Nishie et al., 2004), occasionally coexisting with NCIs in the same neurons. Due to their count number and correlation with disease progression, it has been suggested that NNI formation is an earlier phenomenon than NCI formation (Nishie et al., 2004). One question that remains to be answered is if NCIs and GCIs share mechanistic origins, or if they are originated by independent mechanisms; the shared features between both structures would suggest the former.
The presence of α-syn-positive aggregates within neurons suggests that these cells fail to clear out increased intracellular levels of mis-folded α-syn. It is possible that a significant inhibition of clearance mechanisms is related to the origin of the disease, fueled by the fact that misfolded, aggregated α-syn is also able to inhibit its own degradation (Snyder et al., 2003; Winslow and Rubinsztein, 2011). Supporting this notion, it has been observed that autophagic failure promotes the exocytosis and intercellular transfer of α-syn (Lee et al., 2013). It is possible that the release of α-syn to the extracellular environment is an attempt to reduce its intracellular levels, however more research is needed to elucidate if this is the case. In the past few years, strong evidence has been provided supporting the prion-like behavior of α-syn, which has been confirmed in cellular models (Desplats et al., 2009; Lee et al., 2012b), animal models (Luk et al., 2012; Masuda-Suzukake et al., 2014; Prusiner et al., 2015), and indirectly in patients with PD that received neuronal grafts (Kordower et al., 2008; Li et al., 2008). Moreover, the fact that α-syn accumulation has been observed in cell types other than neurons in animal models of PD and in PD brains (Bruck et al., 2016) further supports this hypothesis.
1.2. Glial neuropathology in MSA
According to the α-syn propagation hypothesis of MSA, oligoden-drocytes would incorporate extracellular α-syn and accumulate it in the form of GCIs (Fig. 1) and nuclear inclusions (Nishie et al., 2004). However, it is unclear why oligodendrocytes preferentially uptake and/or fail to clear α-syn in MSA brains, a neuropathological event not widely observed in other synucleinopathies. One hypothesis is that the incorporation of extracellular, misfolded α-syn may impair the endogenous clearance machinery of the oligodendrocyte, progressively leading to α-syn accumulation (Pukass and Richter-Landsberg, 2015; Schwarz et al., 2012). Another option is that a dysfunction in the clearance machinery is a prerequisite for α-syn uptake and/or accumulation in oligodendrocytes. In both scenarios, the oligodendroglial accumulation of α-syn may be further potentiated by increased expression of its gene (Asi et al., 2014; Djelloul et al., 2015) and oligo-dendrocyte-to-oligodendrocyte propagation. Finally, other suggested mechanisms are the involvement of altered iron metabolism in oligo-dendrocytes (Visanji et al., 2013), and epigenetic and/or environmental factors (Sturm et al., 2016). Furthermore, it is possible that multiple mechanisms combine, leading to the pathological, progressive oligo-dendroglial accumulation of α-syn observed in MSA brains. In light of these observations, it could also be concluded that there may exist a genetic predisposition for oligodendrocytes to develop abnormal α-syn accumulation (Sturm et al., 2016). The genetic risk factors with the most evidence in MSA are variants in the SNCA and COQ2 genes (Collaboration, 2013; Scholz et al., 2009), however genome-wide association studies have failed to find association between common genetic variations in those genes and MSA (Sailer et al., 2016).
