

Abstract
Although inflammation-induced peripheral sensitization oftentimes resolves as an injury heals, this sensitization can be pathologically maintained and contribute to chronic inflammatory pain. Numerous inflammatory mediators increase the production of reactive oxygen (ROS) and nitrogen species (RNS) during inflammation and in animal models of chronic neuropathic pain. Our previous studies demonstrate that ROS/RNS and subsequent DNA damage mediate changes in neuronal sensitivity induced by anticancer drugs and by ionizing radiation in sensory neurons, thus we investigated whether inflammation and inflammatory mediators also could cause DNA damage in sensory neurons and whether that DNA damage alters neuronal sensitivity. DNA damage was assessed by pH2A.X expression and the release of the neuropeptide, calcitonin gene-related peptide (CGRP), was measured as an index of neuronal sensitivity. Peripheral inflammation or exposure of cultured sensory neurons to the inflammatory mediators, LPS and MCP-1, elicited DNA damage. Moreover, exposure of sensory neuronal cultures to LPS or MCP-1 resulted in changes in the stimulated release of CGRP, without altering resting release or CGRP content. Genetically enhancing the expression of the DNA repair enzyme, apurinic/apyrimidinic endonuclease (APE1) or treatment with a small-molecule modulator of APE1 DNA repair activity, both which enhance DNA repair, attenuated DNA damage and the changes in neuronal sensitivity elicited by LPS or MCP-1. In conclusion, our studies demonstrate that inflammation or exposure to inflammatory mediators elicits DNA damage in sensory neurons. By enhancing DNA repair, we demonstrate that this DNA damage mediates the alteration of neuronal function induced by inflammatory mediators in peptidergic sensory neurons.
Keywords: DNA damage, inflammation, TRPV1, dorsal root ganglia, apurinic/apyrimidinic endonuclease 1/redox effector factor 1, E3330
Introduction
Inflammatory mediators that are released from injured tissue and immune cells during damage can have acute and chronic effects on the sensitivity of nociceptive primary sensory neurons. Prostaglandins, bradykinin, serotonin, tryptases, cytokines, ATP, and other mediators can alter the sensitivity of these neurons to various stimuli through posttranslational modifications of proteins that contribute to the excitability of sensory neurons (see Richardson and Vasko, 2002). Numerous inflammatory mediators also increase the production of reactive oxygen (ROS) and nitrogen species (RNS) during inflammation and in animal models of chronic neuropathic pain, which can alter excitability of sensory neurons (Bauerova and Bezek, 1999, Babior, 2000, Kim et al., 2004, Wang et al., 2004, Remans et al., 2005, Fidanboylu et al., 2011, Salvemini et al., 2011). Furthermore, several studies demonstrate that antioxidants can reverse changes in neuronal sensitivity (Khattab, 2006, Keeble et al., 2009, Fidanboylu et al., 2011, Duggett et al., 2016). The mechanisms by which ROS and RNS alter the sensitivity of sensory neurons remain to be determined.
One important consequence of ROS/RNS production in sensory neurons is oxidative DNA damage. The repair of DNA damage is critical to maintain neuronal homeostasis (Brooks, 2002, McMurray, 2005, Fishel et al., 2007a, Hetman et al., 2010) and neurons contain multiple repair pathways including base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), direct damage repair (DDR), and nonhomologous end-joining (NHEJ) or homologous recombination (HR), however BER is the predominant neuronal DNA repair pathway (Fishel et al., 2007b, Barzilai et al., 2008, Fortini and Dogliotti, 2010). The BER pathway repairs DNA damage in the nucleus and mitochondria, caused by oxidative damage to bases as well as alkylation of bases and is likely an important repair pathway for protecting neurons. A major step in the BER pathway involves the enzyme, apurinic/apyrimidinic endonuclease/redox effector factor (APE1), which hydrolyzes the phosphodiester backbone immediately 5′ to an apurinic/apyrimidinic (AP) site. This generates a normal 3′-hydroxyl group and an abasic deoxyribose-5-phosphate, which is processed by subsequent enzymes of the BER pathway. As such, compromising the repair activity of APE1 likely augments oxidative DNA damage, whereas enhancing APE1 repair activity could diminish damage. Indeed, our previous studies demonstrate that ROS/RNS and subsequent DNA damage mediate changes in neuronal sensitivity induced by anticancer drugs and by ionizing radiation in sensory neurons and that increasing APE1 expression reverses the changes in sensitivity of peptidergic sensory neurons (Jiang et al., 2008, Vasko et al., 2011, Kelley et al., 2014).
Because oxidative stress can produce DNA damage in sensory neurons, we determined whether inflammation and inflammatory mediators cause DNA damage in sensory neurons. We also examined whether persistent changes in the sensitivity of sensory neurons secondary to exposure to inflammatory mediators are reversed by enhancing the DNA BER pathway. We demonstrate that peripheral inflammation induces DNA damage in the soma of neurons of the lumbar DRG and recapitulate this DNA damage in DRG cultures exposed to the inflammatory mediators, LPS or MCP-1. We also establish that DNA damage mediates changes in neuronal sensitivity, as determined by capsaicin-stimulated neuropeptide release by exogenously enhancing DNA repair via the overexpression of the enzyme APE1 and use of a small molecule APE1 DNA repair enhancer, E3330 (Vasko et al., 2011, Kelley et al., 2014, Georgiadis et al., 2016, Kelley et al., 2016). These data identify a novel pathway by which inflammatory mediators sustain changes in neuronal sensitivity and highlight the enhancement of neuronal DNA repair as a pharmacological target to alleviate inflammatory or chronic pain.
Experimental Procedures
Materials
Unless otherwise indicated, tissue culture supplies were purchased from Thermo Fisher Scientific (Waltham, MA). Poly-D-lysine, laminin, mouse monoclonal anti-vinculin antibody, 1-methyl-2-pyrrolidone (MPL), complete Freund’s adjuvant (CFA), Lipopolysaccharides (LPS) from Escherichia coli 0111:B4, RS 504393, and routine chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Nerve growth factor was purchased from Envigo (Indianapolis, IN), Normocin and LPS-RS were obtained from InvivoGen (San Diego, CA), and Neuroporter was purchased from Genlantis (San Diego, CA). Mouse monoclonal antihuman APE1 antibodies were raised in our laboratory and are available from Novus Biologicals (Littleton, CO), mouse monoclonal anti-phospho-H2AX antibody was acquired from EMD Millipore (Billerica, MA), and anti-Hemagglutinin (HA) antibody conjugated to horseradish peroxidase was purchased from Miltenyi Biotec (San Diego, CA). Chemiluminescence secondary antibodies were acquired from Roche Diagnostics Corp. (Indianapolis, IN). siRNA molecules were purchased from GE Dharmicon (Lafayette, CO).
E3330, also called APX3330, was synthesized per previously published methods (Nyland et al., 2010), dissolved in N,N-dimethylformamide (Sigma-Aldrich) and stored as a 40mM stock at −80°C. LPS was dissolved in MPLand stored as a 50 mM stock at −20°C for a month. Recombinant rat CCL2/MCP-1 protein was purchased from R&D Systems (Minneapolis, MN), dissolved in PBS and stored at −20°C for up to a month. The TLR4 antagonist, LPS-RS, was dissolved in MPL and stored at −80°C. The CCR2 antagonist, RS 504393, was dissolved in MPL and stored at −20°C for a month. Before drug treatment, the stocks were diluted in F-12 growth medium and added to cultures and incubated for 2–96 hours as indicated.
Hindpaw inflammation
Adult male (150–175 g) Sprague-Dawley rats (Envigo, Indianapolis, IN) were used in all experiments. The Animal Care and Use Committee at Indiana University School of Medicine, Indianapolis, IN approved all procedures used in these studies. Rats were transiently anesthetized with isoflurane and injected subcutaneously with 150 μl of a 1:1 (v/v) solution of complete Freund’s adjuvant (CFA) and 0.9% saline into the plantar surface of the right hind paw. The induction of inflammation was confirmed by redness and swelling; only animals with an increase in the injected paw thickness of 3.5 mm or greater were used in experiments.
Cell culture
Dorsal root ganglia (DRG) were dissected from all spinal levels of adult male (150–175 g) Sprague-Dawley rats (Envigo, Indianapolis, IN) and the cells were dissociated as previously described (Kelley et al., 2014). Briefly, rats were euthanized by CO2 asphyxiation and DRGs were collected, trimmed, and then transferred into collagenase solution (1 mg/ml) for incubation at 37°C for 1 hr. The digested DRGs were rinsed with growth medium, centrifuged and dissociated by mechanical agitation. The cells (~30,000/well) were plated into each well of 12-well culture plates precoated with poly-D-lysine and laminin. Cells were maintained in F-12 media supplemented with 10% horse serum, 2 mM glutamine, 100 μg/ml Normocin, 50 μg/ml penicillin, 50 μg/ml streptomycin, 50 μM 5-fluoro-2′-deoxyuridine, 150 μM uridine, and 30 ng/ml of NGF in 3% CO2 at 37°C. Growth medium was changed every other day.
