

Abstract
Bacillithiol is a compound produced by several Gram-positive bacterial species including the human pathogens Staphylococcus aureus and Bacillus anthracis. It is involved in maintaining cellular redox balance as well as the destruction of reactive oxygen species and harmful xenobiotic agents, including the antibiotic fosfomycin. BshA, BshB, and BshC are the enzymes involved in bacillithiol biosynthesis. BshA is a retaining glycosyltransferase responsible for the first committed step in bacillithiol production, namely the addition of N-acetylglucosamine to l-malate. Retaining glycosyltransferases like BshA are proposed to utilize an SNi-like reaction mechanism in which leaving group departure and nucleophilic attack occur on the same face of the hexose. However, significant questions persist regarding the details of how BshA and similar enzymes accommodate their substrates and facilitate catalysis. Here we report X-ray crystallographic structures of BshA from Bacillus subtilis 168 bound with UMP and/or GlcNAc-mal at resolutions of 2.15 and 2.02 Å, respectively. These ligand-bound structures, along with our functional and computational studies, provide clearer insight into how BshA and other retaining GT-B glycosyltransferases operate, corroborating the substrate-assisted, SNi-like reaction mechanism. The analyses presented herein can serve as the basis for the design of inhibitors capable of preventing bacillithiol production and subsequently, help combat resistance to fosfomycin in various pathogenic Gram-positive microorganisms.
Table of contents
Low-molecular weight thiols such as glutathione (GSH) are prevalent in eukaryotes and Gram-negative bacteria, where they are involved in maintenance of redox homeostasis and detoxification of reactive oxygen species and electrophilic reagents (Scheme 1).1 Gram-positive microorganisms such as Mycobacterium spp. utilize mycothiol (MSH) rather than glutathione for these purposes.2 Other Gram-positive bacteria including Bacillus anthracis, Staphylococcus aureus, and Deinococcus radiodurans manufacture neither glutathione nor mycothiol. Instead, these organisms produce and utilize bacillithiol (BSH) as the major low-molecular weight thiol involved in these processes.3–8 BSH is the preferred cosubstrate for FosB, an enzyme produced by certain species of Gram-positive bacteria that enables resistance to the FDA-approved antibiotic fosfomycin.9–11 Sold under the trade name Monurol, fosfomycin is the only clinically-used drug that inhibits MurA, the enzyme that catalyzes the committed step in bacterial cell wall biogenesis.12 Fosfomycin-resistant organisms in which the BSH biosynthesis genes have been disrupted demonstrate increased sensitivity to fosfomycin,3 suggesting that inhibition of BSH-producing enzymes is an effective therapeutic avenue for the treatment of infections caused by fosfomycin-resistant Gram-positive organisms.
Scheme 1.

Selected low-molecular-weight thiols
BSH, like MSH, consists of a glucosamine core with a cysteinyl (BSH) or N-acetylcysteinyl (MSH) moiety attached via an amide linkage at the 2-position. At the sugar’s anomeric carbon in MSH is an inositol group, whereas BSH has an l-malyl group in this place. The biosynthesis pathways of both compounds are evolutionarily related, as evidenced by similarities in the structures and mechanisms of some, though not all, of the enzymes in the two pathways. The BSH biosynthesis pathway consists of the retaining glycosyltransferase BshA, the zinc-dependent deacetylase BshB, and a putative cysteine ligase BshC (Scheme 2), which differs in several ways from the corresponding MshC enzyme involved in MSH production.3, 13 BshA catalyzes the first committed step in BSH production, utilizing UDP-N-acetylglucosamine (UDP-GlcNAc) as an activated sugar donor and l-malate as an acceptor to form N-acetylglucosaminyl-malate (GlcNAc-mal) and the by-product UDP. BshA, like the corresponding MshA in the MSH biosynthesis pathway, is a GT-4 class retaining glycosyltransferase consisting of the GT-B fold.14, 15 Enzymes of this type are hypothesized to utilize an SNi-like reaction mechanism in which the departure of the UDP leaving group and attack by the malate hydroxyl group at the anomeric position occur at the same face of the sugar in a concerted, asynchronous fashion via a short-lived oxocarbenium-like intermediate.16–18 Deprotonation of the malate hydroxyl group and protonation of the UDP leaving group are hypothesized to proceed via substrate-assisted catalysis. The features within the active site of BshA that accommodate the substrates are currently not well understood, and it is unclear if the substrates bind the enzyme in the manner required for a substrate-assisted SNi-like mechanism.
Scheme 2.

The bacillithiol biosynthesis pathway
Apo and ligand-bound structures of BshA from B. anthracis have been previously determined,14, 19 but they are of low resolution (3.1 and 3.3 Å, respectively) and unambiguous placement of ligands and active site features is not possible. Ligand-bound structures of other GT-B glycosyltransferases, such as MshA20 from the MSH biosynthesis pathway, OtsA17 from the trehalose biosynthesis pathway, and VldE21 from the validamycin A biosynthesis pathway, have been determined at moderate to good resolution. These structures provide some insight into the active site features critical for catalysis, and they help corroborate the SNi-like reaction mechanism for this type of enzyme, but there is limited information on how these enzymes bind their true substrates or products. To further characterize the manner in which BshA and other GT-B retaining glycosyltransferases accommodate their substrates and catalyze their reactions, we embarked upon a combined structural, functional, and computational study of the BshA enzyme. Here we present X-ray crystallographic structures of BshA from Bacillus subtilis strain 168 complexed with UMP and/or the enzyme’s hexose product GlcNAc-mal at resolutions of 2.15 and 2.02 Å, respectively. They represent the first hexose-bound structures of enzymes within the BSH or MSH biosynthesis pathways, and they provide critical insight into the post-catalytic and product-bound states of BshA, MshA, and similar retaining glycosyltransferases. Along with the functional data and computational simulations presented, these structures identify active site features critical for substrate binding and catalysis, and they demonstrate that this class of enzymes most likely proceeds via a substrate-assisted SNi-like reaction mechanism.
EXPERIMENTAL PROCEDURES
Cloning of the gene encoding BshA
The gene encoding BshA (ypjH) was PCR-amplified from B. subtilis strain 168 genomic DNA using the primers: 5’-AAACATATGAGAAAACTAAAAATAGGAATCACATGCTATCCGAGCG-3’ and 5'-AAACTCGAGTCACTCCGGTTCTGCTAAATCGGCATAAATCTG-3', which introduced 5' NdeI and 3' XhoI sites. The PCR product was digested with NdeI and XhoI, purified, and ligated into a pET28b(+) modified to yield a protein product with a recombinant tobacco etch virus protease (rTEV) recognition site. The resulting plasmid was amplified in XL1-blue E. coli cells and sequenced at the Vanderbilt University DNA Sequencing Core Facility. Site-directed mutant genes were produced using the Stratagene site-directed mutagenesis kit and sequenced at the Vanderbilt University DNA Sequencing Core Facility or the University of Michigan DNA Sequencing Core.
Expression and purification
A pET28(b)-BshA clone of the appropriate sequence was used to transform BL21 (DE3) Escherichia coli cells. The cells were grown at 37°C in Lennox L broth containing 25 µg/mL kanamycin until an OD600 of 0.8 was achieved. IPTG was added to a final concentration of 1 mM, and the cells were incubated for 18 hours at 16°C. The cells were harvested by centrifugation and stored at −80°C. The cell paste was resuspended in Lysis Buffer (20 mM tris-HCl pH 7.5, 300 mM NaCl, 10 mM imidazole) and the cells were lysed with lysozyme, DNase and sonication. The lysate was clarified by centrifugation and BshA was purified via nickel affinity chromatography (Thermo) in accordance with the manufacturer's instructions. The BshA preparation was then incubated for 24 hours at room temperature in the presence of 0.5 mM DTT and rTEV protease in a ~50:1 molar ratio (BshA:rTEV) to cleave the nickel affinity tag. The rTEV protease was removed via nickel affinity chromatography. The resulting BshA preparation was dialyzed against 20 mM HEPES, pH 7.5 with 50 mM NaCl and 5 mM MgCl2 and concentrated to 15 mg/mL on the basis of the A280 (ε=22,500 M−1 cm−1, as determined by ProtParam22). Site-directed mutant BshA enzymes were expressed and purified in the same manner with identical results.
