

Abstract
Background
About 40–50% of patients with familial microscopic hematuria (FMH) caused by thin basement membrane nephropathy (TBMN) inherit heterozygous mutations in collagen IV genes (COL4A3, COL4A4). On long follow-up, the full phenotypic spectrum of these patients varies a lot, ranging from isolated MH or MH plus low-grade proteinuria to chronic renal failure of variable degree, including end-stage renal disease (ESRD).
Methods
Here, we performed Whole Exome Sequencing (WES) in patients of six families, presenting with autosomal dominant FMH, with or without progression to proteinuria and loss of renal function, all previously found negative for severe collagen IV mutations. Hierarchical filtering of the WES data was performed, followed by mutation prediction analysis, Sanger sequencing and genetic segregation analysis.
Results
In one family with four patients, we found evidence for the contribution of two co-inherited variants in two crucial genes expressed in the glomerular basement membrane (GBM); LAMA5-p.Pro1243Leu and COL4A5-p.Asp654Tyr. Mutations in COL4A5 cause classical X-linked Alport Syndrome, while rare mutations in the LAMA5 have been reported in patients with focal segmental glomerulosclerosis. The phenotypic spectrum of the patients includes hematuria, proteinuria, focal segmental glomerulosclerosis, loss of kidney function and renal cortical cysts.
Conclusions
A modifier role of LAMA5 on the background of a hypomorphic Alport syndrome causing mutation is a possible explanation of our findings. Digenic inheritance is another scenario, following the concept that mutations at both loci more accurately explain the spectrum of symptoms, but further investigation is needed under this concept. This is the third report linking a LAMA5 variant with human renal disease and expanding the spectrum of genes involved in glomerular pathologies accompanied by familial hematurias. The cystic phenotype overlaps with that of a mouse model, which carried a Lama5 hypomorphic mutation that caused severely reduced Lama5 protein levels and produced kidney cysts.
Electronic supplementary material
The online version of this article (10.1186/s12882-018-0906-5) contains supplementary material, which is available to authorized users.
Keywords: Digenic inheritance, Modifier gene, Familial hematuria, Renal cysts, Collagen IV, FSGS, Thin Basement Membrane Nephropathy (TBMN), Kidney disease, Laminin alpha 5, Alport syndrome, Synaptopodin, Metalloproteinase
Background
Hereditary hematuric diseases comprise a genetically and clinically heterogeneous group of conditions, a common feature of which is microscopic hematuria (MH) since early childhood. The most frequent pathological entity is thin basement membrane nephropathy (TBMN) [1, 2], with an estimated population prevalence of 0.3–1% [3, 4]. About 40–50% of familial TBMN are explained by heterozygous mutations in the COL4A3 and COL4A4 genes, which encode for the α3 and α4 chains of collagen IV, the most abundant component of GBM [5]. A much more severe and progressive glomerulopathy, which also presents with MH since childhood is Alport syndrome, caused by either mutations in the COL4A3/A4 genes or mutations in the X-linked COL4A5 gene [2, 6]. Mutations in CFHR5, a gene playing a role in the regulation of the alternative pathway of complement, in FN1 (fibronectin 1), or in MYH9 (heavy chain of myosin 9), comprise rarer genetic causes of hereditary hematurias [1, 6–8].
Although TBMN, caused by COL4A mutations, was considered for many years to be a benign condition accompanied by excellent prognosis on long follow-up, several publications, including ours as early as 2007, convincingly showed that this is not the case [9–11]. This is based on results showing that a variable subset of patients will develop proteinuria and focal segmental glomerulosclerosis (FSGS) and progress to chronic renal failure, even end-stage renal disease (ESRD) [12]. In a Cypriot cohort, up to 30% of TBMN patients with known mutations reached ESRD, at an average age of 56-years. To date, these results have been verified by multiple groups [13–17]. The exact mechanisms for this adverse outcome remain unknown but the role of genetic modifiers has been implicated [18–22]. A significant feature of heterozygous COL4A mutations is the broad phenotypic heterogeneity, while the clinical outcome is at times better described as later-onset Alport-related nephropathy (LOAN) [23]. Some authors also use the diagnosis of autosomal dominant Alport syndrome.
