

Abstract
Peripheral neuropathy (PN) is a debilitating and dose-limiting side effect of treatment with the chemotherapeutic agent, paclitaxel. Understanding the effects of paclitaxel on sensory neuronal function and the signaling pathways which mediate these paclitaxel-induced changes in function are critical for the development of therapies to prevent or alleviate the PN. The effects of long-term administration of paclitaxel on the function of sensory neurons grown in culture, using the release of the neuropeptide calcitonin gene-related peptide (CGRP) as an endpoint of sensory neuronal function, were examined. Dorsal root ganglion cultures were treated with low (10 nM) and high (300 nM) concentrations of paclitaxel for 1, 3, or 5 days. Following paclitaxel treatment, the release of CGRP was determined using capsaicin, a TRPV1 agonist; allyl isothiocyanate (AITC), a TRPA1 agonist; or high extracellular potassium. The the effects of paclitaxel on the release of CGRP were stimulant-, concentration-, and time-dependent. When neurons were stimulated with capsaicin or AITC, a low concentration of paclitaxel (10 nM) augmented transmitter release, whereas a high concentration (300 nM) reduced transmitter release in a time-dependent manner; however, when high extracellular potassium was used as the evoking stimulus, all concentrations of paclitaxel augmented CGRP release from sensory neurons. These results suggest that paclitaxel alters the function of sensory neurons in vitro, and suggest that the mechanisms by which paclitaxel alters neuronal function may include functional changes in TRP channel activity. The described in vitro model will facilitate future studies to identify the signaling pathways by which paclitaxel alters neuronal sensitivity.
Keywords: chemotherapy-induced peripheral neuropathy, paclitaxel, neurotoxicity, peripheral sensory neuron, dorsal root ganglion culture, neuropeptide release, CGRP, TRPV1, TRPA1
Introduction
Paclitaxel is a chemotherapeutic microtubule targeting agent that is commonly used in the treatment of breast, ovarian, and lung cancers (Rowinsky et al., 1991). Although paclitaxel is an effective antineoplastic, its use is often limited by severe side effects including peripheral neuropathy (PN). Paclitaxel-induced PN is characterized by burning pain, numbness, tingling in the hands and feet, and loss of proprioception (Dougherty et al., 2004; Forsyth et al., 1997; Lipton et al., 1989; Wiernik et al., 1987). Although these symptoms resolve in some patients following cessation of treatment, they may be irreversible and can persist as chronic neuropathic pain (Connelly et al., 1996). There are no known treatment options which specifically prevent or reverse the neuropathy, and most patients resort to scaling back the bolus of paclitaxel per treatment or to discontinuing administration of paclitaxel entirely to minimize symptoms of the neuropathy (Capri et al., 1996). Although it is clear that chronic administration of paclitaxel alters the function of sensory neurons, the questions remain whether the drug augments or reduces neuronal sensitivity and what are the cellular mechanisms by which the drug affects neuronal function. Indeed, it is critical to understand how paclitaxel alters the function of sensory neurons and whether it differentially affects subpopulations of sensory neurons before therapies can be developed to alleviate paclitaxel-induced PN.
There are several animal models to examine the in vivo effects of paclitaxel on neuronal activity. A low-dose model, whereby paclitaxel (cumulative doses of 4, 8, or 16 mg/kg) is injected systemically over the course of 7 days, results in mechanical hyperalgesia and allodynia, as well as cold allodynia, without causing overt nerve damage, alterations in nerve conduction velocity, or changes in neuronal survival (Flatters and Bennett, 2006; Matsumoto et al., 2006; Polomano et al., 2001). These data suggest that the sensitivity of sensory nerve fibers (unmyelinated C-fibers and lightly myelinated Aδ-fibers) is enhanced after paclitaxel treatment. In contrast, using vasodilatation induced by activation of peripheral endings of sensory neurons as an endpoint to indirectly measure the activity of small diameter sensory nerve fibers innervating vascular smooth muscle in the dermis, we showed that the same low-dose paclitaxel treatment that produces hyperalgesia reduced vasodilatation induced by the activation of sensory neurons by capsaicin (Gracias et al., 2011). In an alternative animal model, paclitaxel (cumulative doses of 80–135 mg/kg) is injected systemically over the course of 5–9 weeks. This model produces impairments in pain-like behaviors using the tail-flick test and thermal withdrawal latencies as endpoints, decreases in coordination as measured by the rota-rod test, and decreases in nerve conduction velocity in the tail nerve with variable axonal damage, suggesting a reduced sensitivity of sensory neurons (Authier et al., 2000; Cavaletti et al., 1997). These data suggest that, dependent upon the endpoint measured and the dosing paradigm administered, both increases and decreases in neuronal sensitivity occur; however, investigators still do not have a clear understanding of what population of neurons is affected and the mechanisms by which these changes in sensitivity develop.