The principal consequences of α-syn-induced oligodendroglial degeneration are the loss of trophic support to neurons and demyelination (Ettle et al., 2016; Stefanova and Wenning, 2016; Ubhi et al., 2011; Wong et al., 2014), which in turn lead to further neurodegeneration. This secondary neurodegeneration may explain the lack of response to L-DOPA observed in MSA patients and the fast progression of this devastating disease. One of the most relevant characteristics of MSA is the selective neuronal loss and axonal degeneration in the central autonomic, striatonigral and olivopontocerebellar networks, with cell loss also present in autonomic brain stem nuclei (Jellinger, 1998; Kuzdas-Wood et al., 2014; Wakabayashi et al., 2010). Moreover, the presence of misfolded α-syn in the extracellular compartment can result not only in oligodendroglial dysfunction, but also in the overstimulation of as-troglia and microglia (Bae et al., 2012; Lee et al., 2010; Vieira et al., 2015) (Fig. 1). Both astrogliosis and microgliosis have been observed in MSA brains (Schwarz et al., 1996), and in transgenic mouse models of MSA (Ubhi et al., 2012; Valera et al., 2015; Valera et al., 2014). As-trocytes are able to accumulate α-syn in MSA and in tg mouse models (Mandler et al., 2015; Nakamura et al., 2016), and stimulate the release of pro-inflammatory cytokines (Lee et al., 2010). MSA brains exhibit widespread astrogliosis (Schwarz et al., 1996) correlated to the presence of nearby GCI-positive oligodendrocytes (Radford et al., 2015), suggesting that localized presence of extracellular α-syn may underlie the astrocytic pathology in MSA. Additionally, microglia phagocytize α-syn (Lee et al., 2008b; Park et al., 2008) and also release pro-in-flammatory factors and reactive oxygen species in response to extracellular α-syn (Beraud et al., 2013; Fellner et al., 2013). Microgliosis has been described and identified as one of the main features of the disease process in MSA (Ishizawa et al., 2004; Stefanova et al., 2007). However, there is evidence of both neuroprotective and detrimental effects of microglial activation in MSA and MSA models. This dual role seems to be associated with the capacity of microglia to both remove extracellular α-syn and produce neurotrophic factors, and their ability to release pro-inflammatory mediators (Fellner et al., 2013; Stefanova et al., 2011). These neuroinflammatory mechanisms would create a hostile environment for neurons in the MSA brain.
Finally, it is worth mentioning that, although the principal component of GCIs is fibrillar α-syn, other proteins such as p25α, tau, ubiquitin, tubulin, Cdk5 and MAP2 can also be found (Cairns et al., 1997; Chiba et al., 2011; Gai et al., 1999; Nakamura et al., 1998; Wakabayashi et al., 1998b). Interestingly, p25α is an oligodendroglial protein that can induce aggregation of α-syn (Hasegawa et al., 2010). Changes in the cellular interactions between the myelin protein MBP and p25α occur early in MSA and contribute to abnormalities in myelin and subsequent α-syn aggregation (Song et al., 2007).
2. Therapeutic opportunities based on the MSA neuropathology
The neuropathological features of MSA suggest that targeting the oligodendroglial α-syn accumulation, neuronal α-syn accumulation, or common mechanisms leading to α-syn accumulation in both cell types, may be potential therapeutic alternatives. In this sense, it is likely that approaches that lead to a reduction in α-syn accumulation in both oligodendrocytes and neurons may be more effective that cell-specific approaches. Moreover, the important component of α-syn propagation and the pathological accumulation of α-syn within two different cell types, and its orphan disease status make MSA a strong synucleinopathy candidate for accelerated drug discovery (Krismer et al., 2014).
Most of the research aimed at developing new therapeutic candidates for MSA has been primarily focused on targeting oligodendroglial α-syn accumulation. The use of transgenic models that express α-syn directly in oligodendroglial cells, under the control of the PLP (Kahle et al., 2002), MBP (Shults et al., 2005), or CNP (Yazawa et al., 2005) promoters, is a reflection of this trend. However, this approach does not cover an important part of the neuropathological landscape: the presence of α-syn within neurons, and its pathological propagation from neurons to oligodendrocytes. Another important limitation of these models lays on the fact that the mechanisms leading to α-syn accumulation within oligodendrocytes in MSA are still unknown, thus limiting the possibility of finding suitable targets or preventing α-syn accumulation from early stages (Stefanova and Wenning, 2015). In this scenario, approaches limited to reducing α-syn accumulation in oligo-dendrocytes may not provide enough disease modification to be able to stop or delay the progression of the disease.