Based on available data there must be more genes that confer FMH when mutated [11, 24]. Whole-exome sequencing (WES) is one of the best modern genetic approaches for gene discovery. Here we report the analysis of 6 families by (WES) and the finding of two variants co-inherited in two genes, the COL4A5 and the LAMA5 genes. Invoking this digenic inheritance can better explain the spectrum of symptoms observed in some patients, than the variant at each one locus alone [25].
Methods
Six families segregating FMH in at least two generations (autosomal dominant inheritance) were thoroughly studied. Some patients also had proteinuria, renal impairment or ESRD. Renal biopsy showed FSGS and TBMN in two of six families (Families CY5372 and CY5381). At first, the index patient of each family underwent Next Generation Sequencing (NGS) that included the parallel analysis of a 5-gene panel, COL4A3, COL4A4, COL4A5, CFHR5, FN1 (Ion Torrent, PGM, Life Sciences) [11]. Subsequently, WES was performed in the index patients plus one unrelated healthy subject by Macrogen (Kyoto, Japan), using the Illumina HiSeq platform (San Diego, CA, USA). The analysis included all exons, splice junctions and 5’-UTR/3’-UTR. Mean DNA fragment size was 101 bp. In total, 51.189.318 bases of genomic DNA were selected. The filtering algorithm of the data (Fig. 1) gave special attention to glomerulus specific genes [26]. Selected variants were validated by Sanger DNA re-sequencing using the ABI BigDye Terminator v1.1 Cycle Sequencing Kit and the ABI PRISM 3130xl genetic analyzer.
Fig. 1.

Hierarchical filtering that we followed for mining the Whole Exome Sequencing data
The study was approved by the Cyprus National Bioethics Committee and all participants gave a signed consent.
Results
WES analysis
NGS analysis for the 5-gene panel was negative. In family CY5500 (Table 1) a variant in the COL4A5 gene, p.Asp654Tyr, was not considered as pathogenic and was overlooked as it did not involve a conserved glycine residue and was thought inadequate to explain the phenotype. WES resulted to a mean of 9000 non-synonymous SNPs and indels in coding regions, per individual (Additional file 1 After applying the analytical filtering algorithm (Fig. 1), we excluded about ~ 8500 variants and ended with a list of 549 candidate variants. When checked for any relation with the glomerular function, for animal models, or functional studies available in the literature, seven variants were considered of high risk, confirmed by Sanger re-sequencing (Tables 2 and 3). Two variants were predicted by five of six software to be damaging/disease causing. Specifically, variant COL4A5-p.Asp654Tyr was not found in 54 samples of the Cypriot general population nor in 305 samples with FMH tested at our setting. Also this variant was never reported in the ExAC genomes database [27]. Variant LAMA5-p.Pro1243Leu was neither found in 81 healthy individuals of the Cypriot general population nor was it present in any of 159 TBMN patients’ DNA. Also, according to the ExAC genomes database, it was found in European (non-Finish) populations with a frequency of ~ 1/21,123.
Table 1.
Clinical data of CY5500 family
| Patient | Gender | Micr. Hematuria | Proteinuria | CRF | Kidney Cysts | ESRD | Renal biopsy result (age range performed) |
|---|---|---|---|---|---|---|---|
| UCY2069 | Male | YES | YES | YES | YES | YES, dialysis | FSGS – thick GBM (50–55 yrs) |
| UCY2075 | Male | YES | YES | YES | YES | YES, transplanted | FSGS - no EM (50–55 yrs) |
| UCY2074 | Female | YES | YES | NO | Not known | NO | FSGS (20–25 yrs) |
| UCY4041 | Female | YES | YES | NO | NO | NO | FSGS – thinning and thickening of GBM (25–30 yrs) |
CRF Chronic Renal Failure, ESRD End Stage of Renal Disease, EM Electron Microscope, FSGS Focal Segmental Glomerulosclerosis, GBM Glomerular Basement Membrane
Table 2.