Modulation of APE1 expression
As described previously, small interfering RNA to APE1 (APE1siRNA) and a scrambled siRNA (SCsiRNA) control were used to decrease APE1 protein expression in sensory neuronal cell cultures (Vasko et al., 2005, Jiang et al., 2008). On day 3 in culture, the growth media was replaced with 0.5 ml of Opti-MEM 1 media containing 100 nM of APE1siRNA (5′-GUCUGGUAAGACUGGAGUACC-3′) or SCsiRNA (5′-CCAUGAGGUCAGCAUGGUCUG-3′; and 10 μl of the transfection reagent, Neuroporter. On the next day, 0.5 ml of the growth media without antibiotics was added to each well. The next day, the siRNA-containing media was replaced with normal growth media.
Lentiviral constructs containing (1) the CMV promoter, HA-tagged APE1, IRES, and enhanced green fluorescent protein (EGFP); or (2) CMV, IRES, and EGFP were used to enhance APE1 protein expression in neuronal cell cultures, as previously described (Vasko et al., 2011). On day 5 in culture, 150 pfu/cell of the lentivirus was added to the growth media. Two days later, the virus was removed and the cells were grown for an additional 5 days in normal growth media. In these studies, we selectively reduced APE1 expression in the cultures with siRNA designed against rat APE1 mRNA and added back human APE1 transgenes that were not recognized by the rat siRNA, since the human APE1 homolog has a different nucleic acid sequence at the siRNA binding site (Vasko et al., 2005).
Immunoblotting
Tissues or cells were harvested, lysed in RIPA buffer (Santa Cruz Biotechnology; Dallas, TX), sonicated, and cleared of cellular debris by centrifuging at 4000 RPM for 2 minutes. The protein concentration in lysates was quantified using Lowry assay. Protein aliquots were electrophoresed in a 12% SDS-polyacrylamide gel, transferred to a PVDF membrane, and blocked with Tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% nonfat dry milk for 1 h at room temperature under gentle agitation. Mouse monoclonal antihuman APE1 antibodies (1:1000), mouse monoclonal anti-phospho H2A.X (pH2A.X) antibodies (1:1000), mouse monoclonal anti-vinculin antibody (1:1000), and anti-Hemagglutinin (HA) antibody were added to the blocking solution and incubated for 2 h at room temperature with gentle agitation. Antibody binding was detected following appropriate secondary antibody methods using chemiluminescence. The density of the bands was measured using Quantity One software from Bio-Rad (Hercules, CA) and data were expressed as density of the protein of interest normalized to vinculin.
Measurement of calcitonin gene-related peptide release
To assess the stimulated release of treated neuronal cultures, the cultures were washed once with HEPES buffer consisting of (in mM) 25 HEPES, 135 NaCl, 3.5 KCl, 2.5 CaCl2, 1 MgCl2, 3.3 D-glucose, and 0.1% bovine serum albumin, pH 7.4 and maintained at 37°C. Cultures were then incubated for successive 10-min intervals with 0.4 ml of HEPES buffer alone (to assess basal release), with buffer containing 30 nM capsaicin (to assess stimulated release), then with buffer alone (to assess return to basal release). After each incubation, the buffer was removed and immunoreactive calcitonin gene-related peptide (CGRP) in each sample was measured using radioimmunoassay, as previously described (Chen et al., 1996). After the release experiment, the cells in each well were lysed in 0.4 ml of 0.1 M HCl for 20 min and an aliquot was taken to measure total CGRP content in each well. Total content (fmol/well) was calculated by adding the total amount of CGRP released in all incubations to the total amount of CGRP measured from the lysed cells. The release data is calculated as fmol released/well/10 min and expressed as the % of CGRP release normalized to the total CGRP content.
Statistical analysis
Data are expressed as the mean ± SEM from at least three repeats of each experiment. Differences in pH2A.X expression and CGRP release in DRG cultures were determined using one- or two-way analysis of variance (ANOVA) and Dunnett’s post hoc test. Differences in pH2A.X expression in DRG tissues were determined using Student t-tests. In all cases, significance was established as p < 0.05, comparing treated versus controls.
Results
Hindpaw inflammation elicits DNA damage in the L4/L5 DRG
The ability of neurons to repair DNA is critically important in maintaining neuronal homeostasis (Brooks, 2002, McMurray, 2005, Fishel et al., 2007a, Hetman et al., 2010). The question remains, however, whether peripheral inflammation produces DNA damage. To determine whether inflammation produces DNA damage within the DRG, CFA (1:1 with saline) was injected unilaterally into the plantar hindpaw of the rat to elicit inflammation. The average increase in paw thickness in hindpaws injected with saline was 0.99 ± 0.03 mm, whereas the increase in hindpaws injected with CFA was 5.51 ± 0.12 mm 5 days following injection (n=7). One, two and five days following injection, the animals were sacrificed and the lumbar DRG were collected bilaterally. In this manner, DNA damage and protein expression from tissue ipsilateral to the inflammation could be compared to the contralateral control. As can be seen in the graph in Figure 1A, inflammation induced an increase in DNA damage, as indicated by a 58% increase in the phosphorylation of H2A.X (Rogakou et al., 1998) and this increase was first observed at the 5 day timepoint (representative blot illustrated in Figure 1B). To ascertain whether DNA damage occurred within the sensory neuronal soma in the DRG, immunohistochemistry was performed. As illustrated in representative images in Figure 1C, immunoreactivity for pH2A.X was localized to the nuclei of both neurons and support cells.
Figure 1.
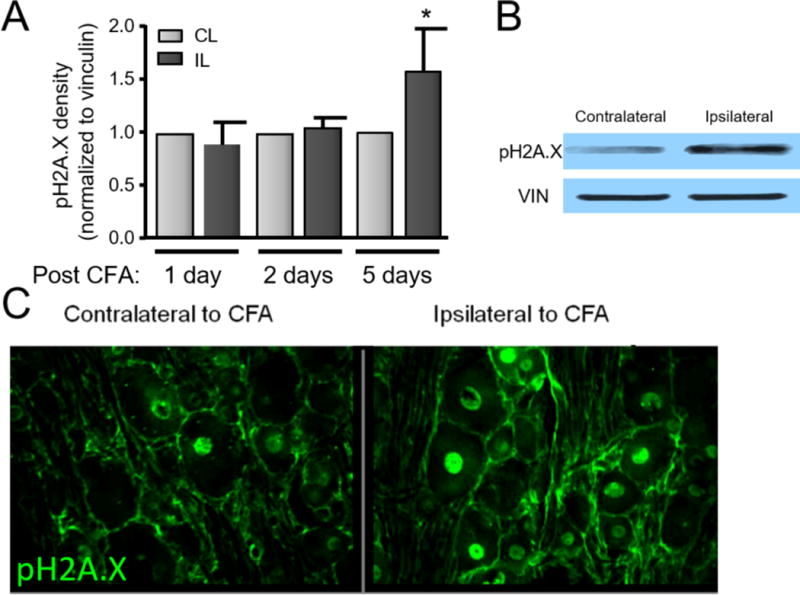
DNA damage is enhanced in the lumbar DRG following hindpaw inflammation. A. Representative western blot of pH2A.X and vinculin (loading control) expression in contralateral and ipsilateral L4/L5 DRG 5 days following unilateral CFA injection into the rat hindpaw. B. Each column represents the mean ± SEM of the density of pH2A.X from 6 experiments normalized to the amount of vinculin. An asterisk indicates a statistically significant increase in the DRG ipsilateral to CFA injection compared to those contralateral to the injection using Student t-test. C. Photomicrographs (20X) of pH2A.X in L5 DRG from a rat 5 days after CFA injection. Green fluorescence indicates the immunoreactivity to pH2A.X.
The inflammatory mediators, LPS and MCP-1, enhance DNA damage in a time-dependent manner
To ascertain whether exposing sensory neurons to inflammatory mediators can cause DNA damage, we utilized cultures of sensory neurons. We exposed the cultures to LPS or MCP-1 and then determined DNA damage, as measured by expression of pH2A.X. As can be seen in Figure 2, exposure of neuronal cultures to LPS (1 μg/ml) resulted in a time-dependent increase in the levels of pH2A.X, apparent within 8 h of treatment and peaking at 24 h. In a similar manner, exposure to MCP-1 (100 ng/ml) induced pH2A.X expression, with an onset of 4 h and peak effects at 24 h (Figure 2). Because the peak effects of the inflammatory mediators on DNA damage were observed at 24 hours, all subsequent experiments were performed at that time point.