Expanding the MSA therapeutic landscape beyond the oligoden-drocyte, the neuronal pathology of MSA has not been so extensively explored. While neuronal α-syn accumulation has been greatly studied (and targeted) in PD and other synucleinopathies, less is known of its potential as a target for MSA, and its interest has been mostly limited to act as α-syn source for oligodendrocytes. The involvement of neuronal α-syn accumulation as a main pathological event in MSA remains to be investigated. Nevertheless, according to the propagation model of MSA pathology, a reduction in the neuronal expression, release and accumulation of α-syn may translate in a decrease in oligodendroglial α-syn levels and lead to disease modification. Moreover, supporting and repairing neuronal function by restoring trophic support (e.g. BDNF, GDNF) (Ubhi et al., 2010), myelination (Ettle et al., 2016), and by the use of regenerative therapies (Lee et al., 2012a) are therapeutic alternatives to consider for MSA.
Recently, therapies aimed at reducing α-syn propagation have been extensively explored. That is the case in immunotherapies, which not only block toxic propagation of α-syn species, but are also able to reduce intracellular α-syn accumulation (Games et al., 2014; Mandler et al., 2015; Mandler et al., 2014). The Austrian company AFFiRiS recently completed an active immunotherapy Phase I clinical trial with the α-syn vaccine PD03A. Both low and high doses were well tolerated and no serious adverse events were reported. PD03A induced a dose-dependent immune response against both the vaccine itself and the α-syn epitope over time. An α-syn passive immunotherapy approach using a humanized monoclonal antibody against α-syn (Prothena, PRX002) has also been tested in Phase Ia and Ib clinical trials. In both trials, free serum α-syn levels were drastically reduced (Schenk et al., 2017). A dose-dependent increase in PRX002 levels in cerebrospinal fluid was observed, without serious adverse events. PRX002 has move forward to Phase II trials in patients with early PD. A Phase I passive immunotherapy trial using the anti α-syn antibody BIIB-054 (Biogen) is also ongoing. Preliminary reports suggest that this antibody was well tolerated in healthy volunteers, and was detectable in the cerebrospinal fluid (Brundin et al., 2017). Additional clinical trials to commence soon include the α-syn antibodies BAN0805 (BioArctic & AbbVie), targeting oligomeric forms of α-syn, and MEDI1341 (AstraZeneca & Takeda). Moreover, the therapeutic potential of stimulating α-syn degradation pathways, such as autophagy, is also being investigated at the preclinical level (Xilouri et al., 2013). Neuroinflammation induced by extracellular α-syn contributes to MSA pathology, thus therapies reducing the overactivation of glial cells and the production of pro-inflammatory cytokines are also being explored (Stefanova et al., 2012; Stefanova et al., 2007; Vieira et al., 2015). Finally, using strategic drug combinations or multi-target drugs might increase the efficiency of therapeutic treatments for MSA (Valera and Masliah, 2016). Therapies aimed at reducing α-syn accumulation and cell-to-cell transfer, such as immunotherapy, could be combined with agents that reduce neuroin-flammation with synergistic outcomes (Valera et al., 2017).