Crystallization of BshA/GlcNAc-mal complex
Crystallization conditions were surveyed by the hanging drop method of vapor diffusion using commercially-available crystallization screens (Hampton Research) along with various combinations of relevant ligands. Single crystals suitable for X-ray diffraction analysis were grown by mixing 2 µL dialyzed rTEV-cleaved BshA solution with 2 µL Hampton Research PEG-ion 2 #22 (0.2 M ammonium citrate pH 7.0, 20% (w/v) PEG 3,350) in the presence of 5 mM UDP-N-acetylglucosamine (Sigma) and 5 mM l-malate. Crystals grew to a maximum dimension of 0.1 × 0.2 × 0.3 mm within 1 week after microseeding from the screen. Crystals were transferred to a cryoprotectant solution consisting of 75% well solution and 25% ethylene glycol with 5 mM each of UDP-N-acetylglucosamine and l-malate and flash frozen in liquid nitrogen. The crystals belonged to the C2221 space group with unit cell dimensions of 89.1 × 163.3 × 97.2 Å.
Crystallization of BshA/GlcNAc-mal + UMP complex
Crystals were grown under similar conditions as they were for the BshA/GlcNAc-mal complex, but grew in Hampton Research PEG-ion 2 #15 (4% (v/v) tascimate (which contains l-malate) pH 7.0, 12% (w/v) PEG 3,350) with the addition of 5 mM UDP-GlcNAc to ensure GlcNAc-mal formation. Crystals grew to a maximum dimension of 0.1 × 0.2 × 0.3 mm within 2 weeks. Crystals were transferred to a cryoprotectant solution consisting of 75% well solution and 25% ethylene glycol with the addition of 5 mM UMP, where they were incubated for approximately 10 minutes before being flash frozen in liquid nitrogen. The crystals belonged to the C2221 space group with unit cell dimensions of 90.1 × 164.6 × 97.7 Å.
Structural analysis
X-ray diffraction data were collected at 100 K on the LS-CAT 21-ID-D beamline at the Advanced Photon Source of Argonne National Labs. Data were processed with XDS23 in the AutoPROC24 program and scaled with Scala.25 Relevant data collection statistics are presented in Table 1. The structure of BshA bound with GlcNAc- mal was determined via molecular replacement with Phaser26 using a polyalanine model based on BshA from B. anthracis (PDB entry 3MBO14) as the search model. An initial solution produced a model with an R-factor of 38%. Maximum-likelihood refinement with Refmac27 within the CCP4 Program Suite28 and manual model building with Coot29 produced a final model with an overall R-factor of 19.6 %. The structure of the GlcNAc-mal + UMP complex was solved via molecular replacement using our product-bound structure as a search model. Refinement with Refmac and model building with Coot produced a final model with an R-factor of 18.7%. Relevant refinement statistics are presented in Table 2.
Table 1.
X-ray data collection statistics
| BshA/GlcNAc-mal | BshA/GlcNAc-mal + UMP | |
|---|---|---|
| Wavelength (Å) | 1.07805 | 1.07813 |
| Resolution limitsa (Å) | 97.25 – 2.02 (2.13–2.02) | 93.73–2.15 (2.27–2.15) |
| Number of independent reflections | 46645 (6697) | 40122 (5780) |
| Completeness (%) | 99.9 (99.6) | 100 (100) |
| Redundancy | 7.4 (7.5) | 9.6 (9.9) |
| Avg I/Avg σ(I) | 19.5 (2.5) | 15.0 (3.7) |
| Rmergeb (%) | 5.7 (70.9) | 9.3 (69.0) |
Values in parentheses are for the highest resolution shell
Rmerge = Σ(|I – Ī|)/ ΣI X 100
Table 2.
Refinement statistics
| BshA/GlcNAc-mal (PDB ID 5D01) | BshA/GlcNAc-mal + UMP (PDB ID 5D00) |
|
|---|---|---|
| Resolution limits (Å) | 81.63–2.02 | 82.31–2.15 |
| a Roverall (%)/number of reflections | 19.6/44312 | 18.7/38084 |
| Rwork (%)/number of reflections | 19.3/41999 | 18.4/36074 |
| Rfree (%)/number of reflections | 25.3/2313 | 23.4/2010 |
| Number of protein atoms | 5530b | 5555 |
| Number of heteroatoms | 227c | 161d |
| Average B values | ||
| Protein atoms (Å2) | 45.3 | 42.2 |
| Ligands (Å2) | 32.7 | 44.2 |
| Solvent (Å2) | 42.1 | 36.3 |
| RMSD values | ||
| Bond lengths (Å) | 0.016 | 0.017 |
| Bond angles (°) | 1.74 | 1.82 |
| Planar groups (Å) | 0.008 | 0.008 |
R-factor = Σ(|Fo – Fc|)/ ΣFo X 100, where Fo is the observed structure factor amplitude and Fc is the calculated structure factor amplitude.
Protein atoms include multiple conformations for Tyr 160 in chain A and Ser 364 in chains A and B.
Heteroatoms include 2 GlcNAc-mal molecules and 181 water molecules.
Heteroatoms include 1 phosphate ion, 1 UMP molecule, 2 GlcNAc-mal molecules, and 89 water molecules.
Gel permeation assay
The molar mass of the rTEV-cleaved BshA WT enzyme was estimated via gel permeation chromatography using a BioRad BioLogic LP protein purification system fitted with a 2 × 120 cm Sephacryl S-200 column. The mobile phase was 20 mM HEPES pH 7.5 and 100 mM NaCl and the flow rate was 0.5 mL/min. The column was calibrated using gel filtration standards (Sigma) and the BshA enzyme eluted with an apparent molar mass of 83 kDa, approximately two times its native monomeric molecular weight.
Enzyme assays
Steady-state kinetic parameters for wild-type and mutant BshA enzymes were determined at 21°C utilizing a continuous coupled assay that links UDP formation to the oxidation of NADH via the enzymes pyruvate kinase (PK) and lactate dehydrogenase (LDH). Typical reactions consisted of varying concentrations of UDP-N-acetylglucosamine (0–4 mM) variable concentrations of malate (0–25 mM), 0.2 mM phosphoenolpyruvate, 0.15 mM NADH, 8 U PK, 11 U LDH, and BshA (77 nM, wild-type; 160 nM, S17A and Y95F) in Reaction Buffer (50 mM HEPES pH 7.5, 50 mM NaCl, 5 mM MgCl2). The decrease in absorbance at 340 nm was monitored on an Agilent Cary-60 UV/visible spectrophotometer. Reaction velocities were determined and fit to Equation 1, which describes a sequential ordered bi-bi reaction.
| (Eqn. 1) |
Metal ion dependence was analyzed utilizing a discontinuous HPLC-based assay since the coupled assay requires the presence of magnesium ions. A preparation of rTEV-cleaved BshA WT was subjected to extensive dialysis in the presence of EDTA (50 mM HEPES pH 7.5, 50 mM NaCl, 2 mM EDTA), followed by extensive dialysis in the absence of EDTA (50 mM HEPES pH 7.5, 50 mM NaCl). A control (no treatment) preparation of rTEV-cleaved BshA was dialyzed in the absence of EDTA for the same duration as the EDTA-treated sample. Typical reactions were conducted at 21°C and consisted of 0.5 mM UDP-N-acetylglucosamine, 0.5 mM malate, metal ion (5 mM MgCl2 or 100 µM ZnCl2) or 2 mM EDTA, and 160 nM BshA WT in Reaction Buffer (50 mM HEPES pH 7.5, 50 mM NaCl). Samples of each reaction were removed and quenched by chloroform extraction at 0, 1, 2, and 3 minutes. The reactions were analyzed utilizing an Agilent 1100 HPLC fitted with a 1 mL Resource-Q anion exchange column (GE Healthcare). The column was equilibrated with Buffer A (100 mM NH4HCO3, pH 8.5), after which 10 µL of the quenched reaction mixture was injected onto the column, washed, and eluted with a linear gradient to 60% Buffer B (500 mM NH4HCO3, pH 8.5). UDP peak areas were compared to a standard curve and reaction rates were determined.