Treating sensory neuron cultures or explants with paclitaxel induces shortening of neurite lengths (Melli et al., 2008; Scuteri et al., 2006; Yang et al., 2009); however, mechanistic in vitro studies to examine the effects of paclitaxel on the sensitivity of subpopulations of sensory neurons are limited. Several investigators have isolated the lumbar dorsal root ganglia from paclitaxel-treated animals to examine the effects of the chemotherapeutic on electrical excitability and intracellular calcium signaling within the soma of the lumbar neurons, which predominantly innervate the hindpaws (Kawakami et al., 2012; Mo et al., 2012; Zhang et al., 2013). While this experimental paradigm works well for measuring excitability and intracellular calcium signaling in the soma, it does not permit investigation of the effects of paclitaxel on an integrated neuronal response, such as the stimulated release of neurotransmitters from the neurites and soma of sensory neurons. Therefore, we examined the effects of paclitaxel treatment on the function of sensory neurons grown in culture. Previous studies have demonstrated a direct excitatory effect of paclitaxel on neuropeptide release (Materazzi et al., 2012; Miyano et al., 2009); however, most patients do not feel pain during paclitaxel infusion. Since the clinical onset of symptoms occurs 3–6 weeks following the first dose of paclitaxel (Forsyth et al., 1997), we examined the effects of paclitaxel at timepoints distant from the start of drug exposure: 1, 3 and 5 days following treatment with paclitaxel.
We determined the effects of paclitaxel on the basal and evoked release of the nociceptive neuropeptide, calcitonin gene-related peptide (CGRP), from sensory neuron cultures as an indicator of sensory neuronal function (Vasko et al., 1994). We evoked the release of CGRP from the sensory neurons with 3 different stimulatory agents in order to gain mechanistic insight into the paclitaxel-induced changes within our cultures. We used a TRPV1 agonist, capsaicin, since we previously showed that capsaicin-induced bloodflow was altered by paclitaxel treatment (Gracias et al., 2011) and because systemic administration of a TRPV1 antagonist has been shown to attenuate thermal hyperalgesia following paclitaxel treatment (Chen et al., 2011). We also used a TRPA1 agonist, allyl isothiocyanate (AITC), since systemic administration of a TRPA1 antagonist reduces paclitaxel-induced mechanical and cold allodynia (Chen et al., 2011; Materazzi et al., 2012). Finally, we utilized a general depolarizing stimulus, high extracellular potassium, to examine the effects of paclitaxel treatment on the sensitivity of neurons independent of TRP channel activation. We found that paclitaxel alters the sensitivity of isolated sensory neurons in a concentration- and time-dependent manner. Paclitaxel can enhance or reduce capsaicin- and AITC-evoked release of CGRP depending on the magnitude and duration of exposure to the chemotherapeutic. In contrast, paclitaxel augments CGRP release evoked by high extracellular potassium, regardless of the concentration or duration of paclitaxel exposure. Our findings support a role for small diameter sensory neurons in paclitaxel-induced neurotoxicity and suggest that the mechanisms by which paclitaxel alters neuronal function may include functional changes in TRP channel activity. Furthermore, the described in vitro model will facilitate future studies to identify the signaling pathways by which paclitaxel alters neuronal sensitivity.