Figure 2.
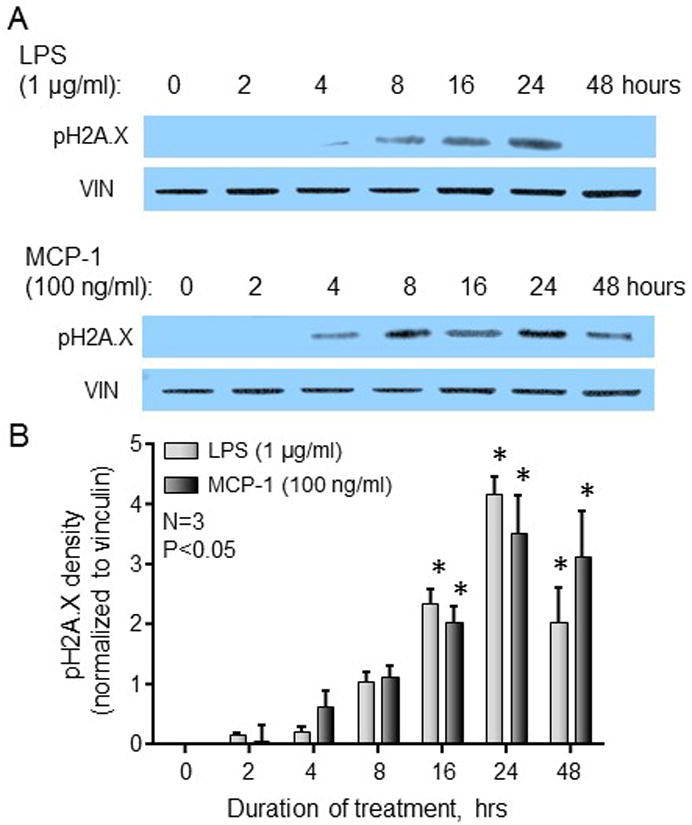
DNA damage is enhanced in neuronal cultures in a time-dependent manner following exposure to inflammatory mediators. A. Representative western blots for pH2A.X and vinculin (loading control) from cultures grown in the absence or presence of LPS or MCP-1 for the indicated time periods. B. Each column represents the mean ± SEM of pH2A.X band density from 3 experiments normalized to that of vinculin following treatment with 1 μg/ml LPS (light bars) or 100 ng/ml MCP-1 (dark bars). An asterisk indicates a significant difference from expression at time zero using one-way ANOVA with Dunnett’s post-test.
The effects of LPS and MCP-1 to induce DNA damage and alter CGRP release are reversed by antagonists of the TLR4 and CCR2 receptors
To demonstrate inflammatory mediator-induced changes in the sensitivity of neurons within DRG cultures, we exposed the cultures to increasing concentrations of each of the inflammatory mediators for 24 h and then examined the basal and capsaicin-stimulated release of the putative nociceptive neuropeptide, CGRP. As illustrated in Figures 3A and 3B, exposure to 30nM capsaicin for 10 min increases CGRP release to approximately 10% of the total content of CGRP. Exposing sensory neurons to a low concentration of LPS (1.0 μg/ml) enhanced the capsaicin-stimulated release of CGRP to 14.4 ± 1.2 % of total content. In contrast, treatment with higher concentrations of LPS significantly decreased the release of CGRP to 6.3 ± 0.4 % of total content in cultures treated with 10.0 μg/ml LPS (Figure 3A). The effects of MCP-1 were analogous to those of LPS; exposure of cultures to low concentrations of MCP-1 for 24 h augmented the release of CGRP to 13.0 ± 0.8 and 15.0 ± 1.0 % of total content in cultures treated with 0.3 and 1.0 μg/ml MCP-1, respectively, whereas higher concentrations of MCP-1 (3.0 – 10.0 μg/ml) decreased CGRP release to 7.1 ± 0.3 % and 6.3 ± 1.0 % of total content, respectively (Figure 3B). Exposure to various concentrations of LPS or MCP-1 did not alter the basal release of CGRP (data not shown). Furthermore, the changes in release of CGRP were not secondary to an altered content of CGRP in the neurons, as the total content of CGRP was similar in cultures treated with vehicle, LPS, and MCP-1 (data not shown).
Figure 3.
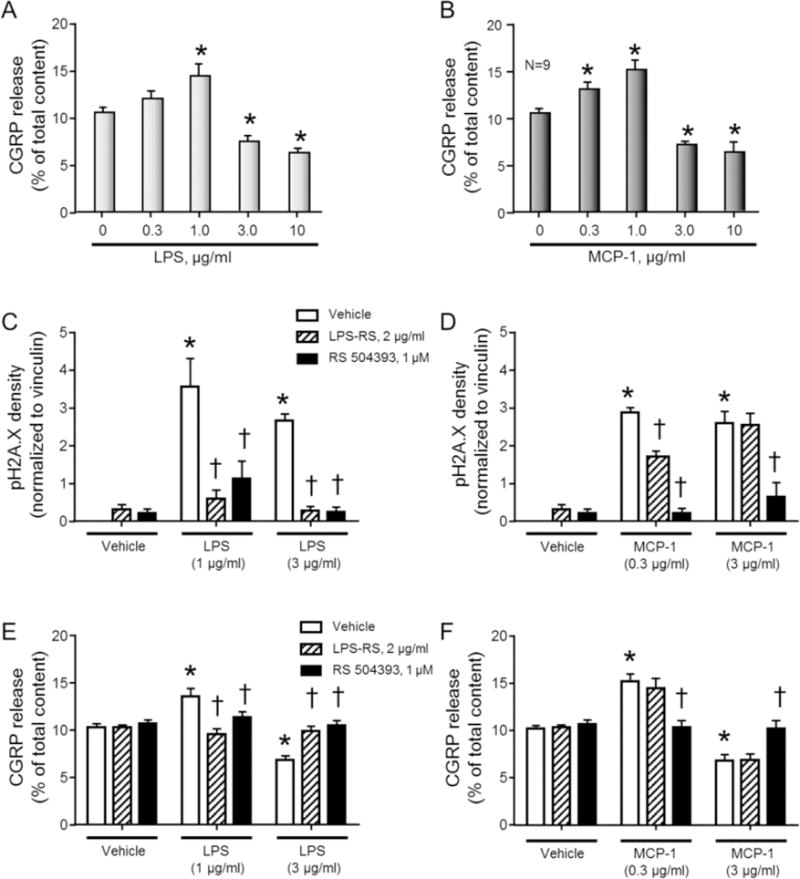
CGRP release from neuronal cultures is altered following exposure to inflammatory mediators and the changes in DNA damage or stimulated CGRP release following exposure to LPS or MCP-1 are reversed by antagonists to the TLR4 (LPS) and CCR2 (MCP-1 and LPS). A B. Columns represent the mean ± SEM of CGRP release stimulated by a 10-minute exposure to 30 nM capsaicin following a 24 hr exposure to increasing concentrations of LPS (A) or MCP-1 (B). An asterisk indicates a significant difference from release in the absence of LPS or MCP-1 using one-way ANOVA with Dunnett’s post-test. C–D. Columns represent the mean ± SEM of the density of pH2A.X from 3–4 experiments normalized to the amount of vinculin following a 24hr exposure to LPS (C) or MCP-1 (D) in the absence or presence of LPS-RS or RS 50493, as indicated. E–F. Columns represent the mean ± SEM of CGRP release stimulated by a 10-minute exposure to 30 nM capsaicin following a 24hr exposure to LPS (E) or MCP-1 (F) in the absence or presence of LPS-RS or RS 50493, as indicated. An asterisk indicates a significant difference from expression/release in the presence of vehicle treatment in the absence of antagonists, whereas a cross indicates a significant difference from expression/release in the presence of inflammatory mediator treatment compared to inflammatory mediator ± antagonist using two-way ANOVA with Dunnett’s post-test.