Effect prediction of the studied variants and general population information
| Gene | Exon | DNA change | Aminoacid change | SNPs3D | SIFT | POLYPHEN2 | Grantham | Mutation Taster | AlignGVGD | General population |
|---|---|---|---|---|---|---|---|---|---|---|
| LAMA5 | 30 | c.3728 C > T | p.P1243L | 1.19 | 0.02 (Damaging) | 0.635 (Probably Damaging) |
98 | Disease Causing | C65 (High) | rs756101090 4/84492 (ExAC database) |
| LAMB2 | 27 | c.4082 C > G | p.S1361 W | 0.07 | 0.00 (Damaging) |
0.999 (Probably Damaging) |
177 | Disease Causing | C65 (High) | – |
| MMP24 | 2 | c.355 G > A | p.G119R | −1.84 | 0.07 (Tolerated) |
0.999 (Probably Damaging) |
125 | Disease Causing | C65 (High) | 3/60382 (ExAC database) |
| SYNPO2L | 3 | c.473 G > A | p.R158H | −0.70 | 0.01 (Damaging) |
0.999 (Probably Damaging) |
29 | Disease Causing | C25 (Medium) | rs200006608 12/9758 (ExAC database) |
| NID1 | 14 | c.2809 G > A | p.V937 M | 1.61 | 0.04 (Damaging) |
0.999 (Probably Damaging) |
21 | Polymorphism | C15 (Low) | rs200467845 11/59184 (ExAC database) |
| TJP1 | 5 | c.428 G > A | p.R143Q | 1.22 | 0.08 (Tolerated) |
0.999 (Probably Damaging) |
43 | Polymorphism | C35 (Medium) | rs377122303 7/60349 (ExAC database) |
| COL4A5 | 26 | c.1960 G > T | p.D654Y | −0.56 | 0.11 (Tolerated) |
0.999 (Probably Damaging) |
160 | Disease Causing | C65 (High) | – |
Table 3.
Direct DNA re-sequencing results (AD: autosomal dominant). Variants not following AD inheritance have not been studied further
| Family | Gene | Variant | WES result confirmed? | Ref. codon | New codon | Family analysis | Inheritance pattern compatible with AD | General population analysis |
|---|---|---|---|---|---|---|---|---|
| CY5500 | LAMA5 | p.P1243L | YES | CCG | CTG | YES | YES | 0/81 0/159a |
| COL4A5 | p.D654Y | YES | GAT | TAT | YES | YES | 0/54 0/305a |
|
| CY5372 | LAMB2 | p.S1361 W | YES | TCG | TGG | YES | NO | – |
| CY5381 | MMP24 | p.G119R | YES | GGG | GGA | YES | More samples needed | 0/54 |
| CY5394 | SYNPO2L | p.R158H | YES | CGC | CTG | YES | More samples needed | – |
| NID1 | p.V937 M | YES | GTG | GTA | YES | NO | – | |
| CY5417 | TJP1 | p.R143Q | YES | CGG | CAG | YES | NO | – |
aResults from patients’ cohorts tested in our lab
Family CY5500
Both variants on LAMA5 and COL4A5 genes are co-inherited by all four patients in family CY5500. The LAMA5 variant has been inherited by male UCY2067 that he is presently healthy. There is oral information that the grandfather (generation I) was suffering by chronic kidney disease. Below there is analytical clinical description for all mutation carriers of family CY5500 (Fig. 2).
Fig. 2.
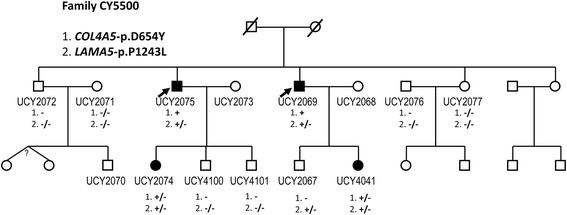
Pedigree of family CY5500. For family members with a UCY code, there is available a DNA sample. Four patients of the family carry both variants (patients UCY2075 and UCY2069 are actually hemizygous for the COL4A5 variant). Family member UCY2067 carries only the LAMA5 variant in heterozygosity, having normal kidney function and being negative for microscopic hematuria, aged 27
UCY2069
CA is a 57-yr-old male, diagnosed with, and treated for, hypertension at the age of 36-yrs, associated with MH, minimal proteinuria and impaired renal function (MDRD eGFR = 66.0 ml/min/1.73m2). At the age of 54-yrs he developed heavy proteinuria of nephrotic range. Despite treatment with steroids and cyclosporine, his kidney function deteriorated and he developed ESRD at the age of 57-yrs. At the age of 54-yrs he had a kidney biopsy showing FSGS. The GBM was folded and appeared thick in many areas, while the podocytes appeared vacuolated and with marked fusion. Renal ultrasound showed multiple renal cortical cysts bilaterally, well before he reached ESRD (Fig. 3).