It can be concluded that more research is needed to elucidate how neurons, oligodendrocytes and other glial types interplay at the origin of the MSA pathology and during the progression of the disease. The pathological production, accumulation and propagation of α-syn between different cell types may be a significant therapeutic target not only limited to the oligodendroglial aspect of the disease. Investigating the role of neurons on the pathology as source and accumulators of toxic protein species may lead to more effective therapies for reducing neurodegeneration in MSA.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant NS092803-01, and a MSA Coalition Research Grant.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, Weiner M, Lipton A, Powers JM, White CL, 3rd, Dickson DW. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha-synuclein. Acta Neuropathol. 2015;130:93–105. doi: 10.1007/s00401-015-1442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, Houlden H, Holton JL. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia. 2014;62:964–970. doi: 10.1002/glia.22653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular alpha-synu-clein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraud D, Hathaway HA, Trecki J, Chasovskikh S, Johnson DA, Johnson JA, Federoff HJ, Shimoji M, Mhyre TR, Maguire-Zeiss KA. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein alpha-synuclein. J NeuroImmune Pharmacol. 2013;8:94–117. doi: 10.1007/s11481-012-9401-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck D, Wenning GK, Stefanova N, Fellner L. Glia and alpha-synuclein in neurodegeneration: a complex interaction. Neurobiol Dis. 2016;85:262–274. doi: 10.1016/j.nbd.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol. 2017 doi: 10.1016/j.expneurol.2017.10.003. http://dx.doi.org/10.1016/j.expneurol.2017.10.003. pii: S0014-4886(17)30245-5. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Cairns NJ, Atkinson PF, Hanger DP, Anderton BH, Daniel SE, Lantos PL. Tau protein in the glial cytoplasmic inclusions of multiple system atrophy can be distinguished from abnormal tau in Alzheimer’s disease. Neurosci Lett. 1997;230:49–52. doi: 10.1016/s0304-3940(97)00474-6. [DOI] [PubMed] [Google Scholar]
- Chiba Y, Takei S, Kawamura N, Kawaguchi Y, Sasaki K, Hasegawa-Ishii S, Furukawa A, Hosokawa M, Shimada A. Immunohistochemical localization of aggresomal proteins in glial cytoplasmic inclusions in multiple system atrophy. Neuropathol Appl Neurobiol. 2011;38:559–571. doi: 10.1111/j.1365-2990.2011.01229.x. [DOI] [PubMed] [Google Scholar]
- Collaboration, T.M.-S.A.R. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med. 2013;369:233–244. doi: 10.1056/NEJMoa1212115. [DOI] [PubMed] [Google Scholar]
- Cykowski MD, Coon EA, Powell SZ, Jenkins SM, Benarroch EE, Low PA, Schmeichel AM, Parisi JE. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain J Neurol. 2015;138:2293–2309. doi: 10.1093/brain/awv114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Lin W, Liu WK, Yen SH. Multiple system atrophy: a sporadic synucleinopathy. Brain Pathol. 1999;9:721–732. doi: 10.1111/j.1750-3639.1999.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung MS, Goldwurm S, Frisen J, Deierborg T, Roybon L. Alpha-synuclein expression in the oligo-dendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Reports. 2015;5:174–184. doi: 10.1016/j.stemcr.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettle B, Kerman BE, Valera E, Gillmann C, Schlachetzki JC, Reiprich S, Buttner C, Ekici AB, Reis A, Wegner M, Bauerle T, Riemenschneider MJ, Masliah E, Gage FH, Winkler J. Alpha-synuclein-induced myelination deficit defines a novel interventional target for multiple system atrophy. Acta Neuropathol. 2016;132:59–75. doi: 10.1007/s00401-016-1572-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372:1375–1376. doi: 10.1056/NEJMc1501657. [DOI] [PubMed] [Google Scholar]
- Farrer M, Gwinn-Hardy K, Hutton M, Hardy J. The genetics of disorders with synuclein pathology and parkinsonism. Hum Mol Genet. 1999;8:1901–1905. doi: 10.1093/hmg/8.10.1901. [DOI] [PubMed] [Google Scholar]
- Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai WP, Power JH, Blumbergs PC, Culvenor JG, Jensen PH. Alpha-sy-nuclein immunoisolation of glial inclusions from multiple system atrophy brain tissue reveals multiprotein components. J Neurochem. 1999;73:2093–2100. [PubMed] [Google Scholar]
- Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, Patrick C, Ubhi K, Nuber S, Sacayon P, Zago W, Seubert P, Barbour R, Schenk D, Masliah E. Reducing C-terminal-truncated alpha-synuclein by im-munotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci. 2014;34:9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Dürr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa T, Baba T, Kobayashi M, Konno M, Sugeno N, Kikuchi A, Itoyama Y, Takeda A. Role of TPPP/p25 on alpha-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy. Neurochem Int. 2010;57:857–866. doi: 10.1016/j.neuint.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Ishizawa K, Komori T, Sasaki S, Arai N, Mizutani T, Hirose T. Microglial activation parallels system degeneration in multiple system atrophy. J Neuropathol Exp Neurol. 2004;63:43–52. doi: 10.1093/jnen/63.1.43. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Neuropathology of movement disorders. Neurosurg Clin N Am. 1998;9:237–262. [PubMed] [Google Scholar]
- Jin H, Ishikawa K, Tsunemi T, Ishiguro T, Amino T, Mizusawa H. Analyses of copy number and mRNA expression level of the alpha-synuclein gene in multiple system atrophy. J Med Dent Sci. 2008;55:145–153. [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Spooren W, Fuss B, Mallon B, Macklin WB, Fujiwara H, Hasegawa M, Iwatsubo T, Kretzschmar HA, Haass C. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002;3:583–588. doi: 10.1093/embo-reports/kvf109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisos H, Pukaß K, Ben-Hur T, Richter-Landsberg C, Sharon R. Increased neuronal α-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling α-synucleinopathies. PLoS One. 2012;7:e46817. doi: 10.1371/journal.pone.0046817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucken J, Poehler AM, Ebrahimi-Fakhari D, Schneider J, Nuber S, Rockenstein E, Schlotzer-Schrehardt U, Hyman BT, McLean PJ, Masliah E, Winkler J. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy. 2012;8:754–766. doi: 10.4161/auto.19371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno M, Hasegawa T, Baba T, Miura E, Sugeno N, Kikuchi A, Fiesel FC, Sasaki T, Aoki M, Itoyama Y, Takeda A. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener. 2012;7:38. doi: 10.1186/1750-1326-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Krismer F, Jellinger KA, Scholz SW, Seppi K, Stefanova N, Antonini A, Poewe W, Wenning GK. Multiple system atrophy as emerging template for accelerated drug discovery in alpha-synucleinopathies. Parkinsonism Relat Disord. 2014;20:793–799. doi: 10.1016/j.parkreldis.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzdas-Wood D, Stefanova N, Jellinger KA, Seppi K, Schlossmacher MG, Poewe W, Wenning GK. Towards translational therapies for multiple system atrophy. Prog Neurobiol. 2014;118:19–35. doi: 10.1016/j.pneurobio.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavandeira A. Orphan drugs: legal aspects, current situation. Haemophilia. 2002;8:194–198. doi: 10.1046/j.1365-2516.2002.00643.x. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008a;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee SJ. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun. 2008b;372:423–428. doi: 10.1016/j.bbrc.2008.05.045. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes in-flammatory responses in synucleinopathies. J Biol Chem. 2010;285:9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PH, Lee JE, Kim HS, Song SK, Lee HS, Nam HS, Cheong JW, Jeong Y, Park HJ, Kim DJ, Nam CM, Lee JD, Kim HO, Sohn YH. A rando-mized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol. 2012a;72:32–40. doi: 10.1002/ana.23612. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Desplats P, Lee HJ, Spencer B, Masliah E. Cell-to-cell transmission of α-synuclein aggregates. Methods Mol Biol. 2012b;849:347–359. doi: 10.1007/978-1-61779-551-0_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med. 2013;45:e22. doi: 10.1038/emm.