Although the cognate receptor pathways that are activated by LPS and MCP-1 are the TLR4 receptor pathway and the CCR2 receptor pathway, respectively (Charo et al., 1994, Poltorak et al., 1998), recent reports suggest that these inflammatory agents can modulate other targets (Meseguer et al., 2014). Therefore, we determined whether blocking the activation of the TLR4 or CCR2, using the TLR4 antagonist, LPS-RS (2 μg/ml), or the CCR2 antagonist, RS 50493 (1 μM), inhibited the effects of the inflammatory mediators to enhance pH2A.X expression and alter capsaicin-stimulated CGRP release. In these experiments, we chose to induce DNA damage with both low and high concentrations of LPS and MCP-1. Recent studies have demonstrated that LPS treatment of sensory neurons in culture can upregulate the endogenous production of CCL2 (Miller et al., 2015), therefore we also examined whether the CCR2 antagonist would block the effects of LPS on neuronal DNA damage and neuropeptide release. The cultures were treated with the receptor antagonists 1 hr prior to the introduction of the inflammatory mediators and maintained in the media throughout the exposure. Both low and high concentrations of LPS and MCP-1 induced the expression of pH2A.X. The LPS-induced increases in expression were reversed by both the TLR4 antagonist and by the CCR2 antagonist (Figure 3C). Interestingly, the increases in pH2A.X expression induced by both concentrations of MCP-1 were reversed by the CCR2 antagonist (Figure 3D) and the TLR4 antagonist had a modest effect to attenuate DNA damage elicited by low concentrations of MCP-1, but did not affect damage induced by the higher concentration of MCP-1 (Figure 3D).
The effects of the antagonists to block inflammatory mediator-induced changes in neuropeptide release also were examined. As observed previously, 1.0 μg/ml LPS increased the stimulated release of CGRP from neuronal cultures by 31.9%, whereas 3.0 μg/ml LPS decreased the release by 33.6%. Treatment with either the TLR4 or CCR2 antagonist blocked the changes in release induced by LPS (Figure 3E). Exposing neuronal cultures to 0.3 μg/ml MCP-1 for 24 hr enhanced CGRP release by 49.1% compared to vehicle control. This augmentation was prevented by treatment with RS 50493 (Figure 3F), but not the TLR4 antagonist. As with higher concentrations of LPS, a 3 μg/ml MCP-1 treatment decreased the release of CGRP by 33.1%, a decrease that was reversed by the CCR2 antagonist, but unaffected by the TLR4 antagonist (Figure 3F).
LPS-induced DNA damage and alteration of CGRP release are reversed by overexpression of wildtype APE1 or a repair competent APE1 mutant, but not by an APE1 repair-deficient transgene
We next examined whether enhancing or diminishing the activity of APE1, a critical enzyme in the BER pathway, altered the DNA damage and changes in capsaicin-stimulated CGRP release induced by LPS and MCP-1 treatment. For these studies, cultures were treated as illustrated in Figure 4A. Cultures were transfected with SCsiRNA or rat APE1siRNA on days 4–6 in culture and then exposed to lentivirus containing expression constructs for vector control, human wildtype APE1, mutant c65 APE1, or mutant 226/117 APE1 on days 6–8 in culture. The C65 APE1 mutant has impaired redox, but normal repair function whereas the 226/117 APE1 mutant has impaired DNA repair function but normal redox activity (Luo et al., 2008, Vasko et al., 2011). In one set of cultures, the neurons were treated with E3330 on days 10–14 days in culture. E3330 is a small molecule compound that modulates APE1 activity to enhance DNA repair in sensory neurons (Luo et al., 2008, Kelley et al., 2014). Finally, cultures were treated with LPS (3 μg/ml) for the 24 h immediately prior to experiments.
Figure 4.
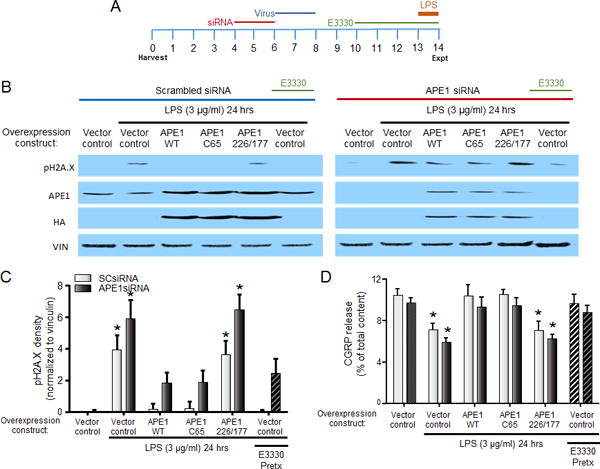
The effects of LPS to induce DNA damage and inhibit CGRP release are reversed by increasing APE1-mediated DNA repair. A. Treatment schema. B. Representative western blots for pH2A.X, APE1, HA tag, and vinculin (loading control) from cultures grown in the absence or presence of LPS for 24hr following the indicated pretreatments. C. Each column represents the mean ± SEM of pH2A.X band density from 3 experiments normalized to vinculin induced by treatment with 3 μg/ml LPS following the indicated pretreatments in conjunction with SCsiRNA (light bars) or APE1siRNA (dark bars). An asterisk indicates a significant difference from expression compared to SCsiRNA-treated vector control using two-way ANOVA with Dunnett’s multiple comparisons post-test. D. Each column represents the mean ± SEM of CGRP release (expressed as % of total content) stimulated by capsaicin following treatment with 3 μg/ml LPS in the absence and presence of APE1 overexpression, as indicated, in conjunction with SCsiRNA (light bars) or APE1siRNA (dark bars). An asterisk indicates a significant difference from release in the absence of LPS using two-way ANOVA with Dunnett’s multiple comparisons post-test.
When cultures treated with SCsiRNA and viral vector control were exposed to LPS for 24 h, there was a significant induction of pH2A.X expression (Figures 4B and 4C). Exogenous expression of either wildtype APE1 or C65 APE1 (repair-competent), at levels ~175% of wildtype endogenous expression as indicated by the expression of HA tag (Figure 4B), ameliorated the ability of LPS to induce DNA damage, decreasing the density of pH2A.X by 95% and 94%, respectively. In contrast, exogenous expression of the 227/177 APE1 mutant (repair-deficient) had no effect on LPS-induced pH2A.X levels. Similar effects were observed in cultures treated with APE1siRNA, which decreased APE1 expression to ~ 20% of wildtype expression (Figures 4B and 5B). Exposure to LPS in cultures treated with APE1siRNA and viral vector control increased the expression of pH2A.X and this trended to be more extensive compared to the SCsiRNA-treated cultures (Figure 4C). As in cells exposed to SCsiRNA, exogenous expression of either wildtype APE1 or C65 APE1 (repair-competent), attenuated the ability of LPS to induce DNA damage, whereas overexpression of 226/177 APE1 had no effect Figures 4B and 4C). Interestingly, the enhancement of the DNA repair activity of APE1 by E3330 mimicked the effects of exogenously expressing wildtype APE1. Pretreatment with E3330 (20 μM) reduced the induction of pH2A.X in both SCsiRNA- and APE1siRNA- treated cultures (Figure 4C).
Figure 5.
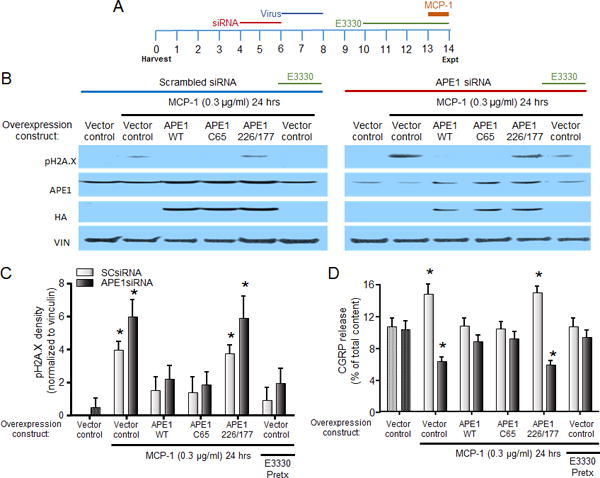
The effects of MCP-1 to induce DNA damage and augment CGRP release are reversed by increasing APE1-mediated DNA repair. A. Treatment schema. B. Representative western blots for pH2A.X, APE1, HA tag, and vinculin (loading control) from cultures grown in the absence or presence of MCP-1 for 24hr following the indicated pretreatments. C. Each column represents the mean ± SEM of pH2A.X band density from 4 experiments normalized to vinculin induced by treatment with 0.3 μg/ml MCP-1 following the indicated pretreatments in conjunction with SCsiRNA (light bars) or APE1siRNA (dark bars). An asterisk indicates a significant difference from expression compared to SCsiRNA-treated vector control using two-way ANOVA with Dunnett’s multiple comparisons post-test. D. Each column represents the mean ± SEM of CGRP release (expressed as % of total content) stimulated by capsaicin following treatment with 0.3 μg/ml MCP-1 in the absence and presence of APE1 overexpression, as indicated, in conjunction with SCsiRNA (light bars) or APE1siRNA (dark bars). An asterisk indicates a significant difference from release in the absence of MCP-1 using two-way ANOVA with Dunnett’s multiple comparisons post-test.