Computational simulations
Calculations were carried out using CHARMM v. 40b2 (explicit solvent) or CHARMM v. 37b2 (implicit solvent) with the all-atom C36 additive force field.29 Explicit solvent simulations used particle mesh Ewald electrostatics with TIP3P water under rhombic dodecahedron periodic boundary conditions. Implicit solvent simulations used the generalized Born implicit solvent model (GBSW).30, 31 Parameters for l-malate and the glycosidic linkages for GlcNAc-mal and UDP-GlcNAc were modeled by analogy using ParamChem.32, 33 The BshA protein was modeled using the crystal structure coordinates, modified to add all disordered loops and terminal residues not resolved in the crystal structure. For those residues not present in either chain, i.e., loop 42–47, loop 177–183, and the terminal residues 1, 376, and 377, conformations were built using crystallographic α-carbon positions and standard dihedral angles followed by energy minimization. All hydrogens were added. Histidine protonation states were selected using PROPKA 3.134, 35 and by examination of the crystal structure environment. His 36, 50, and 54 were protonated, His 33, 99, 193, and 204 were deprotonated at the side chain δ position, and His 56, 92, 94, and 121 were deprotonated at the ɛ position.
To model UDP, hydrogens and a β-phosphate were added to the crystal coordinates of UMP in the chain A active site and placed in the same position in chain B. Conformers of UDP were generated by a conformational search of three dihedral angles: C5’-O5’-Pα-O, where O is the oxygen bridging the α- and β-phosphorus, O5’-Pα-O-Pβ, and Pα-O-Pβ-O−, where O− is the terminal oxygen of the β-phosphate. Each angle was varied in 60° increments, generating a total of 72 unique conformers. The five conformers with a terminal oxygen within 2.5 Å of the anomeric carbon of GlcNAc were minimized, both in the presence and absence of the crystallographic GlcNAc-mal. To model the substrate complex, the two lowest energy conformers, called poses 98 and 104, were chosen for additional modification. Pose 52 was also chosen, as this conformer had its α-phosphate in the same orientation as those in the UMP crystallographic structure. These UDP conformers were placed in the active site of BshA and linked to the anomeric carbon of GlcNAc, whose coordinates were taken from the crystallographic GlcNAc-mal but with the malyl group removed. The UDP-GlcNAc models were then energy minimized. Malate, the second substrate, was added to the active site, using the coordinates for the malyl group from the crystal structure and building in hydrogens. This substrate complex was energy minimized in the active site of BshA.
Molecular dynamics (MD) were run starting with the minimized UDP-GlcNAc modeled from pose 98 in the A subunit and that from pose 104 in the B subunit. Malate was also in the active sites of each subunit. The system was equilibrated for 20 ps with a 2 fs time step at 300 K, followed by 1300 ps (explicit solvent, 2 fs time step) or 1125 ps (implicit solvent, 1.5 fs time step) of production dynamics at 300 K.
RESULTS AND DISCUSSION
Overall structure of B. subtilis BshA
The crystals of BshA with GlcNAc-mal bound contained two polypeptides in the asymmetric unit. Electron density is good throughout the model, with the exception of regions at the N- and C-termini and residues 42–48, 178–183, and 263–265 (Subunit A). These regions were too disordered for unambiguous placement of amino acid residues, but positions of α-carbons could be estimated (Figure 1). These regions are in loops at the surface of the enzyme near the interdomain cleft. Loops 42–48 and 178–183 are several angstroms away from the active site and do not interact with active site ligands. However, the 263–265 loop is ordered in our UMP-bound structure and is involved in nucleotide binding. The Ramachandran statistics determined by Molprobity36 for the GlcNAc-mal bound structure are good with 94.8% of residues in favored regions and 5.1% in additional allowed regions. Tyr 95 in both subunits is in the disallowed region, but the electron density corresponding to this residue is unambiguous and supports the given φ and ψ angles. Tyr 95 is within the active site, and its side chain interacts with the bound product, making this strained set of angles likely the result of ligand binding. The crystals containing GlcNAc-mal + UMP contained two polypeptides in the asymmetric unit. Electron density is good throughout the model with the exception of disordered regions at the N-and C-termini and residues 44–48 and 178–184 (Subunit B). Ramachandran analysis of this structure reveals 95.9% of residues are in favored regions and 3.8% are in additional allowed regions. Tyr 95 in both subunits is in the disallowed region, but again electron density corresponding to these residues is unambiguous.
Figure 1. Overall structure of the B. subtilis BshA enzyme.

(A) Cartoon representation of the BshA monomer shown in stereo. Bound ligands are shown as spheres. The locations of the 61–70 loop and the N- and C-termini are indicated with arrows. Estimated positions of the disordered regions consisting of residues 44–48 and 178–184 are shown by dashed lines. (B) Cartoon representation of the BshA dimeric assembly in the closed conformation, colored by chain. The product molecules are shown in spheres. Macromolecular graphics of X-ray crystallographic structures were prepared using PyMOL.46
BshA adopts the typical GT-B fold that consists of two distinct N- and C- terminal Rossmann fold domains with the active site present within the resulting cleft (Figure 1 Panel A). These enzymes can exist in open or closed conformations, but both structures in our study are ligand bound and in the closed conformation. The monomers of the two structures superimpose with a root-mean-square deviation of 0.2 Å, indicating that the presence of UMP did not cause any major changes in enzyme conformation. GT-B glycosyltransferases typically form dimers in solution via interactions between their N-terminal domains. The asymmetric units of both structures in our study revealed a likely dimer interface between the N-terminal domains (Figure 1 Panel B). The interface buries approximately 3800 Å2 of surface area with an average ΔG of −39 kcal/mol as calculated by PDBePISA.37 No relevant additional interfaces were observed within the unit cell or by PDBePISA. Previous gel permeation and X-ray crystallographic analyses of the B. anthracis BshA enzyme suggest the enzyme forms dimers in solution.14 However, another study suggested that B. subtilis BshA enzymes form tetramers.15 In order to confirm the native quaternary structure of B. subtilis BshA, we utilized a gel permeation assay in addition to our structural analysis and PDBePISA calculations. A comparison with the standard curve (R2 = 0.99) revealed that BshA eluted with an oligomeric molar mass of 83 kDa, approximately two times its native monomeric molar mass of 42 kDa. Taken together, our analyses strongly suggest that B. subtilis BshA exists as a dimer under the conditions assayed.