Fig. 3.
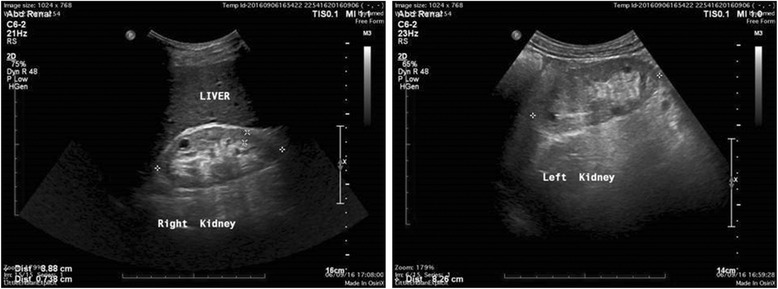
Ultra-sound of the kidneys of patient CA (UCY2069 on Fig. 1), aged 57-yrs, presently on ESRD. Both Kidneys are smaller than normal, measuring Rt kidney 8.88 cm in long diameter and the Lt kidney 9.51 cm, with increased echogenicity and cortical thinning, cortex measuring about 0.88 cm. There is no polycystic kidney disease (pelvocalyceal dilatation). Multiple cortical cysts are noted in both kidneys, about 6–8 cysts in each kidney, the bigger to the Rt about 4.5 cm and to the Lt about 3.6 cm. The cysts however have been present at least since 4-yrs, according to the patient’s medical record, at the time his renal function was much better (MDRD 50 ml/min)
UCY4041
SCA, the daughter of patient CA, is a 25-yr-old girl with MH, borderline proteinuria, normal blood pressure and normal renal function (MDRD eGFR = 130 ml/min/1.73m2). A recent kidney biopsy showed FSGS. Mean width of the GBM was 218.73 nm, thinner than normal. Two measurements showed thickening alternating with thinning. In the most part the podocyte processes were maintained normal but there were a few segments with effacement. Folding of the GBM was observed in a percentage of 5% and in two areas thickening was observed. There were no clear pathognomonic features for Alport syndrome. Electron microscopy showed some deposits which most probably were IgM precipitates, recognized by immunofluorescent microscopy, localized in the periphery and the mesangium. Renal ultrasound was negative for renal cysts.
UCY2075
Male patient SA, brother of CA, aged 60-yrs, was diagnosed with heavy proteinuria, in the nephrotic range, associated with MH and renal impairment (MDRD eGFR = 43.0 ml/min/1.73m2) at the age of 51-yrs. At age 57-yrs he developed ESRD and had a cadaveric kidney transplantation. At the age of 51-yrs he had a kidney biopsy that revealed secondary FSGS, which at the time was attributed to pre-existing arterial hypertension and renal function impairment due to proteinuria. Electron microscope analysis is not available. Renal ultrasound showed multiple renal cortical cysts.
UCY2074
CSA, the daughter of patient SA, is a 34-yr-old girl, with MH and proteinuria. Her kidney function was normal at the age of 29-yrs, taking corticosteroids therapy (MDRD eGFR = 118.0 ml/min/1.73m2). She had a kidney biopsy at the age of 24-yrs, due to MH and proteinuria, revealing FSGS. Renal ultrasound was not available.
UCY2067
EA, the son of CA, aged 27, has inherited only the variant LAMA5-p.Pro1243Leu, and he is presently healthy.