To discover whether a reversal in DNA damage also reversed the effects of LPS on the sensitivity of peptidergic neurons, we again examined the capsaicin-stimulated release of CGRP. In a manner analogous to the data illustrated in Figure 3A, LPS (3 μg/ml) treatment attenuated the release of CGRP stimulated by capsaicin (Figure 4D). In cultures treated with SCsiRNA and a viral vector control, the stimulated release of CGRP from vehicle-treated wells was 10.4 ± 0.6 % of total content, whereas release from cells treated with LPS for 24 h was decreased to 7.1 ± 0.6 % of total content. Exogenous expression of either wildtype APE1 or C65 APE1 (repair-competent) reversed the effects of LPS, so that the stimulated release of CGRP was 10.4 ± 1.1 and 10.5 ± 0.4 % of total content in the presence of APE1 wildtype and C65 mutant, respectively. Exogenous expression of the repair-deficient APE1 mutant did not reverse the effects of LPS, as release was decreased to 7.0 ± 0.9 % of total content. Finally, treatment with E3330 also protected against the effects of LPS on CGRP release; release following E3330 treatment was 9.7 ± 0.8 % of total content, which was no different from release in the absence of LPS treatment. These results support the role of APE1’s DNA repair activity and not it’s redox signaling function in preventing LPS-induced changes in neuronal sensitivity.
MCP-1-induced DNA damage and alteration of CGRP release are reversed by overexpression of wildtype APE1 or a repair competent APE1 mutant, but not by an APE1 repair-deficient transgene
Using the same methods described for experiments with LPS, we manipulated APE1 expression and activity and then treated cultures with MCP-1 (0.3 μg/ml) for the 24 h immediately prior to experiments (Figure 5A). As observed in non-treated cultures (Figure 2), sensory neurons treated with SCsiRNA and a control viral vector then exposed to MCP-1 had enhanced expression of pH2A.X (Figures 5B and 5C). Increasing the exogenous expression of wildtype or repair-competent APE1 (as indicated by expression of HA) prevented the ability of MCP-1 to increase pH2A.X; levels of pH2A.X were decreased to 36.9 and 33.6% of the MCP-1-induced increase in the presence of wildtype and C65 APE1, respectively. In contrast, exogenous expression of the repair-deficient APE1 (226/177) did not prevent the MCP-1-induced expression of pH2A.X. Similar effects were observed in cultures treated with APE1siRNA; MCP-1 induced pH2A.X and this trended to be more extensive compared to the SCsiRNA-treated cultures (Figures 5B and 5C). As in cells exposed to SCsiRNA, exogenous expression of either wildtype APE1 or C65 APE1 (repair-competent), attenuated the ability of MCP-1 to induce DNA damage, whereas overexpression of 226/177 APE1 had no effect (Figures 5B and 5C). As observed with LPS, treatment of sensory neurons exposed to SCsiRNA or APE1siRNA with E3330 reduced the induction of pH2A.X by MCP-1 (Figure 5C)
To determine whether these changes in pH2A.X expression correlated with changes in the neuronal sensitivity of peptidergic sensory neurons, we examined the release of CGRP stimulated by capsaicin (Figure 5D). Following treatment with SCsiRNA, MCP-1 (0.3 μg/ml) enhanced the release of CGRP. This enhancement was not observed when APE1 expression was upregulated exogenously with either the wildtype APE1 or a repair-competent/redox deficient APE1 (C65 APE1). Exogenous expression of the repair-deficient APE1 (226/177); however, did not prevent the MCP-1 induced sensitization of CGRP release (Figure 5D, light gray columns). In cultures treated with APE1siRNA, MCP-1 treatment caused a decrease in CGRP release, suggesting that the response to MCP-1 is shifted leftwards, based on the concentration response curve presented in Figure 3B, in cultures with reduced DNA repair activity. This decrease was reversed by exogenous expression of wildtype or repair-competent APE1, but unaffected by expression of repair-deficient APE1 (Figure 5D, dark gray columns). As observed with the induction of pH2A.X expression, treatment of cultures with E3330 prevented the change in CGRP release induced by MCP-1 exposure (Figure 5D). Collectively, these data support the notion that MCP-1 induces DNA damage in neuronal nuclei and that this DNA damage mediates changes in neuronal sensitivity.
Discussion
Our results demonstrate that peripheral inflammation enhances DNA damage, as indicated by an increase in pH2A.X expression, within the soma of sensory neurons innervating the inflamed tissue. An increase in pH2A.X expression also is observed in sensory neuronal cultures, following exposure to LPS or MCP-1. In addition to DNA damage, exposure of sensory neuronal cultures to LPS or MCP-1 results in changes in the sensitivity of the neurons, as indicated by the stimulated release of the neuropeptide, CGRP, without altering resting release or the total content of CGRP. Genetic manipulation of APE1 expression or treatment with a small-molecule modulator of APE1 DNA repair activity, E3330, to enhance DNA repair via the BER pathway attenuates DNA damage elicited by LPS or MCP-1. Enhancement of APE1 DNA repair activity reverses the inflammatory mediator-induced changes in neuronal sensitivity. Additionally, the inhibition of APE1 redox signaling function did not impact the protection of the cells from inflammation induced cellular alterations. These data support our previous findings demonstrating that it is the DNA repair and not the redox signaling component of APE1 that is necessary for protection of sensory neuronal function and extends these findings into inflammation-induced alterations of sensory neurons (Jiang et al., 2008, Vasko et al., 2011, Kelley et al., 2014). Of interest, we also demonstrate that DNA damage and changes in neuronal sensitivity induced by LPS are inhibited by the CCR2 antagonist, suggesting that long-term sensitization induced by TLR4 activation might be mediated through an increase in the production and putative autocrine activity of CCL2/MCP-1.
Injection of CFA into the hindpaw of a rat elicits behavioral hypersensitivity to thermal and mechanical stimuli (Stein et al., 1988, Woolf et al., 1994), and this hypersensitivity has been attributed to the enhancement of local inflammatory mediators within the damaged tissue (Ferreira et al., 1988, Williams and Higgs, 1988, Cunha et al., 1992, Ferreira et al., 1993, Safieh-Garabedian et al., 1995) or in the soma of the neurons (Jeon et al., 2008). The signaling pathways by which inflammation alters the sensitivity of primary afferent neurons have been investigated extensively and include posttranslational modifications to reversibly alter the function of receptors, ion channels, or associated regulatory proteins and transcriptional regulation to alter the expression of proteins that contribute to excitability of sensory neurons (Neumann et al., 1996). In the latter case, DNA damage could alter the expression of various proteins and/or induce novel expression of mutated proteins to chronically alter the function of sensory neurons.
Although posttranslational modifications clearly alter the sensitivity of sensory neurons, these effects are readily reversible when the evoking stimulus is removed. In contrast, DNA damage, unless repaired can result in permanent alterations in the phenotype of post-mitotic cells. Thus, to identify a causative role for DNA damage in maintaining neuronal sensitization induced by inflammation, we utilized neuronal cultures derived from DRG. The cultures were treated with the TLR4 or CCR2 ligands, LPS or MCP-1/CCL2, respectively, to mimic the effects of inflammation on neurons in culture. LPS is expressed on the outer membrane of gram negative bacteria, including the inactivated Mycobacterium tuberculosis present in complete Freund’s adjuvant used in our in vivo inflammation studies. LPS is an exogenous ligand for the TLR4 receptor, which is expressed in sensory neurons (Diogenes et al., 2011, Li et al., 2014, Li et al., 2015) and exposure to LPS enhances the expression of TNFα, IL-1β, COX-2 and MCP-1 in sensory neurons (Tse et al., 2014, Miller et al., 2015), thus recapitulating the activation of multiple pathways elicited by inflammation. Furthermore, LPS acutely enhances the sensitivity of sensory neurons as demonstrated by nociceptive behaviors following injection into the hindpaw of rodents (Ferreira et al., 1993, Calil et al., 2014) and by in vitro experiments, where LPS enhances the excitability and exocytotic activity of sensory neurons (Hou and Wang, 2001, Diogenes et al., 2011, Meseguer et al., 2014).
MCP-1 is a cytokine that is upregulated in DRG by inflammation (Jeon et al., 2008), and released from DRG or dorsal spinal cord via stimulation of sensory neurons (Dansereau et al., 2008). MCP-1 exposure has been shown to upregulate the neuronal expression of TRPV1 and NaV1.8 (Kao et al., 2012), potentially mediated by the activation of NFκB (Tse et al., 2014, Zhao et al., 2014). MCP-1 also enhances the sensitivity of sensory neurons as evidenced by an increase in nociceptive behaviors following hindpaw injection (Dansereau et al., 2008) and by a direct stimulation of CGRP release from cultures derived from neonatal DRG (Qin et al., 2005b). MCP-1 is a ligand for the CCR2 receptor. Although the CCR2 is not expressed in DRG neurons derived from naïve animals, the CCR2 is expressed in DRG following inflammation or nerve injury (White et al., 2005, Miller et al., 2012, Zhang et al., 2013). Furthermore, the CCR2 is functionally active in cultures derived from DRG (Qin et al., 2005b, Kao et al., 2012).