Active site of BshA/GlcNAc-mal complex
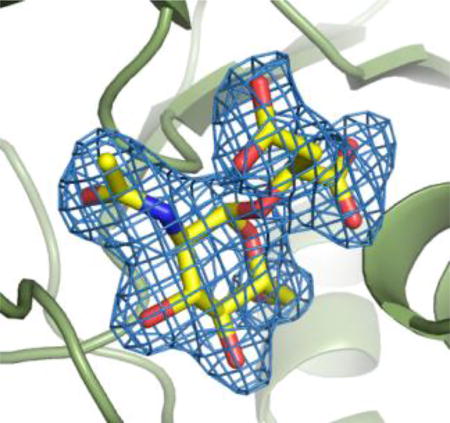
The BshA active site exists within a deep cleft formed between the N- and C-terminal domains, as is the case with all GT-B glycosyltransferases. The active site comprises residues primarily from its host subunit and a loop consisting of residues 61–70 from the other subunit (Figure 1, Panel B). Although in our crystallographic structures residues from this loop do not appear to interact directly with the active site ligands, they do partially define one “wall” of the active site, and our computational simulations (described later) do suggest this loop is important in glycosyl acceptor binding. The crystals of the BshA structure with GlcNAc-mal bound were grown in the presence of the enzyme’s substrates, UDP-N-acetylglucosamine and l-malate, at high concentrations that exceeded their KM values. These compounds were also present in the cryoprotectant solutions at similar concentrations. Significant peaks in the Fo – Fc map were apparent in the active sites of both subunits immediately after molecular replacement. After subsequent refinements to the protein model, it was clear that the density within the active site was that of a hexose, with hydroxyl and the N-acetyl groups fitting nicely within the observed electron density (Figure 2 Panel A). Additional density emanating from the α-face of the hexose anomeric carbon most closely resembled a malyl moiety, suggesting that the enzyme’s product, GlcNAc-mal, was present within the active site. The hexose substrate, UDP-N-acetylglucosamine, would contain a phosphate in this position, and the size and shape of the electron density emanating from the α-face of the hexose anomeric carbon does not suggest the presence of such a group. The other product of the reaction, UDP, was not observed within the active site. However, some minor peaks in the Fo - Fc map occupied the region near the α-face of the hexose, but they are most likely water molecules and have been modeled in as such. Our model suggests that the enzyme was active within the crystallization drop and converted the substrates into products. The resulting GlcNAc-mal product was at a high enough concentration to remain bound within the active site, but the region around the anomeric carbon of the hexose may have been too crowded to allow UDP to remain bound. Although we attempted various soaking and/or cocrystallization experiments with UDP or UDP-N-acetylglucosamine, neither UDP nor UDP-N-acetylglucosamine was ever observed within the BshA active site.
Figure 2. Structure of BshA bound with GlcNAc-mal.

(A) Stereo representation of electron density corresponding to the bound GlcNAc-mal product. The map was calculated with coefficients of the form Fo - Fc with the atoms of the product excluded from the coordinate file and is contoured at 2.5 σ. (B) Close-up stereo view of the active site with the bound product. Amino acid residues lying within ~3.4 Å of the product are shown. Potential hydrogen bonds are highlighted with dashed lines. Water molecules are shown as red spheres. (C) Surface rendering of BshA highlighting the active site cleft and GlcNAc-mal. Subunit A is colored green, whereas Subunit B is colored blue. GlcNAc-mal is shown in sticks with the malyl moiety pointing toward the viewer and the α-face of the hexose pointing into the cleft.
The electron density for GlcNAc-mal is somewhat better in Subunit A, so the following discussion refers to this subunit. The hexose moiety of GlcNAc-mal adopts the 4C1 conformation and is held in the BshA active site via side chain and main chain hydrogen bonding interactions (Figure 2, Panel B). Two regions of the enzyme, the Lys 282 – Gly 286 loop and Ser 17, provide many of the groups that interact with GlcNAc-mal. The hexose hydroxyl groups (3-OH, 4-OH, and 6-OH) are accommodated by main chain and side chain hydrogen bonding interactions with Asn 174, His 121, Glu 283, Phe 285, and Gly 286. The N-acetyl moiety forms hydrogen bonds via its carbonyl oxygen to the side chains of Lys 282 and Ser 284. The 1-carboxylate group of the malyl moiety is accommodated via hydrogen bonding interactions between the main chain amide group from Ser 17 and side chain interactions with Ser 17 and Tyr 95. The 4-carboxylate group of the malyl moiety is involved in a possible electrostatic interaction with Arg 209. Asn 207 is in close proximity to the 4-carboylate group in the crystal structure, but the distance and geometry do not support a hydrogen bonding interaction in the observed conformation. However, our computational simulations suggest that Asn 207 is involved in substrate binding (explained later). The features of the enzyme that interact with GlcNAc-mal are largely on the β-side of the hexose, leaving a large, solvent-filled cleft on the other side of GlcNAc-mal that faces the surface of the enzyme (Figure 1 Panel C).
Active site of BshA/GlcNAc-mal + UMP complex
The crystals of the BshA structure containing GlcNAc-mal and UMP in the active site were grown in the presence of UDP-N-acetylglucosamine. The crystallization solution contained tascimate, a mixture of several carboxylate-containing compounds, including l-malate. Again, the active enzyme produced GlcNAc-mal, which remained bound within the active site. Electron density for the hexose portion of this compound was clearly observed within the maps (Figure 3 Panel A). The malyl group projects from the α-face of the hexose anomeric carbon, although the electron density for the malyl moiety in this structure is not as strong as it is in the BshA complex containing only GlcNAc-mal. Prior to cryoprotection, UMP at 5 mM was soaked into the crystals. Electron density for the UMP molecule is clearly visible within the Fo - Fc omit map in Subunit B (Figure 3 Panel A). However, when UMP was built into the model at 100% occupancy and refined in Refmac, negative peaks in the Fo - Fc map were observed around the UMP molecule, particularly near the phosphorus atom. Subsequently, we built the UMP molecule into our model at 80% occupancy, which resulted in no peaks in the Fo - Fc map after refinement and acceptable B-factors. In Subunit A, UMP is likely present at a very low occupancy such that only the phosphate group is readily apparent. We have chosen to model in only a phosphate group at 50% occupancy in this position since realistic placement of the rest of the UMP molecule is not possible. Attempts to cocrystallize with or soak in the UMP at higher concentrations or for longer times did not improve its occupancy in our structures or were too detrimental to the crystals. Nonetheless, despite being present at partial occupancy, the positioning of the UMP molecule in Subunit B is unambiguous and provides useful insight into how BshA accommodates it.
Figure 3. Structure of BshA bound with GlcNAc-mal + UMP.

(A) Stereo representation of electron density corresponding to the bound GlcNAc-mal product and UMP. The map was calculated with coefficients of the form Fo - Fc with the atoms of the product excluded from the coordinate file and is contoured at 2.5 σ. (B) Close-up stereo view of the active site with the bound product and UMP. Amino acid residues lying within ~3.4 Å of the ligands are shown. Potential hydrogen bonds are highlighted with dashed lines. Water molecules are shown as red spheres. (C) Surface rendering of BshA highlighting the active site cleft and ligands. Subunit B is colored green, whereas Subunit A is colored blue. GlcNAc-mal and UMP are shown in sticks.