Discussion
Miner et al. [28] described Lama5 (laminin alpha 5 chain) in 1995, member of the vertebrate subfamily of laminin chains, widely expressed in adult tissues, with highest levels in lung, heart, and kidney. In mature glomeruli, collagen α1α2α1(IV) and laminin α1β1γ1 (LN-111) interacting networks are replaced by collagen α3α4α5(IV), laminin α5β1γ1 (LN-511), and laminin α5β2γ1 (LN-521), as glomerular capillary loops expand [29]. It is documented that the maintenance of glomerular filtration barrier integrity requires laminin alpha5 [30]. Despite the fact that Lama5 mouse knockouts are fatal [31], mice with a hypomorphic Lama5 mutation (Lama5neo) that reduces laminin α5 expression, exhibit proteinuria, hematuria and cystic kidneys [32]. Podocyte-specific inactivation of Lama5 in mice, resulted in varying degrees of proteinuria and rates of progression to nephrotic syndrome. The GBM of the proteinuric mice appeared thickened and “moth-eaten,” and podocyte foot processes were effaced [30]. These facts, added to the population data mentioned above, enhance the likelihood that DNA variant LAMA5-p.Pro1243Leu is a mutation, probably a hypomorphic one, since the patients we identified exhibit a great phenotypic similarity with these animal models, presenting with hematuria, proteinuria, renal impairment, thinning and thickening of the GBM, podocyte foot processes effacement and multiple renal cysts.
In the absence of proper functional experiments it is impossible to know how exactly this Lama5 variant affects GBM structure and integrity. We can speculate however that the interaction of the laminin network with the collagen IV α3α4α5 network might be disturbed, resulting in distortion of the mature GBM meshwork and the glomerular filtration barrier. It is also reasonable to suspect an additive effect regarding the damage of GBM in the presence of a defective alpha 5 (Col-IV) chain, where if a dosage threshold is exceeded then a pathogenic phenotype appears. Even though analytical functional experiments are beyond the scope of this paper, we propose that the phenotype is better described by taking into consideration the variants at both loci than each one on its own [25].
It is equally worth mentioning that this is the third report presenting data supporting a probable contribution of the LAMA5 gene to human disease. In a previous report the authors found novel LAMA5 mutations, predicted to be deleterious, in three out of five patients with FSGS or presumed FSGS, also co-inherited with mutations in other genes [33]. Similar results were reported by another study [13]. No renal cysts were described in those patients. An additional argument in support of our hypothesis is the fact that laminin alpha 5 is an important protein of the glomerulus and an interacting partner of collagen IV, while animal models and patients exhibit similar phenotypes with our patients. While the evidence presented is valid, more studies are needed and more patients must be studied before this link is unequivocally documented. Probably, the easiest way is screening for mutations or for modifier variants in the LAMA5 gene in multiple FMH and Alport syndrome families. Confirmation of these results will be of high clinical importance, expanding the spectrum of genes involved in glomerular pathologies. Finally, in recent years other examples have been reported of patients with COL4A5 mutations and Alport syndrome, and phenotypes exacerbated by co-inheritance of mutations in other genes. These include mutations in other collagen IV genes (COL4A3/A4) and in the MYO1E gene [34, 35]. It should not escape our attention that high throughput analysis, including NGS technologies are enabling us to detect and connect clearly pathogenic or hypomorphic mutations in different genes and thus elucidating previously complex phenotypes. Often times though, it is difficult to discriminate cases of true non-Mendelian digenic inheritance from monogenic Mendelian inheritance of phenotypes exacerbated by serendipitous co-inheriting of a second non-allelic mutation or from the effect of otherwise neutral genetic modifiers, or even from the serendipitous co-inheritance of two independent clinical entities.
Conclusions
In conclusion, we found evidence that digenic inheritance in patients of one family presenting with FMH, renal failure/ESRD, FSGS and cystic kidneys, can more clearly explain the phenotypic spectrum than one gene alone. The suspected genes are COL4A5 and LAMA5. Existing animal models and two previous reports for rare mutations in LAMA5 gene support this observation. These results need further confirmation, as LAMA5 could explain a percentage of patients with complex phenotypes.