It is appreciated that inflammation and the inflammatory mediators, LPS and MCP-1 acutely activate signaling pathways that contribute to hypersensitivity, which are likely independent of DNA damage. Indeed, changes in neuronal excitability induced by exogenous administration of LPS and MCP-1 can be observed at timepoints prior to the development of DNA damage (<2 hrs, Figure 2) (Hou and Wang, 2001, Qin et al., 2005b, Diogenes et al., 2011, Meseguer et al., 2014). In addition to the acute sensitization of neurons, inflammatory mediators also can enhance the generation of ROS/RNS via enzymatic (NADPH oxidase) and autooxidation reactions (metabolism-induced increases in electron transport chain leakage) (Bauerova and Bezek, 1999, Babior, 2000, Qin et al., 2005a, Remans et al., 2005, Ibi et al., 2008, Hackel et al., 2013). Because we found that the effects of LPS could be attenuated by a CCR2 antagonist, we suspect that the maintenance of hypersensitivity induced by LPS is mediated through activation of TLR4 and subsequent upregulation of MCP-1/CCL2. This finding was intriguing because activation of TLR4 elicits the generation of ROS/RNS in macrophages (Qin et al., 2005a, Zhang et al., 2015), yet in neurons TLR4 activation cannot maintain sensitivity without activation of the CCR2. Therefore, we propose that the quantitative, spatial and temporal aspects of ROS/RNS generation are critical for inducing DNA damage and will be studied further. Two recent publications suggest that NADPH oxidase plays a large role in modulating the sensitivity of sensory neurons in the context of inflammation (Ibi et al., 2008, Ding et al., 2016).
ROS/RNS function as agonists for the TRPV1 and TRPA1 channels (Andersson et al., 2008, Sawada et al., 2008, Keeble et al., 2009, Ito et al., 2013, Lin et al., 2015). In addition to the acute effects of ROS to enhance TRPV1 and TRPA1 sensitivity, an intracellular increase in free radical moieties can lead to the oxidation of molecules, including nucleic acids, proteins, and lipids, leading to potentially serious consequences for sensory neurons. One potential long-term consequence of this oxidative stress is oxidative DNA damage. Although sensory neurons have endogenous antioxidant mechanisms and DNA repair pathways to combat excessive production of ROS/RNS, excessive oxidative stress can overwhelm the endogenous antioxidants and DNA repair mechanisms. Although sensory neurons are post-mitotic, DNA damage can still have critical consequences on the integrity of gene transcription and for the maintenance of neuronal homeostasis (Fishel et al., 2007b). Indeed, we recently demonstrated that DNA damage was a causative factor in altering the sensitivity of neurons following treatment with chemotherapies that cause oxidative DNA damage (Vasko et al., 2005, Jiang et al., 2008, Jiang et al., 2009, Kelley et al., 2014). Our studies identified that changes in neuronal sensitivity could be reversed by repair of oxidative lesions induced by cisplatin or ionizing radiation, suggesting an important role for ROS/RNS in modulating neuronal sensitivity by damaging DNA.
Inflammation-induced DNA damage in sensory ganglia are not restricted to the sensory neurons. There is abundant evidence to support inflammation- or inflammatory mediator-induced activation of satellite glial cells, as evidenced by an increase in the expression of glial fibrillary acid protein (GFAP) and phosphorylation of ERK (Villa et al., 2010, Matsuura et al., 2013, Blum et al., 2014, Lukacs et al., 2015). Mechanistic studies have identified putative signaling pathways by which the activation of satellite cells enhances the excitability or sensitivity of sensory neurons and these include: coupling between the glial and neuronal cells via gap junctions (Dublin and Hanani, 2007) and enhanced release of cytokines, PGE2 and glutamate from the glial cells (Takeda et al., 2008, Capuano et al., 2009, Rozanski et al., 2013). In our experiments, we did not distinguish whether alteration in DNA damage or repair was limited to sensory neurons. It is likely that inflammation-induced DNA damage also is present within the satellite glial cells and it is conceivable that this contributes to the sustained hypersensitivity of neurons. Consequently, further experiments are warranted to examine this possibility.
Our current findings demonstrating that DNA damage is critical for the maintenance of changes in peptidergic neuronal function induced by inflammation expand on our previous studies with anticancer drugs (Jiang et al., 2008, Kelley et al., 2014, Kelley and Fehrenbacher, 2017). Reducing the expression of APE1 increases the neurotoxicity produced by LPS and MCP-1 exposure, whereas augmenting the DNA repair activity of APE1, whether through enhanced expression or with E3330, lessened the neurotoxicity. In addition to the AP endonuclease function of APE1, the enzyme also functions as a protein-protein redox signaling molecule modulating the redox status of transcription factors to regulate their function (Fishel and Kelley, 2007, Luo et al., 2008, Jiang et al., 2010, Fishel et al., 2011, Kelley et al., 2012). Our findings that overexpression of the DNA repair-competent APE1, but not the redox-competent APE1, suggest that the DNA repair component of APE1 is essential to reverse sensitization induced by inflammatory mediators. These results parallel our earlier findings, that the neuroprotective effect of APE1 to prevent cisplatin-induced toxicity required its DNA repair function, but not its redox signaling function (Jiang et al., 2008, Vasko et al., 2011). The implication, therefore, is that exposure of sensory neurons to inflammation can elicit hypersensitivity through a variety of signaling pathways; however, the maintenance of this sensitization is dependent on DNA damage and this damage is the crux of the problem.
What is still unclear is how seemingly random DNA damage elicited by inflammation or inflammatory mediators can elicit such a reproducible phenotype to sustain neuronal hypersensitivity. The major oxidative DNA lesion formed by oxidative stress, 8oxoG, has been suspected to contribute to the development of inflammation and aging (Shigenaga et al., 1994, David et al., 2007). Further experiments examining the role of OGG1 in neuronal function are ongoing to discern how 8oxoG affects sensory neurons. The redox function of APE1 already has been recognized as contributing to an inflammatory response in other cell types (Jedinak et al., 2011), but our findings are the first to implicate a protective role for the DNA repair function of APE1 and not the redox function. We hypothesize that posttranslational and transcriptional effects of inflammatory mediators can mediate the induction of hypersensitivity in neurons, but DNA damage maintains these changes due to the impact of oxidative DNA lesions on transcriptional activity. Thus, inflammation could cause functional changes in neurons that are reproducible and that enhanced DNA repair could reverse the functional changes in neurons induced by the damage. Oxidative damage to DNA is known to alter the ability of transcription factors to recognize and bind promoter regions (Ziel et al., 2004, Gillespie et al., 2009, Pastukh et al., 2015), thus the DNA damage induced by inflammation might be reproducible because of damage to specific promoter/repressor regions of genes or transcription factors that are already activated by inflammation (Ruchko et al., 2009). Oxidative DNA damage, and specifically 8oxoG, has surfaced as a significant signaling mechanism for gene activation in promoter regions (Fleming et al., 2017).
In conclusion, our studies demonstrate that inflammation or exposure to inflammatory mediators elicits DNA damage in sensory neurons. By enhancing BER, we demonstrate that this DNA damage mediates the maintenance of neuronal hypersensitivity induced by inflammatory mediators. Although we have established that DNA damage is critical for the maintenance of changes in neuronal sensitivity, the specific pathways by which inflammatory mediators generate DNA damage, the specific types of DNA damage, and how DNA damage results in reproducible changes in neuronal sensitivity are still unknown and will be explored in future studies.
Acknowledgments
Financial support for this work was provided by the National Institutes of Health, [R21NS091667 (M.R. Vasko, M.R. Kelley and J.C. Fehrenbacher)] by the National Cancer Institute CA205166 (M.R. Kelley and J.C. Fehrenbacher) and CA138798 (M.R. Kelley). Additional financial support was provided by: a Research Support Funds Grant to J.C. Fehrenbacher from the IUPUI Office of the Vice Chancellor for Research, the Earl and Betty Herr Professor in Pediatric Oncology Research, the Jeff Gordon Children’s Foundation, and the Riley Children’s Foundation (M.R. Kelley).