The hexose product is bound within the active site in this structure in nearly same manner as it is in the structure containing only GlcNAc-mal. The only appreciable difference is that the side chain of Lys 282 moved away from the N-acetyl moiety and no longer forms a hydrogen bond with the carbonyl oxygen of this group. The UMP molecule is bound in the same cleft but is closer to the surface of the enzyme with its phosphate group near the hexose product (Figure 3 Panels B and C). The enzyme accommodates the UMP molecule by expulsion of water molecules rather than significant conformation changes. However, the 263–265 loop, which is disordered in our structure containing only GlcNAc-mal, is ordered in our structure containing both GlcNAc-mal and UMP. The uracil base forms hydrogen bonding interactions with the backbone carbonyl oxygen and amide nitrogen of Gln 263 within this loop. Interestingly, the uracil base in chains C and E of the BshA structure from B. anthracis14 is rotated nearly 180° about the χ angle of the nucleotide as compared to our structure, resulting in unlikely hydrogen bonding interactions with Gln 263 backbone atoms. Given the higher resolution in our study (2.15 Å vs. 3.3 Å) and the high quality of our Fo - Fc map, we believe the uracil base is correctly modeled in our structure. The ribose moiety in our structure adopts the C2'-endo conformation, and its hydroxyl groups interact with Glu 291. The α-phosphoryl group sits at the δ+ end of a helix dipole and interacts with the backbone amide nitrogens of Leu 287 and Val 288 as well as the C4-hydroxyl group of GlcNAc-mal. The phosphate group also interacts with three water molecules, two of which are situated near the α-face of GlcNAc-mal. These water molecules (w 57 and w 82) are within hydrogen bonding distance to the malyl carboxylate groups and hexose amido group, and they are within close proximity to the hexose anomeric carbon. Therefore, these water molecules approximate the positions where two of the β-phosphoryl oxygens of UDP would be bound in the post-catalytic state of the enzyme. This positioning suggests that UDP-GlcNAc binds the enzyme in a “bent-back” conformation that provides space within the active site for malate to bind. The resulting orientation of substrates allows the malate hydroxyl group to be deprotonated by a phosphoryl oxygen and access the anomeric carbon during the proposed SNi-like mechanism. In addition, the main chain carbonyl oxygen of His 121 points toward the hexose in both structures and is within 3.2 – 3.4 Å from the hexose anomeric carbon and ring oxygen (Figure 2, Panel B). This group may provide stabilization of the partial positive charge that develops on the oxocarbenium intermediate.
Comparison with other GT-B glycosyltransferases
MshA is the retaining glycosyltransferase that catalyzes the analogous reaction within the MSH biosynthesis pathway, namely the formation of N-acetylglucosaminyl-inositol-1-phosphate (GlcNAc-Ins-P) from UDP- N-acetylglucosamine and 1-l-myo-inositol-1-phosphate.20, 38 The MshA enzyme from Corynebacterium glutamicum adopts the GT-B fold and shares 22% amino acid sequence identity with BshA from B. subtilis. Structures of C. glutamicum MshA bound with UDP and UDP + 1-l-myo-inositol-1-phosphate have been determined to resolutions of 2.8 and 2.6 Å, respectively. These structures revealed many of the active site features important for binding their respective ligands and provide evidence for the substrate-assisted SNi-like reaction mechanism. However, the MshA structures lack the donor hexose moiety that is at the core of the MSH molecule, preventing full characterization of the features critical for substrate binding and limiting the ability to make conclusions about the reaction mechanism.
B. subtilis BshA and C. glutamicum MshA (PDB Accession Number 3C4V) superimpose with a root-mean-squared deviation of 1.9 Å for 314 structurally-equivalent core α-carbons. Both enzymes are in their closed conformations, and their secondary structural elements closely match with the notable exception of the 61–70 loop of BshA and a nearby α-helix (Figure 4 Panel A). These elements are within the N-terminal domain and are involved in dimerization in both enzymes. In BshA the 61–70 loop interacts with the active site of the neighboring subunit. In MshA this so-called β4/α2 loop is composed of residues 71–79 and interacts with the active site of its own subunit. Within the active sites of the two enzymes, the nucleotide ribosyl and α-phosphoryl groups are closely aligned, and a minor rotation of the nucleotide χ angle is observed that changes the orientation of the uracil base slightly (Figure 4 Panel B). The inositol-phosphate molecule of MshA and the malyl moiety of BshA occupy similar locations within their respective active sites. The β-phosphoryl group of the MshA UDP extends toward the α-face of the BshA hexose anomeric carbon and occupies the position where water 57 and water 82 are located (Figure 3 Panel B). Thus the β-phosphoryl group of the MshA UDP is consistent with where the same feature would bind within the BshA active site.
Figure 4. Superposition of BshA and MshA structures.

(A) Cartoon representation of BshA (green) and MshA (blue) monomers, shown in stereo. The dimeric interfaces for both enzymes occur along the helices and loops at the bottom of the image. (B) Close-up view of the ligands bound in the active sites. The UMP and GlcNAc-mal ligands from BshA are shown with green carbons and the inositol-phosphate and UDP ligands from MshA are shown with blue carbons. (C) Close-up view of the active sites highlighting polar side chains near the hexose anomeric carbon. Residues from MshA (blue) are labeled with an asterisk. BshA ligands are shown as green carbons, but MshA ligands have been omitted for clarity.
OtsA is a GT-B retaining glycosyltransferase that produces the α,α-1,1-glucose disaccharide trehalose,39 which is involved in several biological processes including insect energy storage, resistance to cellular desiccation, and glycolipid formation in mycobacteria.40 The structure of OtsA was determined to a resolution of 2.2 Å with UDP and a validoxylamine-derived pseudo-disaccharide product analog (VDO).41 The structure mimics the transition state and product-bound state of the enzyme, and it gives clues to how the donor hexose moiety is accommodated within the active site. However, the molecular structure of the VDO analog is different than a true disaccharide product, since it contains a double bond that mimics a C5-O double bond rather than O-C1 double bond, as would occur in the oxocarbenium-like transition state. Nonetheless, the OtsA structure provides insight into how this group of enzymes accommodates its substrates. A structural superposition of the ligands within the active sites of OtsA and our BshA reveals close alignment of the OtsA UDP and BshA UMP (Figure S1 of the Supporting Information). The positioning of the β-phosphoryl group in OtsA indicates the binding site of this group within the BshA active site, and it closely aligns with the β-phosphoryl position in the MshA structure.
A double displacement mechanism is unlikely
Retaining GT-B glycosyltransferases like BshA utilize α-anomeric substrates and release α-anomeric products. Initially, it was suspected that such glycosyltransferases could utilize a double displacement mechanism in which an inverted glycosyl-enzyme intermediate is produced. However, subsequent structural studies of GT-B glycosyltransferases revealed that no conserved nucleophilic group is present within the active sites of these enzymes.42 In BshA, the only amino acid side chains close enough to the anomeric carbon of the product to serve as a nucleophile in such a double displacement mechanism are Ser 17, Tyr 95, His 121, and Thr 123. On the basis of amino acid sequence alignments, these four residues are strictly conserved among BshA enzymes from various bacillithiol-producing organisms including B. subtilis, B. anthracis, S. aureus, Staphylococcus saprophyticus, and Deinococcus radiodurans (Figure S2 of the Supporting Information). Structural comparison with MshA reveals that many features within the active sites are conserved, including the position and orientation of His 121 (His 134 in MshA) and the position but not the orientation of Tyr 95 (Tyr 110 in MshA) (Figure 4 Panel C). Thr 123 (Thr 134 in MshA) is shifted 2–3 Å away from Tyr 95 and Ser 17 (Met 24 in MshA) is not conserved. To explore the roles of these residues in BshA, we produced and analyzed site-directed mutants. The S17A and Y95F mutant enzymes exhibited only mildly reduced catalytic function (Table 3). Both mutations result in increases in the KM values for UDP-GlcNAc and l-malate, but only mild reductions in kcat (95% of WT kcat for S17A and 35% of WT kcat for Y95F). Our crystal structures demonstrate that the hydroxyl groups of Ser 17 and Tyr 95 interact with one of the malate carboxylate groups. These interactions no longer occur in the mutant enzymes, which perturbs substrate binding and requires higher concentrations of the substrates to catalyze the reaction. Although the kcat values are reduced in both mutants, such a mild reduction is inconsistent with the mutation of a critical active site nucleophile.
Table 3.