Additional file
WES statistics. Number of genetic variants called after the WES analysis. (DOCX 15 kb)
Acknowledgements
The authors express their gratitude to all patients and relatives who participated and made this study possible.
Funding
The work was supported from the Cyprus Research Promotion Foundation through the grant NEW INFRASTRUCTURE/STRATEGIC/0308/24 to CD (co-funded by the European Regional Development Fund and the Republic of Cyprus). The funding body did not contribute to the design of study, collection, analysis and interpretation of data, or in manuscript writing.
Availability of data and materials
COL4A5 transcript: NM_000495.4.
COL4A5: c.1960G > T (p.Asp654Tyr) has been registered in ClinVar with accession number 430716.
LAMA5 transcript: NM_005560.4.
LAMA5: c.3728C > T (p.Pro1243Leu) has been registered in ClinVar with accession number 430574
Authors’ contributions
CD conceived and led the study and revised the manuscript critically for important intellectual content. KV performed the WES data filtering, finalized the list of candidate variants, supervised genotyping, drafted and revised the manuscript and interpreted the data. GP contributed to WES data analysis and supervised molecular analysis of the families. IS performed clinical examination of patients, interpreted clinical information and collected blood samples. CS performed clinical examination of patients of family CY5500 and collected all the needed clinical information for interpretation of the inheritance pattern. DH and IP performed part of the general population and of the family genotyping. DH performed part of the family segregation analysis. AE, YA, AP1, MK, MH and AP2 performed part of the clinical examination and of sample collection. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Cyprus National Bioethics Committee and all participants gave a signed consent.
Consent for publication
All patients have provided written consent for their data to be used in a research publication and that this has also been verified and approved by the Cyprus National Bioethics Committee.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12882-018-0906-5) contains supplementary material, which is available to authorized users.
Contributor Information
Konstantinos Voskarides, Phone: 357-22-895219, Email: kvoskar@ucy.ac.cy.
Gregory Papagregoriou, Email: papagregoriou@gmail.com.
Despina Hadjipanagi, Email: hadjipanagi.despina@ucy.ac.cy.
Ioanelli Petrou, Email: petrou.ioanelli@ucy.ac.cy.
Isavella Savva, Email: savva.isavella@gmail.com.
Avraam Elia, Email: avraamelia@hotmail.com.
Yiannis Athanasiou, Email: avraamelia@hotmail.com.
Androulla Pastelli, Email: apastelli@cytanet.com.cy.
Maria Kkolou, Email: kkoloum@gmail.com.
Michalis Hadjigavriel, Email: dr.mhadjigavriel@gmail.com.
Christoforos Stavrou, Email: stavrou-nephrodial@cytanet.com.cy.
Alkis Pierides, Email: a.pierides@hippocrateon.com.
Constantinos Deltas, Phone: +357-22-892882, Email: deltas@ucy.ac.cy.
References
- 1.Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol Dial Transplant. 2013;28(12):2946–2960. doi: 10.1093/ndt/gft253. [DOI] [PubMed] [Google Scholar]
- 2.Kashtan CE, Segal Y. Genetic disorders of glomerular basement membranes. Nephron Clinical practice. 2011;118(1):c9–c18. doi: 10.1159/000320876. [DOI] [PubMed] [Google Scholar]
- 3.Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169–1178. doi: 10.1046/j.1523-1755.2003.00234.x. [DOI] [PubMed] [Google Scholar]
- 4.Gregory MC. The clinical features of thin basement membrane nephropathy. Semin Nephrol. 2005;25(3):140–145. doi: 10.1016/j.semnephrol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Haas M. Alport syndrome and thin glomerular basement membrane nephropathy: a practical approach to diagnosis. Arch Pathol Lab Med. 2009;133(2):224–232. doi: 10.5858/133.2.224. [DOI] [PubMed] [Google Scholar]