Abbreviations
- AP
apurinic/apyrimidinic
- APE1/Ref-1
apurinic/apyrimidinic endonuclease 1/redox effector factor 1
- BER
base excision repair
- CCR2
CC chemokine receptor type 2
- CFA
complete Freund’s adjuvant
- CGRP
calcitonin gene-related peptide
- DRG
dorsal root ganglia
- HA
Hemagglutinin
- LPS
Lipopolysaccharides
- MCP-1/CCL2
monocyte chemoattractant protein-1/CC chemokine ligand 2
- MPL
methylpyrrolidone
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- TLR4
toll-like receptor 4
- TRPV1
Transient Receptor Potential Vanilloid 1
Footnotes
DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST: Mark R. Kelley has licensed APX3330 through Indiana University Research and Technology Corporation to Apexian Pharmaceuticals LLC. Apexian Pharmaceuticals had neither control nor oversight of the studies, interpretation, or presentation of the data in this manuscript.
References
- Andersson DA, Gentry C, Moss S, Bevan S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci. 2008;28:2485–2494. doi: 10.1523/JNEUROSCI.5369-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babior BM. Phagocytes and oxidative stress. The American journal of medicine. 2000;109:33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- Barzilai A, Biton S, Shiloh Y. The role of the DNA damage response in neuronal development, organization and maintenance. DNA repair. 2008;7:1010–1027. doi: 10.1016/j.dnarep.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Bauerova K, Bezek A. Role of reactive oxygen and nitrogen species in etiopathogenesis of rheumatoid arthritis. General physiology and biophysics. 1999:15–20. 18 Spec No. [PubMed] [Google Scholar]
- Blum E, Procacci P, Conte V, Hanani M. Systemic inflammation alters satellite glial cell function and structure. A possible contribution to pain. Neuroscience. 2014;274:209–217. doi: 10.1016/j.neuroscience.2014.05.029. [DOI] [PubMed] [Google Scholar]
- Brooks PJ. DNA repair in neural cells: basic science and clinical implications. Mutat Res. 2002;509:93–108. doi: 10.1016/s0027-5107(02)00222-1. [DOI] [PubMed] [Google Scholar]
- Calil IL, Zarpelon AC, Guerrero AT, Alves-Filho JC, Ferreira SH, Cunha FQ, Cunha TM, Verri WA., Jr Lipopolysaccharide induces inflammatory hyperalgesia triggering a TLR4/MyD88-dependent cytokine cascade in the mice paw. PLoS One. 2014;9:e90013. doi: 10.1371/journal.pone.0090013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuano A, De Corato A, Lisi L, Tringali G, Navarra P, Dello Russo C. Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol Pain. 2009;5:43. doi: 10.1186/1744-8069-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A. 1994;91:2752–2756. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Barber LA, Dymshitz J, Vasko MR. Peptidase inhibitors improve recovery of substance P and calcitonin gene-related peptide release from rat spinal cord slices. Peptides. 1996;17:31–37. doi: 10.1016/0196-9781(95)02091-8. [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107:660–664. doi: 10.1111/j.1476-5381.1992.tb14503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansereau MA, Gosselin RD, Pohl M, Pommier B, Mechighel P, Mauborgne A, Rostene W, Kitabgi P, Beaudet N, Sarret P, Melik-Parsadaniantz S. Spinal CCL2 pronociceptive action is no longer effective in CCR2 receptor antagonist-treated rats. J Neurochem. 2008;106:757–769. doi: 10.1111/j.1471-4159.2008.05429.x. [DOI] [PubMed] [Google Scholar]
- David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding R, Jiang H, Sun B, Wu X, Li W, Zhu S, Liao C, Zhong Z, Chen J. Advanced oxidation protein products sensitized the transient receptor potential vanilloid 1 via NADPH oxidase 1 and 4 to cause mechanical hyperalgesia. Redox Biol. 2016;10:1–11. doi: 10.1016/j.redox.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res. 2011;90:759–764. doi: 10.1177/0022034511400225. [DOI] [PubMed] [Google Scholar]
- Dublin P, Hanani M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav Immun. 2007;21:592–598. doi: 10.1016/j.bbi.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Duggett NA, Griffiths LA, McKenna OE, de Santis V, Yongsanguanchai N, Mokori EB, Flatters SJ. Oxidative stress in the development, maintenance and resolution of paclitaxel-induced painful neuropathy. Neuroscience. 2016;333:13–26. doi: 10.1016/j.neuroscience.2016.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH, Lorenzetti BB, Bristow AF, Poole S. Interleukin-1 beta as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature. 1988;334:698–700. doi: 10.1038/334698a0. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Lorenzetti BB, Poole S. Bradykinin initiates cytokine-mediated inflammatory hyperalgesia. Br J Pharmacol. 1993;110:1227–1231. doi: 10.1111/j.1476-5381.1993.tb13946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidanboylu M, Griffiths LA, Flatters SJ. Global inhibition of reactive oxygen species (ROS) inhibits paclitaxel-induced painful peripheral neuropathy. PLoS One. 2011;6:e25212. doi: 10.1371/journal.pone.0025212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007a;13:260–267. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- Fishel ML, Jiang Y, Rajeshkumar NV, Scandura G, Sinn AL, He Y, Shen C, Jones DR, Pollok KE, Ivan M, Maitra A, Kelley MR. Impact of APE1/Ref-1 redox inhibition on pancreatic tumor growth. Mol Cancer Ther. 2011;10:1698–1708. doi: 10.1158/1535-7163.MCT-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don’t divide what’s to repair? Mutat Res. 2007b;614:24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Fleming AM, Ding Y, Burrows CJ. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc Natl Acad Sci U S A. 2017;114:2604–2609. doi: 10.1073/pnas.1619809114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini P, Dogliotti E. Mechanisms of dealing with DNA damage in terminally differentiated cells. Mutat Res. 2010;685:38–44. doi: 10.1016/j.mrfmmm.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Georgiadis MM, Chen Q, Meng J, Guo C, Wireman R, Reed A, Vasko MR, Kelley MR. Small molecule activation of apurinic/apyrimidinic endonuclease 1 reduces DNA damage induced by cisplatin in cultured sensory neurons. DNA repair. 2016;41:32–41. doi: 10.1016/j.dnarep.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie MN, Pastukh V, Ruchko MV. Oxidative DNA modifications in hypoxic signaling. Ann N Y Acad Sci. 2009;1177:140–150. doi: 10.1111/j.1749-6632.2009.05036.x. [DOI] [PubMed] [Google Scholar]
- Hackel D, Pflucke D, Neumann A, Viebahn J, Mousa S, Wischmeyer E, Roewer N, Brack A, Rittner HL. The connection of monocytes and reactive oxygen species in pain. PLoS One. 2013;8:e63564. doi: 10.1371/journal.pone.0063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Vashishta A, Rempala G. Neurotoxic mechanisms of DNA damage: focus on transcriptional inhibition. J Neurochem. 2010;114:1537–1549. doi: 10.1111/j.1471-4159.2010.06859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Wang X. PKC and PKA, but not PKG mediate LPS-induced CGRP release and [Ca(2+)](i) elevation in DRG neurons of neonatal rats. J Neurosci Res. 2001;66:592–600. doi: 10.1002/jnr.1249. [DOI] [PubMed] [Google Scholar]
- Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, Nakagawa T, Sango K, Shirai Y, Yokoyama T, Kaneko S, Saito N, Yabe-Nishimura C. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci. 2008;28:9486–9494. doi: 10.1523/JNEUROSCI.1857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y, Takeda S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat Med. 2013;19:101–106. doi: 10.1038/nm.3019. [DOI] [PubMed] [Google Scholar]
- Jedinak A, Dudhgaonkar S, Kelley MR, Sliva D. Apurinic/Apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer Res. 2011;31:379–385. [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Lee KM, Park ES, Jeon YH, Cho HJ. Monocyte chemoattractant protein-1 immunoreactivity in sensory ganglia and hindpaw after adjuvant injection. Neuroreport. 2008;19:183–186. doi: 10.1097/WNR.0b013e3282f3c781. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Guo C, Fishel ML, Wang ZY, Vasko MR, Kelley MR. Role of APE1 in differentiated neuroblastoma SH-SY5Y cells in response to oxidative stress: use of APE1 small molecule inhibitors to delineate APE1 functions. DNA repair. 2009;8:1273–1282. doi: 10.1016/j.dnarep.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Zhou S, Sandusky GE, Kelley MR, Fishel ML. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Invest. 2010;28:885–895. doi: 10.3109/07357907.2010.512816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao DJ, Li AH, Chen JC, Luo RS, Chen YL, Lu JC, Wang HL. CC chemokine ligand 2 upregulates the current density and expression of TRPV1 channels and Nav1.8 sodium channels in dorsal root ganglion neurons. J Neuroinflammation. 2012;9:189. doi: 10.1186/1742-2094-9-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeble JE, Bodkin JV, Liang L, Wodarski R, Davies M, Fernandes ES, de Coelho CF, Russell F, Graepel R, Muscara MN, Malcangio M, Brain SD. Hydrogen peroxide is a novel mediator of inflammatory hyperalgesia, acting via transient receptor potential vanilloid 1-dependent and independent mechanisms. Pain. 2009;141:135–142. doi: 10.1016/j.pain.2008.10.025. [DOI] [PubMed] [Google Scholar]