Steady-state kinetic parameters for BshA from B. subtilis
| BshA forma | KM(UDP-GlcNAc) (mM) | KM(malate) (mM) | kcat (s−1) | kcat/KM(UDP-GlcNAc) (M−1s−1) |
|---|---|---|---|---|
| Wild-type | 0.16 ± 0.02 | 0.054 ± 0.005 | 2.63 ± 0.07 | 1.6 × 104 |
| S17A | 0.60 ± 0.08 | 10 ± 1 | 2.5 ± 0.1 | 2.1 × 103 |
| Y95F | 0.35 ± 0.05 | 0.7 ± 0.1 | 0.92 ± 0.03 | 2.6 × 103 |
H121N, T123A, T123V, K212A, E283A, and E291A mutant enzymes exhibited no detectable catalytic activity.
Mutating His 121 or Thr 123 resulted in enzymes with no detectable catalytic activity. In MshA, the hydroxyl group of the threonine occupying the similar position (Thr 134) points away from the location where the hexose anomeric carbon would bind and interacts with the phosphate group of 1-l-myo-inositol-1-phosphate. In our BshA structures, the hydroxyl group of Thr 123 points away from the active site and interacts with two ordered water molecules. Our computational simulations do suggest that Thr 123 may reposition its side chain to interact with l-malate (explained later). The loop containing Thr 123 differs significantly between BshA and MshA (Figure 4 Panel C). Indeed, an examination of several GT-B retaining glycosyltransferase structures, including OtsA and VldE, demonstrate that a polar group is not always present near this position (Ile 155 in OtsA and Ile 183 in VldE). This loop is involved in glycosyl acceptor binding in all of these enzymes and is highly variable in size, orientation, and amino acid composition. Given the variation at this position, it is unlikely that Thr 123 operates as a nucleophile in BshA. It is more likely that mutating Thr 123 caused an alteration in the active site that severely perturbed substrate binding and/or alignment. Although it is uncommon for histidine to act as a nucleophile and form a covalently bound enzyme intermediate, glucose-6-phosphatase does form a phosphohistidine-enzyme intermediate.43 The histidine in position 121 in BshA is highly conserved in terms of position and orientation among several GT-B retaining glycosyltransferases, with some exceptions including replacement with an alanine (Ala 99) in WaaG, a glycosyltransferase involved in lipopolysaccharide biosynthesis.44 Our crystal structures reveal that the His 121 side chain points away from the hexose anomeric carbon and hydrogen bonds with the C-6 hydroxyl group of GlcNAc-mal and the side chain of His 94. Our computational simulations suggest that His 121 is not mobile and remains in this hydrogen bonding configuration. In order for His 121 to act as a nucleophile, these hydrogen bonding interactions would have to be broken and its side chain would need to swing into the active site. It is more likely that the side chain of His 121 is involved in appropriately aligning the hexose during catalysis.
Our mutagenic analysis can rule out Ser 17 and Tyr 95 as active site nucleophiles operating in a double displacement mechanism. Although we cannot rule out His 121 or Thr 123 acting as a nucleophile on the basis of our functional data, their orientations within the active site, interactions with other active site features, and lack of conservation among all GT-B glycosyltransferases make them unlikely candidates. Thus our overall analysis suggests that BshA does not contain a suitable enzymatic nucleophile that would be necessary for a double displacement mechanism.
BshA does not require a divalent cation for activity
Some glycosyltransferases require a divalent metal ion, such as Mg2+, for activity. The metal ion dissipates the negative charge of the phosphate groups of the activated sugar donor. Members of the GT-B fold like BshA typically do not utilize a metal ion in this capacity. Instead, they follow a metal ion-independent mechanism in which negative charges are dissipated by interactions with side chain and main chain formal and partial positive charges. However, the BshA structure from B. anthracis was modeled with UDP and a magnesium ion in the active site.14 Interestingly, this magnesium ion occupies the place where the hexose is bound in our structures (not shown). Despite having Mg2+ present in the dialysis and crystallization solutions in millimolar concentrations, no electron density corresponding to a metal ion was observed in the active sites in our study. The B. anthracis BshA structure was determined at a resolution of 3.3 Å, so caution must be applied to the interpretation of this structure. Given the much higher resolutions in our study (2.15 Å and 2.0 Å) and the quality of our Fo - Fc omit maps (Figures 2 and 3), it is far more likely that the region in question is occupied by a hexose rather than a magnesium ion.
To confirm that magnesium ions are not needed for BshA function, we devised a metal ion-dependence test based upon a discontinuous HPLC assay. In this assay, we dialyzed wild-type BshA extensively in the presence of EDTA to remove divalent cations from the preparation and then dialyzed in the absence of EDTA to ensure no EDTA remained. We then incubated BshA in the presence of its substrates and various metals and determined the rate of UDP formation via HPLC. From this assay it is clear that EDTA does not deactivate the enzyme, the addition of magnesium ions had no significant effect on the reaction rate, and zinc ions had a mild inhibitory effect (Table S1 of the Supporting Information). Thus our study indicates that BshA does not require magnesium ions for enzymatic activity. The structures of MshA from C. glutamicum contained magnesium ions, but they were not within the active site.20 The negative charges of the UDP moiety in the MshA structure are dissipated via interactions with a lysine side chain, main chain amide groups, and the positive end of a nearby helix dipole. Within the active site cleft of BshA, below the sugar’s α face, is Lys 212 (Figure 3, Panel B). This basic residue is appropriately positioned to provide the positive charge needed to bind the phosphoryl groups of the activated glycosyl donor. In addition, the positive end of a nearby helix dipole is present in this region, with the main chain amide group of Gly 16 pointing toward the space where the β-phosphate would be situated. The K212A site-directed mutant enzyme exhibited no enzymatic activity, suggesting that this interaction is absolutely critical for enzyme function or proper active site formation.
Computational simulations
To further validate our hypothesis surrounding the manner in which BshA binds its substrates and products, we undertook a computational study based on the atomic coordinates from our BshA/GlcNAc-mal + UMP complex. The products (UDP and GlcNAc-mal) and substrates (UDP-GlcNAc and malate) were each computationally modeled in the active site of BshA. The conformation of UDP was modeled by adding a phosphate to UMP and varying the diphosphate dihedral angles in 60° increments. Five of the 72 possible conformers of UDP had the terminal β-phosphate oxygen within 2.5 Å of the anomeric carbon of GlcNAc. These conformers were then minimized in the active site in the presence of GlcNAc-mal to create the product complex. Both UDP and GlcNAc-mal products could co-occupy the active site with little to no change relative to the crystal structure.
To model the substrate complex, we rearranged the bonding to link the GlcNAc to UDP rather than the malate, and then minimized the resulting UDP-GlcNAc and malate structures in the active site. After minimization, the diphosphate adopted a very similar conformation for all starting UDP conformations, presumably dictated by the internal hydrogen bond between the α-phosphate O− and GlcNAc’s C4 hydroxyl group (Figure 5, Panel A). When compared to the original crystal structure coordinates (Figure 5, Panel B), the positions of both the GlcNAc ring and the malate shift; the UDP portion of the structure retains a position similar to the UMP crystal structure. In this model, the β-phosphate interacts with Asn 207 and Lys 212, further suggesting that Lys 212 is a critical residue for mitigating the negative charge of the phosphate group.
Figure 5. Substrates of BshA in the active site.