- 6.Kashtan CE. Familial hematurias: what we know and what we don't. Pediatr Nephrol. 2005;20(8):1027–1035. doi: 10.1007/s00467-005-1859-z. [DOI] [PubMed] [Google Scholar]
- 7.Athanasiou Y, Voskarides K, Gale DP, Damianou L, Patsias C, Zavros M, Maxwell PH, Cook HT, Demosthenous P, Hadjisavvas A, et al. Familial C3 glomerulopathy associated with CFHR5 mutations: clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephrol. 2011;6(6):1436–1446. doi: 10.2215/CJN.09541010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, Pusey CD, Pierides A, Kyriacou K, Athanasiou Y, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376(9743):794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voskarides K, Damianou L, Neocleous V, Zouvani I, Christodoulidou S, Hadjiconstantinou V, Ioannou K, Athanasiou Y, Patsias C, Alexopoulos E, et al. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol. 2007;18(11):3004–3016. doi: 10.1681/ASN.2007040444. [DOI] [PubMed] [Google Scholar]
- 10.Pierides A, Voskarides K, Athanasiou Y, Ioannou K, Damianou L, Arsali M, Zavros M, Pierides M, Vargemezis V, Patsias C, et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009;24(9):2721–2729. doi: 10.1093/ndt/gfp158. [DOI] [PubMed] [Google Scholar]
- 11.Papazachariou L, Demosthenous P, Pieri M, Papagregoriou G, Savva I, Stavrou C, Zavros M, Athanasiou Y, Ioannou K, Patsias C, et al. Frequency of COL4A3/COL4A4 mutations amongst families segregating glomerular microscopic hematuria and evidence for activation of the unfolded protein response. Focal and segmental glomerulosclerosis is a frequent development during ageing. PLoS One. 2014;9(12):e115015. doi: 10.1371/journal.pone.0115015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A. Carriers of autosomal recessive Alport syndrome with thin basement membrane nephropathy presenting as focal segmental glomerulosclerosis in later life. Nephron. 2015;130(4):271–280. doi: 10.1159/000435789. [DOI] [PubMed] [Google Scholar]
- 13.Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, Graham N, Venkat-Raman G, Ennis S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2016;31(6):961–970. doi: 10.1093/ndt/gfv325. [DOI] [PubMed] [Google Scholar]
- 14.Xie J, Wu X, Ren H, Wang W, Wang Z, Pan X, Hao X, Tong J, Ma J, Ye Z, et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J Mol Cell Biol. 2015;7(2):184. doi: 10.1093/jmcb/mjv023. [DOI] [PubMed] [Google Scholar]
- 15.Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, Jiang R, Lindsey TB, Wu G, Sparks MA, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253–1259. doi: 10.1038/ki.2014.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Temme J, Peters F, Lange K, Pirson Y, Heidet L, Torra R, Grunfeld JP, Weber M, Licht C, Muller GA, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012;81(8):779–783. doi: 10.1038/ki.2011.452. [DOI] [PubMed] [Google Scholar]
- 17.Hoefele J, Lange-Sperandio B, Ruessmann D, Glockner-Pagel J, Alberer M, Benz MR, Nagel M, Weber LT. Novel heterozygous COL4A3 mutation in a family with late-onset ESRD. Pediatr Nephrol. 2010;25(8):1539–1542. doi: 10.1007/s00467-010-1467-4. [DOI] [PubMed] [Google Scholar]
- 18.Voskarides K, Demosthenous P, Papazachariou L, Arsali M, Athanasiou Y, Zavros M, Stylianou K, Xydakis D, Daphnis E, Gale DP, et al. Epistatic role of the MYH9/APOL1 region on familial hematuria genes. PLoS One. 2013;8(3):e57925. doi: 10.1371/journal.pone.0057925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voskarides K, Arsali M, Athanasiou Y, Elia A, Pierides A, Deltas C. Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr Nephrol. 2012;27(4):675–679. doi: 10.1007/s00467-011-2084-6. [DOI] [PubMed] [Google Scholar]
- 20.Tonna S, Wang YY, Wilson D, Rigby L, Tabone T, Cotton R, Savige J. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr Nephrol. 2008;23(12):2201–2207. doi: 10.1007/s00467-008-0934-7. [DOI] [PubMed] [Google Scholar]