Methods
Animals
All animal experiments were carried out in accordance with the Animal Care and Use Committee at Indiana University School of Medicine, Indianapolis, IN and with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments were performed on primary cultures of sensory neurons derived from the dorsal root ganglia (DRG) of adult male Sprague Dawley rats (150–250 g; Harlan Laboratories, Indianapolis, IN). Prior to sacrifice, animals were housed in group cages in a light controlled room. Food and water were available ad libitum.
Isolation of primary sensory neuron cultures
Dorsal root ganglia (DRG) were harvested and cultured as described previously with slight modifications (Hingtgen and Vasko, 1994). Rats were asphyxiated with CO2 and then decapitated. DRG were harvested from the vertebral column, and the sensory neurons were dissociated by mechanical agitation after incubation for 1 hour in 0.125% collagenase at 37°C. Approximately 30,000 cells were plated into each well of a 12-well plate previously coated with poly-d-lysine and laminin. Cells were grown in F-12 growth media supplemented with 10% heat-inactivated horse serum, 2 mM glutamine, 50 μg/ml penicillin and streptomycin, 50 μM 5-fluoro-2-deoxyuridine, and 150 μM uridine in the presence of 30 ng/ml nerve growth factor (NGF). This concentration of NGF has previously been shown to enhance the expression of CGRP without compromising the ability of neurons to be sensitized by inflammatory mediators (Park et al., 2010). Cultures were maintained in an atmosphere of 3% CO2 at 37°C and media was changed every other day. For paclitaxel treatments, a stock of 10 mM paclitaxel was prepared in 1-methyl-2-pyrrolidinone (MPL) and stored at −20°C. This stock was further diluted in media to appropriate concentrations of paclitaxel. Cultures were treated with paclitaxel in media starting on day 7, 9, or 11 in culture, and experiments were performed on day 12 in culture.
Calcitonin gene-related peptide release
Release experiments were performed as described previously (Hingtgen and Vasko, 1994; Vasko et al., 1994). The wells were rinsed one time with HEPES buffer containing 25 mM HEPES, 135 mM NaCl, 3.5 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 3.3 mM D-glucose, and 0.1% bovine serum albumin, pH 7.4. The cells were maintained at 37°C for 4, 10 min incubations. During the first and second 10 min intervals, cells were incubated with HEPES buffer to establish resting basal release of immunoreactive CGRP (iCGRP), which we will refer to as CGRP.
During the third interval, cells were incubated with HEPES containing 30 nM capsaicin, HEPES containing 30 μM AITC, or HEPES containing 50 mM KCl (substituted for equimolar NaCl) in order to stimulate CGRP release. The fourth incubation was in HEPES to reestablish resting basal release. Supernatants were collected after every interval, and CGRP was measured using radioimmunoassay as previously described (Vasko et al., 1994). At the completion of the release experiment, cells were incubated for 20 min in 0.1 N HCl, and the supernatant was collected to determine total CGRP content.
Viability assay
The viability of sensory neurons following exposure to paclitaxel was assessed by double staining with propidium iodide (PI) and annexin V. Sensory neuronal cultures were exposed to either 300 nM or 1 μM paclitaxel for 5 days. As a positive control, untreated neurons were exposed to 300 μM H2O2 for 1 hour at 37°C. At the end of the 1-hour incubation, the neurons were returned to F-12 media. Approximately 24 hours later, all neurons were incubated with 0.4 ml of the staining solution containing 25μl annexin V-FITC in 10 ml HEPES buffer and 6 μM PI. After washing off the staining solution, cells were visualized. The excitation of PI/annexin V was 530/485 nm and the emitted light was monitored at 645/530 nm. After staining with PI and annexin V, nuclei appear red while cells that are in the initial stages of apoptosis display a green colored ring around the cells. Dead cells display more diffuse staining with annexin V. Neurons in five random fields were counted and scored as either viable (unstained) or non-viable (red or green). The data are expressed as mean ± standard error of the mean (SEM) % of the total number of neurons counted.
Reagents
All materials unless stated otherwise were purchased from Sigma-Aldrich (St. Louis, MO). F-12 media, horse serum, antibiotics, and annexin V-FITC were purchased from Invitrogen (Carlsbad, CA). The AITC was purchased from Fisher Scientific (Pittsburgh, PA), and nerve growth factor was purchased from Harlan Laboratories (Indianapolis, IN).