- Kelley MR, Fehrenbacher JC. Challenges and opportunities identifying therapeutic targets for chemotherapy-induced peripheral neuropathy resulting from oxidative DNA damage. Neural Regen Res. 2017;12:72–74. doi: 10.4103/1673-5374.198986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012;5:36–53. doi: 10.2174/1874467211205010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS One. 2014;9:e106485. doi: 10.1371/journal.pone.0106485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MR, Wikel JH, Guo C, Pollok KE, Bailey BJ, Wireman R, Fishel ML, Vasko MR. Identification and Characterization of new chemical entities targeting Apurinic/Apyrimidinic Endonuclease 1 for the prevention of chemotherapy-induced peripheral neuropathy (CIPN) J Pharmacol Exp Ther. 2016;359:300–309. doi: 10.1124/jpet.116.235283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab MM. TEMPOL, a membrane-permeable radical scavenger, attenuates peroxynitrite- and superoxide anion-enhanced carrageenan-induced paw edema and hyperalgesia: a key role for superoxide anion. European journal of pharmacology. 2006;548:167–173. doi: 10.1016/j.ejphar.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Kim HK, Park SK, Zhou JL, Taglialatela G, Chung K, Coggeshall RE, Chung JM. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116–124. doi: 10.1016/j.pain.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Li Y, Adamek P, Zhang H, Tatsui CE, Rhines LD, Mrozkova P, Li Q, Kosturakis AK, Cassidy RM, Harrison DS, Cata JP, Sapire K, Zhang H, Kennamer-Chapman RM, Jawad AB, Ghetti A, Yan J, Palecek J, Dougherty PM. The Cancer Chemotherapeutic Paclitaxel Increases Human and Rodent Sensory Neuron Responses to TRPV1 by Activation of TLR4. J Neurosci. 2015;35:13487–13500. doi: 10.1523/JNEUROSCI.1956-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain. 2014;15:712–725. doi: 10.1016/j.jpain.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CS, Lee SH, Huang HS, Chen YS, Ma MC. H2O2 generated by NADPH oxidase 4 contributes to transient receptor potential vanilloid 1 channel-mediated mechanosensation in the rat kidney. Am J Physiol Renal Physiol. 2015;309:F369–376. doi: 10.1152/ajprenal.00462.2014. [DOI] [PubMed] [Google Scholar]
- Lukacs M, Haanes KA, Majlath Z, Tajti J, Vecsei L, Warfvinge K, Edvinsson L. Dural administration of inflammatory soup or Complete Freund’s Adjuvant induces activation and inflammatory response in the rat trigeminal ganglion. J Headache Pain. 2015;16:564. doi: 10.1186/s10194-015-0564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, 2nd, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura S, Shimizu K, Shinoda M, Ohara K, Ogiso B, Honda K, Katagiri A, Sessle BJ, Urata K, Iwata K. Mechanisms underlying ectopic persistent tooth-pulp pain following pulpal inflammation. PLoS One. 2013;8:e52840. doi: 10.1371/journal.pone.0052840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray CT. To die or not to die: DNA repair in neurons. Mutat Res. 2005;577:260–274. doi: 10.1016/j.mrfmmm.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Meseguer V, Alpizar YA, Luis E, Tajada S, Denlinger B, Fajardo O, Manenschijn JA, Fernandez-Pena C, Talavera A, Kichko T, Navia B, Sanchez A, Senaris R, Reeh P, Perez-Garcia MT, Lopez-Lopez JR, Voets T, Belmonte C, Talavera K, Viana F. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun. 2014;5:3125. doi: 10.1038/ncomms4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RE, Belmadani A, Ishihara S, Tran PB, Ren D, Miller RJ, Malfait AM. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through Toll-like receptor 4. Arthritis Rheumatol. 2015;67:2933–2943. doi: 10.1002/art.39291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, Malfait AM. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A. 2012;109:20602–20607. doi: 10.1073/pnas.1209294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Doubell TP, Leslie T, Woolf CJ. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature. 1996;384:360–364. doi: 10.1038/384360a0. [DOI] [PubMed] [Google Scholar]
- Nyland RL, Luo M, Kelley MR, Borch RF. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1) J Med Chem. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastukh V, Roberts JT, Clark DW, Bardwell GC, Patel M, Al-Mehdi AB, Borchert GM, Gillespie MN. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1367–1375. doi: 10.1152/ajplung.00236.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, Hong JS, Block ML. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia. 2005a;52:78–84. doi: 10.1002/glia.20225. [DOI] [PubMed] [Google Scholar]
- Qin X, Wan Y, Wang X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J Neurosci Res. 2005b;82:51–62. doi: 10.1002/jnr.20612. [DOI] [PubMed] [Google Scholar]
- Remans PH, van Oosterhout M, Smeets TJ, Sanders M, Frederiks WM, Reedquist KA, Tak PP, Breedveld FC, van Laar JM. Intracellular free radical production in synovial T lymphocytes from patients with rheumatoid arthritis. Arthritis and rheumatism. 2005;52:2003–2009. doi: 10.1002/art.21111. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–845. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rozanski GM, Li Q, Stanley EF. Transglial transmission at the dorsal root ganglion sandwich synapse: glial cell to postsynaptic neuron communication. Eur J Neurosci. 2013;37:1221–1228. doi: 10.1111/ejn.12132. [DOI] [PubMed] [Google Scholar]
- Ruchko MV, Gorodnya OM, Pastukh VM, Swiger BM, Middleton NS, Wilson GL, Gillespie MN. Hypoxia-induced oxidative base modifications in the VEGF hypoxia-response element are associated with transcriptionally active nucleosomes. Free radical biology & medicine. 2009;46:352–359. doi: 10.1016/j.freeradbiomed.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ. Contribution of interleukin-1 beta to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. Br J Pharmacol. 1995;115:1265–1275. doi: 10.1111/j.1476-5381.1995.tb15035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free radical biology & medicine. 2011;51:951–966. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Hosokawa H, Matsumura K, Kobayashi S. Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci. 2008;27:1131–1142. doi: 10.1111/j.1460-9568.2008.06093.x. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Herz A. Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav. 1988;31:445–451. doi: 10.1016/0091-3057(88)90372-3. [DOI] [PubMed] [Google Scholar]
- Takeda M, Takahashi M, Matsumoto S. Contribution of activated interleukin receptors in trigeminal ganglion neurons to hyperalgesia via satellite glial interleukin-1beta paracrine mechanism. Brain Behav Immun. 2008;22:1016–1023. doi: 10.1016/j.bbi.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Tse KH, Chow KB, Leung WK, Wong YH, Wise H. Lipopolysaccharide differentially modulates expression of cytokines and cyclooxygenases in dorsal root ganglion cells via Toll-like receptor-4 dependent pathways. Neuroscience. 2014;267:241–251. doi: 10.1016/j.neuroscience.2014.02.041. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA repair. 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Guo C, Thompson EL, Kelley MR. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA repair. 2011;10:942–952. doi: 10.1016/j.dnarep.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa G, Ceruti S, Zanardelli M, Magni G, Jasmin L, Ohara PT, Abbracchio MP. Temporomandibular joint inflammation activates glial and immune cells in both the trigeminal ganglia and in the spinal trigeminal nucleus. Mol Pain. 2010;6:89. doi: 10.1186/1744-8069-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309:869–878. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, Lamotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KI, Higgs GA. Eicosanoids and inflammation. J Pathol. 1988;156:101–110. doi: 10.1002/path.1711560204. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, Dougherty PM. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2013;14:1031–1044. doi: 10.1016/j.jpain.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Karki R, Igwe OJ. Toll-like receptor 4 signaling: A common pathway for interactions between prooxidants and extracellular disulfide high mobility group box 1 (HMGB1) protein-coupled activation. Biochem Pharmacol. 2015;98:132–143. doi: 10.1016/j.bcp.2015.08.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Pei GX, Cong R, Zhang H, Zang CW, Tian T. PKC-NF-kappaB are involved in CCL2-induced Nav1.8 expression and channel function in dorsal root ganglion neurons. Biosci Rep. 2014;34 doi: 10.1042/BSR20140005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziel KA, Campbell CC, Wilson GL, Gillespie MN. Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. Faseb J. 2004;18:986–988. doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]