- 21.Stefanou C, Pieri M, Savva I, Georgiou G, Pierides A, Voskarides K, Deltas C. Co-inheritance of functional Podocin variants with heterozygous collagen IV mutations predisposes to renal failure. Nephron. 2015;130(3):200–212. doi: 10.1159/000432406. [DOI] [PubMed] [Google Scholar]
- 22.Voskarides K, Stefanou C, Pieri M, Demosthenous P, Felekkis K, Arsali M, Athanasiou Y, Xydakis D, Stylianou K, Daphnis E, et al. A functional variant in NEPH3 gene confers high risk of renal failure in primary hematuric glomerulopathies. Evidence for predisposition to microalbuminuria in the general population. PLoS One. 2017;12(3):e0174274. doi: 10.1371/journal.pone.0174274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papazachariou L, Papagregoriou G, Hadjipanagi D, Demosthenous P, Voskarides K, Koutsofti C, Stylianou K, Ioannou P, Xydakis D, Tzanakis I, et al. Frequent COL4 mutations in familial microhematuria accompanied by later-onset Alport nephropathy due to focal segmental glomerulosclerosis. Clin Genet. 2017; [DOI] [PubMed]
- 24.Savige J. A further genetic cause of thin basement membrane nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2016; [DOI] [PubMed]
- 25.Schaffer AA. Digenic inheritance in medical genetics. J Med Genet. 2013;50(10):641–652. doi: 10.1136/jmedgenet-2013-101713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papagregoriou G, Erguler K, Dweep H, Voskarides K, Koupepidou P, Athanasiou Y, Pierides A, Gretz N, Felekkis KN, Deltas C. A miR-1207-5p binding site polymorphism abolishes regulation of HBEGF and is associated with disease severity in CFHR5 nephropathy. PLoS One. 2012;7(2):e31021. doi: 10.1371/journal.pone.0031021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miner JH, Lewis RM, Sanes JR. Molecular cloning of a novel laminin chain, alpha 5, and widespread expression in adult mouse tissues. J Biol Chem. 1995;270(48):28523–28526. doi: 10.1074/jbc.270.48.28523. [DOI] [PubMed] [Google Scholar]
- 29.Miner JH. Developmental biology of glomerular basement membrane components. Curr Opin Nephrol Hypertens. 1998;7(1):13–19. doi: 10.1097/00041552-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Goldberg S, Adair-Kirk TL, Senior RM, Miner JH. Maintenance of glomerular filtration barrier integrity requires laminin alpha5. J Am Soc Nephrol. 2010;21(4):579–586. doi: 10.1681/ASN.2009091004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143(6):1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon MB, Patton BL, Harvey SJ, Miner JH. A hypomorphic mutation in the mouse laminin alpha5 gene causes polycystic kidney disease. J Am Soc Nephrol. 2006;17(7):1913–1922. doi: 10.1681/ASN.2005121298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatterjee R, Hoffman M, Cliften P, Seshan S, Liapis H, Jain S. Targeted exome sequencing integrated with clinicopathological information reveals novel and rare mutations in atypical, suspected and unknown cases of Alport syndrome or proteinuria. PLoS One. 2013;8(10):e76360. doi: 10.1371/journal.pone.0076360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lennon R, Stuart HM, Bierzynska A, Randles MJ, Kerr B, Hillman KA, Batra G, Campbell J, Storey H, Flinter FA, et al. Coinheritance of COL4A5 and MYO1E mutations accentuate the severity of kidney disease. Pediatr Nephrol. 2015;30(9):1459–1465. doi: 10.1007/s00467-015-3067-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mencarelli MA, Heidet L, Storey H, van Geel M, Knebelmann B, Fallerini C, Miglietti N, Antonucci MF, Cetta F, Sayer JA, et al. Evidence of digenic inheritance in Alport syndrome. J Med Genet. 2015;52(3):163–174. doi: 10.1136/jmedgenet-2014-102822. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
WES statistics. Number of genetic variants called after the WES analysis. (DOCX 15 kb)
Data Availability Statement
COL4A5 transcript: NM_000495.4.
COL4A5: c.1960G > T (p.Asp654Tyr) has been registered in ClinVar with accession number 430716.
LAMA5 transcript: NM_005560.4.
LAMA5: c.3728C > T (p.Pro1243Leu) has been registered in ClinVar with accession number 430574