(A) Energy-minimized conformer of the BshA substrates, malate and UDP-N-acetylglucosamine, generated from the crystal structure ligands using computational methods. The malate hydrogen forms a hydrogen bond to the oxygen of the terminal phosphate of the leaving group, with a distance of 1.8 Å. This conformer is from the B subunit; the A subunit conformer minimized with the malate in a similar position, but with the hydrogen of its OH swung away from the phosphate oxygen (3.2 Å). (Carbons in cyan, oxygen in red, nitrogen in blue, phosphorous in gold, hydrogen in white, hydrogen bonds in dashed lines; nonpolar hydrogens not shown.) (B) Comparison of the energy-minimized UDP-GlcNAc conformer to the UMP and GlcNAc-mal ligands resolved from crystallography (grey). Macromolecular graphics from molecular dynamics simulations were prepared using VMD.47
To assess if the substrate binding is stable, we ran 1300 ps of molecular dynamics in explicit solvent, with the substrate complex in the active site of each subunit. Both substrates appear to be able to coexist stably in the active site. The malate in both subunits remained close to UDP-GlcNAc, and it shows some mobility in the active site. The C1 carboxyl group of malate is consistently anchored by the side chains of Asn 207 and Arg 209, but the C4 carboxyl group interacts with a variety of partners, including the amide protons of Gly 15 and Ser 17, and the side chains of Ser 17, Tyr 95 and Arg 209 (Figure 6). In the implicit solvent simulation, Thr 123 also interacted with the C4 carboxyl group.
Figure 6. Hydroxyl group of malate hydrogen bonds to leaving oxygen of phosphate during molecular dynamics.

Substrate structure and malate hydrogen bonding partners at 289 ps during molecular dynamics, subunit A. Malate’s hydroxyl group is in a 1.9 Å hydrogen bond to the oxygen of the UDP leaving group. Atom colors are shown as they are in Figure 5. For clarity, only the side chains of amino acid residues are shown, with the exception of Ser 17.
The molecular dynamics simulations provide supporting evidence for a substrate-assisted SNi-like mechanism. The malate hydroxyl of both subunits showed stable hydrogen bonding to the oxygen linking UDP to GlcNAc, i.e., the leaving group of UDP. The hydrogen bond persisted for over 300 ps in the active site of Subunit A (Figure 6) and for 100 ps in that of Subunit B. Thus the β-phosphate group is within range to activate the malate by removing its hydroxyl proton. The malate hydroxyl did briefly hydrogen bond to Thr 123 and to Tyr 95, but this was when the group was rotated away from the reactive anomeric carbon of GlcNAc. At one point in the simulation, Ser 17, malate and the phosphate oxygen were in a hydrogen bond network in Subunit A (Figure 6). Based on the kinetics data and the geometry observed during the molecular dynamics simulation, a substrate-assisted SNi-like mechanism is most feasible.
GT-B glycosyltransferases can exist in open and closed conformations. In some cases, the difference in conformation is primarily localized to active site loops, as is the case with VldE from the validamycin A biosynthesis pathway.21 With other enzymes, such as MshA, the conformational change is more dramatic and consists of a major reorientation of the N- and C-terminal domains in addition to changes in active site loops. To explore possible conformational changes in BshA, we carried out implicit solvent molecular dynamics simulations, which can speed up conformational sampling by removing the viscosity effects of explicit water.45 The simulations demonstrated a large conformational shift in Subunit B from a closed to an open conformation much like what is observed in MshA (Figure S3 of the Supporting Information). In molecular dynamics simulations of BshA in the absence of ligands, flexing of the protein between these open and closed conformations was readily observed (not shown), suggesting there is a low barrier for this conformational change. With ligands present, Subunit A remained in the closed conformation throughout our simulations, with Lys 67 (of the 61–70 loop) from the neighboring subunit reaching into the active site and interacting with the malate. The open conformation of Subunit B puts the active site out of reach of the Lys 67 from the neighboring subunit. Both subunits in our crystal structures are in the closed conformation, but the Lys 67 side chain amino group is several Angstroms away from the malate. However, the electron density of the 61–70 loop is somewhat ambiguous in places, and the positioning of the Lys 67 side chain and other portions of the loop differ in the various ligand-bound structures of BshA we have determined, suggesting that this loop is highly mobile and likely undergoes conformational changes upon substrate binding. The MshA structure determined with UDP and 1-l-myo-inositol-1-phosphate has a lysine residue (Lys 78 from the same subunit) in this position, and it interacts with the phosphate group of the 1-l-myo-inositol-1-phosphate acceptor molecule. In the explicit solvent simulation, where both subunits remained in the closed conformation, Lys 67 of Subunit B was within interaction distance of the malate in the Subunit A active site during the most stable hydrogen bonding episode (Figure 6). Therefore, it is likely that Lys 67 in BshA plays a key role in glycosyl acceptor binding and specificity.
Concluding remarks
Evidence obtained through several structural and functional studies suggest that GT-B retaining glycosyltransferases proceed via an SNi-like, substrate-assisted mechanism in which the leaving group (UDP) departs from the same face of the hexose slightly ahead of the nucleophile (malate) via a concerted mechanism in which a short-lived intermediate with distinct oxocarbenium character exists. To facilitate this reaction, the malate hydroxyl group is activated via deprotonation by a phosphoryl oxygen from UDP. Our X-ray crystallographic analysis of BshA has produced the highest resolution structures of this enzyme described to date. The lack of a conserved active site nucleophile among GT-B retaining glycosyltransferases makes it unlikely that these enzymes utilize a double displacement mechanism. Our structures and computational models demonstrate that the substrate and enzyme components are arranged within the BshA active site in a manner that enables the proposed substrate-assisted SNi-like mechanism, serving as additional evidence that GT-B fold retaining glycosyltransferases operate in this manner. Additionally, we have described the first hexose product-bound crystallographic structures within the BSH or MSH biosynthesis pathways. The information gleaned from this study can be used to design, develop, and characterize compounds capable of inhibiting these enzymes in an effort to combat antibiotic resistance in several clinically-relevant Gram-positive microorganisms.
Supplementary Material
Acknowledgments
Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Thank you to Prof. Richard Armstrong for funding the early stages of this investigation and for being a kind person who generously supported the careers of those he trained.
Funding
This research was supported in part by NIH grants (R01GM030910 to Dr. Richard N. Armstrong, Vanderbilt University, and F32GM093507 to P.D.C.) and by a grant from the Center for Scholarly and Creative Excellence at Grand Valley State University.
Abbreviations
- BSH
bacillithiol
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- FDA
United States Food and Drug Administration
- GlcNAc-mal
N-acetylglucosamine-1-l-malate
- GlcN-mal
glucosamine-1-l-malate
- GSH
glutathione
- GT-4
glycosyltransferase family 4
- GT-B
glycosyltransferase fold B
- HEPES
N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
- HPLC
high performance liquid chromatography
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- MSH
mycothiol
- NADH
nicotinamide adenine dinucleotide
- PCR
polymerase chain reaction
- PEG
poly(ethylene glycol)
- rTEV
recombinant tobacco etch virus
- UDP
uridine-5'-monophosphate
- UDP-GlcNAc
uridine-5'-diphospho-N-acetylglucosamine
- UMP
uridine-5'-monophosphate
Footnotes
This paper is dedicated in memory of Prof. Richard N. Armstrong.
X-ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics (Accession numbers: 5D00 and 5D01).