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autop-hagy in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009357. doi: 10.1101/cshperspect.a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, Santic R, Meindl S, Vigl B, Smrzka O, Schneeberger A, Mattner F, Masliah E. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014;127:861–879. doi: 10.1007/s00401-014-1256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, Adame A, Schmidhuber S, Santic R, Schneeberger A, Schmidt W, Mattner F, Masliah E. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. 2015;10:10. doi: 10.1186/s13024-015-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun. 2014;2:88. doi: 10.1186/s40478-014-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DW, Johnson JM, Solano SM, Hollingsworth ZR, Standaert DG, Young AB. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Transm. 2005;112:1613–1624. doi: 10.1007/s00702-005-0378-1. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kawamoto Y, Nakano S, Akiguchi I, Kimura J. Cyclin-dependent kinase 5 and mitogen-activated protein kinase in glial cytoplasmic inclusions in multiple system atrophy. J Neuropathol Exp Neurol. 1998;57:690–698. doi: 10.1097/00005072-199807000-00006. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Mori F, Kon T, Tanji K, Miki Y, Tomiyama M, Kurotaki H, Toyoshima Y, Kakita A, Takahashi H, Yamada M, Wakabayashi K. Accumulation of phosphorylated alpha-synuclein in subpial and periventricular as-trocytes in multiple system atrophy of long duration. Neuropathology. 2016;36:157–167. doi: 10.1111/neup.12243. [DOI] [PubMed] [Google Scholar]
- Nishie M, Mori F, Yoshimoto M, Takahashi H, Wakabayashi K. A quantitative investigation of neuronal cytoplasmic and intranuclear inclusions in the pontine and inferior olivary nuclei in multiple system atrophy. Neuropathol Appl Neurobiol. 2004;30:546–554. doi: 10.1111/j.1365-2990.2004.00564.x. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Okuizumi K, Ikeuchi T, Wakabayashi K, Takahashi H, Tsuji S. Analysis of the expression level of alpha-synuclein mRNA using postmortem brain samples from pathologically confirmed cases of multiple system atrophy. Acta Neuropathol. 2001;102:188–190. doi: 10.1007/s004010100367. [DOI] [PubMed] [Google Scholar]
- Papp MI, Lantos PL. Accumulation of tubular structures in oligodendroglial and neuronal cells as the basic alteration in multiple system atrophy. J Neurol Sci. 1992;107:172–182. doi: 10.1016/0022-510x(92)90286-t. [DOI] [PubMed] [Google Scholar]
- Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocer-ebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- Park JY, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia. 2008;56:1215–1223. doi: 10.1002/glia.20691. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112:E5308–5317. doi: 10.1073/pnas.1514475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukass K, Richter-Landsberg C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents alpha-synuclein aggregate formation by activating the autophagic pathway: implications for multiple system atrophy. Front Cell Neurosci. 2015;9:163. doi: 10.3389/fncel.2015.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford R, Rcom-H’cheo-Gauthier A, Wong MB, Eaton ED, Quilty M, Blizzard C, Norazit A, Meedeniya A, Vickers JC, Gai WP, Guillemin GJ, West AK, Dickson TC, Chung R, Pountney DL. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to alpha-synuclein inclusions. Mol Cell Neurosci. 2015;65:68–81. doi: 10.1016/j.mcn.2015.02.015. [DOI] [PubMed] [Google Scholar]
- Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia. 2014;62:387–398. doi: 10.1002/glia.22611. [DOI] [PubMed] [Google Scholar]
- Sailer A, Scholz SW, Nalls MA, Schulte C, Federoff M, Price TR, Lees A, Ross OA, Dickson DW, Mok K, Mencacci NE, Schottlaender L, Chelban V, Ling H, O’Sullivan SS, Wood NW, Traynor BJ, Ferrucci L, Federoff HJ, Mhyre TR, Morris HR, Deuschl G, Quinn N, Widner H, Albanese A, Infante J, Bhatia KP, Poewe W, Oertel W, Hoglinger GU, Wullner U, Goldwurm S, Pellecchia MT, Ferreira J, Tolosa E, Bloem BR, Rascol O, Meissner WG, Hardy JA, Revesz T, Holton JL, Gasser T, Wenning GK, Singleton AB, Houlden H, European Multiple System Atrophy Study, G, the, U.K.M.S.A.S.G A genome-wide association study in multiple system atrophy. Neurology. 