References
- 1.Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:736–837. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newton GL, Fahey RC. Mycothiol biochemistry. Arch Microbiol. 2002;178:388–394. doi: 10.1007/s00203-002-0469-4. [DOI] [PubMed] [Google Scholar]
- 3.Gaballa A, Newton GL, Antelmann H, Parsonage D, Upton H, Rawat M, Claiborne A, Fahey RC, Helmann JD. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc Natl Acad Sci U S A. 2010;107:6482–6486. doi: 10.1073/pnas.1000928107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton GL, Rawat M, La Clair JJ, Jothivasan VK, Budiarto T, Hamilton CJ, Claiborne A, Helmann JD, Fahey RC. Bacillithiol is an antioxidant thiol produced in Bacilli. Nat Chem Biol. 2009;5:625–627. doi: 10.1038/nchembio.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaballa A, Antelmann H, Hamilton CJ, Helmann JD. Regulation of Bacillus subtilis bacillithiol biosynthesis operons by Spx. Microbiology. 2013;159:2025–2035. doi: 10.1099/mic.0.070482-0. [DOI] [PubMed] [Google Scholar]
- 6.Sharma SV, Arbach M, Roberts AA, Macdonald CJ, Groom M, Hamilton CJ. Biophysical features of bacillithiol, the glutathione surrogate of Bacillus subtilis and other firmicutes. Chembiochem. 2013;14:2160–2168. doi: 10.1002/cbic.201300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton GL, Fahey RC, Rawat M. Detoxification of toxins by bacillithiol in Staphylococcus aureus. Microbiology. 2012;158:1117–1126. doi: 10.1099/mic.0.055715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helmann JD. Bacillithiol, a new player in bacterial redox homeostasis. Antioxid Redox Signal. 2011;15:123–133. doi: 10.1089/ars.2010.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson MK, Keithly ME, Harp J, Cook PD, Jagessar KL, Sulikowski GA, Armstrong RN. Structural and chemical aspects of resistance to the antibiotic fosfomycin conferred by FosB from Bacillus cereus. Biochemistry. 2013;52:7350–7362. doi: 10.1021/bi4009648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamers AP, Keithly ME, Kim K, Cook PD, Stec DF, Hines KM, Sulikowski GA, Armstrong RN. Synthesis of bacillithiol and the catalytic selectivity of FosB-type fosfomycin resistance proteins. Org Lett. 2012;14:5207–5209. doi: 10.1021/ol302327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma SV, Jothivasan VK, Newton GL, Upton H, Wakabayashi JI, Kane MG, Roberts AA, Rawat M, La Clair JJ, Hamilton CJ. Chemical and Chemoenzymatic syntheses of bacillithiol: a unique low-molecular-weight thiol amongst low G + C Gram-positive bacteria. Angew Chem Int Ed Engl. 2011;50:7101–7104. doi: 10.1002/anie.201100196. [DOI] [PubMed] [Google Scholar]
- 12.Dinh A, Salomon J, Bru JP, Bernard L. Fosfomycin: efficacy against infections caused by multidrug-resistant bacteria. Scand J Infect Dis. 2012;44:182–189. doi: 10.3109/00365548.2011.616221. [DOI] [PubMed] [Google Scholar]
- 13.VanDuinen AJ, Winchell KR, Keithly ME, Cook PD. X-ray Crystallographic Structure of BshC, a Unique Enzyme Involved in Bacillithiol Biosynthesis. Biochemistry. 2015;54:100–103. doi: 10.1021/bi501394q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsonage D, Newton GL, Holder RC, Wallace BD, Paige C, Hamilton CJ, Dos Santos PC, Redinbo MR, Reid SD, Claiborne A. Characterization of the N-acetyl-α-d-glucosaminyl l-malate synthase and deacetylase functions for bacillithiol biosynthesis in Bacillus anthracis. Biochemistry. 2010;49:8398–8414. doi: 10.1021/bi100698n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Upton H, Newton GL, Gushiken M, Lo K, Holden D, Fahey RC, Rawat M. Characterization of BshA, bacillithiol glycosyltransferase from Staphylococcus aureus and Bacillus subtilis. FEBS Lett. 2012;586:1004–1008. doi: 10.1016/j.febslet.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gloster TM. Advances in understanding glycosyltransferases from a structural perspective. Curr Opin Struct Biol. 2014;28:131–141. doi: 10.1016/j.sbi.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SS, Hong SY, Errey JC, Izumi A, Davies GJ, Davis BG. Mechanistic evidence for a front-side, SNi-type reaction in a retaining glycosyltransferase. Nat Chem Biol. 2011;7:631–638. doi: 10.1038/nchembio.628. [DOI] [PubMed] [Google Scholar]
- 18.Ardèvol A, Iglesias-Fernández J, Rojas-Cervellera V, Rovira C. The reaction mechanism of retaining glycosyltransferases. Biochem Soc Trans. 2016;44:51–60. doi: 10.1042/BST20150177. [DOI] [PubMed] [Google Scholar]
- 19.Ruane KM, Davies GJ, Martinez-Fleites C. Crystal structure of a family GT4 glycosyltransferase from Bacillus anthracis ORF BA1558. Proteins. 2008;73:784–787. doi: 10.1002/prot.22171. [DOI] [PubMed] [Google Scholar]
- 20.Vetting MW, Frantom PA, Blanchard JS. Structural and enzymatic analysis of MshA from Corynebacterium glutamicum: substrate-assisted catalysis. J Biol Chem. 2008;283:15834–15844. doi: 10.1074/jbc.M801017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavalier MC, Yim YS, Asamizu S, Neau D, Almabruk KH, Mahmud T, Lee YH. Mechanistic insights into validoxylamine A 7'-phosphate synthesis by VldE using the structure of the entire product complex. PLoS One. 2012;7:e44934. doi: 10.1371/journal.pone.0044934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. Humana Press; 2005. [DOI] [PubMed] [Google Scholar]
- 23.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 26.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Im W, Lee MS, Brooks CL. Generalized born model with a simple smoothing function. J Comput Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 32.Vanommeslaeghe K, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model. 2012;52:3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanommeslaeghe K, Raman EP, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model. 2012;52:3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Søndergaard CR, Olsson MH, Rostkowski M, Jensen JH. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J Chem Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 35.Olsson MH, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 36.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 38.Newton GL, Koledin T, Gorovitz B, Rawat M, Fahey RC, Av-Gay Y. The glycosyltransferase gene encoding the enzyme catalyzing the first step of mycothiol biosynthesis (mshA) J Bacteriol. 2003;185:3476–3479. doi: 10.1128/JB.185.11.3476-3479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibson RP, Tarling CA, Roberts S, Withers SG, Davies GJ. The donor subsite of trehalose-6-phosphate synthase: binary complexes with UDP-glucose and UDP-2-deoxy-2-fluoro-glucose at 2 Å resolution. J Biol Chem. 2004;279:1950–1955. doi: 10.1074/jbc.M307643200. [DOI] [PubMed] [Google Scholar]
- 40.Argüelles JC. Why can't vertebrates synthesize trehalose? J Mol Evol. 2014;79:111–116. doi: 10.1007/s00239-014-9645-9. [DOI] [PubMed] [Google Scholar]
- 41.Errey JC, Lee SS, Gibson RP, Martinez Fleites C, Barry CS, Jung PM, O'Sullivan AC, Davis BG, Davies GJ. Mechanistic insight into enzymatic glycosyl transfer with retention of configuration through analysis of glycomimetic inhibitors. Angew Chem Int Ed Engl. 2010;49:1234–1237. doi: 10.1002/anie.200905096. [DOI] [PubMed] [Google Scholar]
- 42.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh A, Shieh JJ, Pan CJ, Sun MS, Chou JY. The catalytic center of glucose-6-phosphatase. HIS176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. J Biol Chem. 2002;277:32837–32842. doi: 10.1074/jbc.M201853200. [DOI] [PubMed] [Google Scholar]
- 44.Martinez-Fleites C, Proctor M, Roberts S, Bolam DN, Gilbert HJ, Davies GJ. Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem Biol. 2006;13:1143–1152. doi: 10.1016/j.chembiol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Anandakrishnan R, Drozdetski A, Walker RC, Onufriev AV. Speed of conformational change: comparing explicit and implicit solvent molecular dynamics simulations. Biophys J. 2015;108:1153–1164. doi: 10.1016/j.bpj.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The PyMOL Molecular Graphics System, version 1.5.0.4. Schrodinger, LLC; Portland, OR: [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.