2016;87:1591–1598. doi: 10.1212/WNL.0000000000003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, Soto J, Atiee G, Ostrowitzki S, Kinney GG. First-in-human assessment of PRX002 an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32:211–218. doi: 10.1002/mds.26878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Del Sorbo F, Schneider S, Bhatia KP, Gasser T. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz J, Weis S, Kraft E, Tatsch K, Bandmann O, Mehraein P, Vogl T, Oertel WH. Signal changes on MRI and increases in reactive microgliosis, astro-gliosis, and iron in the putamen of two patients with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1996;60:98–101. doi: 10.1136/jnnp.60.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz L, Goldbaum O, Bergmann M, Probst-Cousin S, Richter-Landsberg C. Involvement of macroautophagy in multiple system atrophy and protein aggregate formation in oligodendrocytes. J Mol Neurosci. 2012;47:256–266. doi: 10.1007/s12031-012-9733-5. [DOI] [PubMed] [Google Scholar]
- Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, Hashimoto M, Song D, Iwatsubo T, Tsuboi K, Masliah E. Neurological and neurode-generative alterations in a transgenic mouse model expressing human alpha-synu-clein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25:10689–10699. doi: 10.1523/JNEUROSCI.3527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- Song YJ, Lundvig DM, Huang Y, Gai WP, Blumbergs PC, Hojrup P, Otzen D, Halliday GM, Jensen PH. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am J Pathol. 2007;171:1291–1303. doi: 10.2353/ajpath.2007.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG. Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy are alpha-synucleinopathies. Parkinsonism Relat Disord. 1999;5:157–162. doi: 10.1016/s1353-8020(99)00031-0. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- Stefanova N, Wenning GK. Animal models of multiple system atrophy. Clin Auton Res. 2015;25:9–17. doi: 10.1007/s10286-014-0266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova N, Wenning GK. Review: multiple system atrophy: emerging targets for interventional therapies. Neuropathol Appl Neurobiol. 2016;42:20–32. doi: 10.1111/nan.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov Disord. 2007;22:2196–2203. doi: 10.1002/mds.21671. [DOI] [PubMed] [Google Scholar]
- Stefanova N, Bücke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol. 2009;8:1172–1178. doi: 10.1016/S1474-4422(09)70288-1. [DOI] [PubMed] [Google Scholar]
- Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Wenning GK. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol. 2011;179:954–963. doi: 10.1016/j.ajpath.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova N, Georgievska B, Eriksson H, Poewe W, Wenning GK. Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox Res. 2012;21:393–404. doi: 10.1007/s12640-011-9294-3. [DOI] [PubMed] [Google Scholar]
- Sturm E, Fellner L, Krismer F, Poewe W, Wenning GK, Stefanova N. Neuroprotection by epigenetic modulation in a transgenic model of multiple system atrophy. Neurotherapeutics. 2016;13:871–879. doi: 10.1007/s13311-016-0447-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura K, Hashizume Y, Kume A, Takahashi A. Distribution of neuronal cytoplasmic inclusions in multiple system atrophy. Nagoya J Med Sci. 1995;58:117–126. [PubMed] [Google Scholar]
- Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol. 1998;152:367–372. [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, Whitney K, Masliah E. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010;30:6236–6246. doi: 10.1523/JNEUROSCI.0567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Low P, Masliah E. Multiple system atrophy: a clinical and neuro-pathological perspective. Trends Neurosci. 2011;34:581–590. doi: 10.1016/j.tins.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Inglis C, Mante M, Patrick C, Adame A, Spencer B, Rockenstein E, May V, Winkler J, Masliah E. Fluoxetine ameliorates behavioral and neuro-pathological deficits in a transgenic model mouse of α-synucleinopathy. Exp Neurol. 2012;234:405–416. doi: 10.1016/j.expneurol.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera E, Masliah E. Combination therapies: the next logical step for the treatment of synucleinopathies? Mov Disord. 2016;31:225–234. doi: 10.1002/mds.26428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera E, Ubhi K, Mante M, Rockenstein E, Masliah E. Antidepressants reduce neuroinflammatory responses and astroglial alpha-synuclein accumulation in a transgenic mouse model of multiple system atrophy. Glia. 2014;62:317–337. doi: 10.1002/glia.22610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera E, Mante M, Anderson S, Rockenstein E, Masliah E. Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson’s disease. J Neuroinflammation. 2015;12:93. doi: 10.1186/s12974-015-0320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera E, Spencer B, Fields JA, Trinh I, Adame A, Mante M, Rockenstein E, Desplats P, Masliah E. Combination of alpha-synuclein immunotherapy with anti-inflammatory treatment in a transgenic mouse model of multiple system atrophy. Acta Neuropathol Commun. 2017;5:2. doi: 10.1186/s40478-016-0409-1. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vieira BD, Radford RA, Chung RS, Guillemin GJ, Pountney DL. Neuroinflammation in multiple system atrophy: response to and cause of alpha-sy-nuclein aggregation. Front Cell Neurosci. 2015;9:437. doi: 10.3389/fncel.2015.00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji NP, Collingwood JF, Finnegan ME, Tandon A, House E, Hazrati LN. Iron deficiency in parkinsonism: region-specific iron dysregulation in Parkinson’s disease and multiple system atrophy. Journal of Parkinson’s Disease. 2013;3:523–537. doi: 10.3233/JPD-130197. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Hayashi S, Kakita A, Yamada M, Toyoshima Y, Yoshimoto M, Takahashi H. Accumulation of alpha-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol. 1998a;96:445–452. doi: 10.1007/s004010050918. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alpha-synuclein im-munoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998b;249:180–182. doi: 10.1016/s0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Mori F, Tanji K, Orimo S, Takahashi H. Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegen-erative proteinopathies of the brain. Acta Neuropathol. 2010;120:1–12. doi: 10.1007/s00401-010-0706-x. [DOI] [PubMed] [Google Scholar]
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013;110:19555–19560. doi: 10.1073/pnas.1318268110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S, Kollensperger M, Goebel G, Pfeiffer KP, Barone P, Pellecchia MT, Quinn NP, Koukouni V, Fowler CJ, Schrag A, Mathias CJ, Giladi N, Gurevich T, Dupont E, Ostergaard K, Nilsson CF, Widner H, Oertel W, Eggert KM, Albanese A, Sorbo del F, Tolosa E, Cardozo A, Deuschl G, Hellriegel H, Klockgether T, Dodel R, Sampaio C, Coelho M, Djaldetti R, Melamed E, Gasser T, Kamm C, Meco G, Colosimo C, Rascol O, Meissner WG, Tison F, Poewe W, European Multiple System Atrophy Study, G The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol. 2013;12:264–274. doi: 10.1016/S1474-4422(12)70327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Rubinsztein DC. The Parkinson disease protein α-synuclein inhibits autophagy. Autophagy. 2011;7:429–431. doi: 10.4161/auto.7.4.14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JH, Halliday GM, Kim WS. Exploring myelin dysfunction in multiple system atrophy. Exp Neurobiol. 2014;23:337–344. doi: 10.5607/en.2014.23.4.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, Stefanis L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain J Neurol. 2013;136:2130–2146. doi: 10.1093/brain/awt131. [DOI] [PubMed] [Google Scholar]
- Yabe I, Soma H, Takei A, Fujiki N, Yanagihara T, Sasaki H. MSA-C is the predominant clinical phenotype of MSA in Japan: analysis of 142 patients with probable MSA. J Neurol Sci. 2006;249:115–121. doi: 10.1016/j.jns.2006.05.064. [DOI] [PubMed] [Google Scholar]
- Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–859. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]
- Yokoyama T, Kusunoki JI, Hasegawa K, Sakai H, Yagishita S. Distribution and dynamic process of neuronal cytoplasmic inclusion (NCI) in MSA: correlation of the density of NCI and the degree of involvement of the pontine nuclei. Neuropathology. 2001;21:145–154. doi: 10.1046/j.1440-1789.2001.00390.x. [DOI] [PubMed] [Google Scholar]