Statistical Analysis
Data were analyzed by one-way analysis of variance (ANOVA) or two-way ANOVA as indicated, and post-hoc analyses were performed using the Bonferroni’s or Dunnett’s test, as indicated. Statistical calculations were performed with the GraphPad Prism version 6.02 statistical package (GraphPad Software, La Jolla, California USA). Data are presented as mean ± SEM, and differences are considered significant if p < 0.05.
Results
The effects of paclitaxel on basal and stimulated release of CGRP
Our previous results suggest that systemic administration of paclitaxel reduces the sensitivity of small diameter sensory neurons (Gracias et al., 2011), whereas other findings suggest an increase in sensitivity (Flatters and Bennett, 2006; Matsumoto et al., 2006; Polomano et al., 2001). To determine the direct effects of long-term paclitaxel exposure on sensory neurons, we cultured sensory neurons for 5 days in the presence of various concentrations of paclitaxel. We then measured the basal release of CGRP and the release of CGRP upon stimulation with 30 nM capsaicin. Five-day treatment with vehicle (.003% MPL), 10 nM or 300 nM paclitaxel did not alter the resting basal release of CGRP (first B column of each treatment group, Figure 1A). Five-day treatment with 10 nM paclitaxel did enhance the stimulated release of CGRP. Capsaicin stimulated the release of CGRP from a basal level of 29 ± 2 fmol/well/10min to 274 ± 23 fmol/well/10min in vehicle-treated neurons (Figure 1A). The capsaicin-evoked release was augmented to 329 ± 24 fmol/well/10min in neurons treated with 10 nM paclitaxel (C columns, Figure 1A). In contrast, treatment with 300 nM paclitaxel significantly decreased capsaicin-evoked release to 150 ± 9 fmol/well/10min. The changes in release of CGRP were not secondary to an altered content of CGRP in the neurons, as the total content of CGRP was similar in cultures treated with vehicle (1207 ± 80 fmol/well), 10 nM paclitaxel (1359 ± 97), or 300 nM paclitaxel (1165 ± 59; Figure 1B). We also examined other concentrations of paclitaxel: exposure of sensory neurons in culture to 30 nM paclitaxel for 5 days augmented capsaicin-evoked CGRP release by 20%, whereas exposure to 100 nm or 1 μM reduced release by 45% and 75%, respectively (data not shown).
Figure 1.
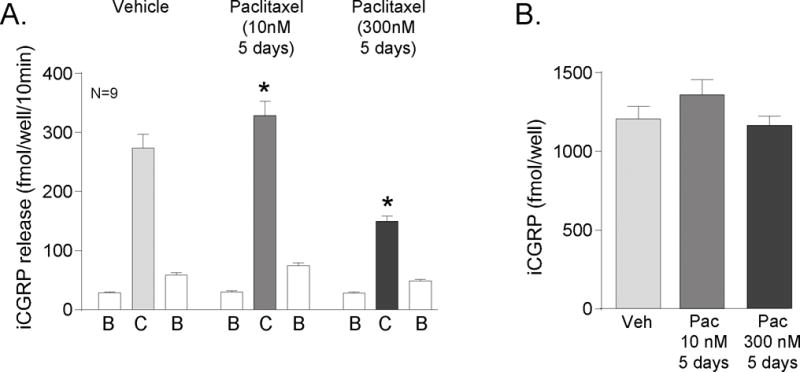
Paclitaxel alters capsaicin-evoked release of CGRP from sensory neurons in culture. A) Each column represents the mean ± SEM of CGRP released in fmol/well/10 min from wells treated with 10 nM or 300 nM paclitaxel for 5 days. The first open column of each group represents basal release (B), the shaded column represents release in the presence of 30 nM capsaicin (C), and the second open column of each group represents the recovery of basal release following stimulation (B). An asterisk indicates a significant difference in capsaicin-evoked release compared to release from the vehicle-treated neurons (p<0.05, N=9) using a two-way ANOVA with Bonferroni’s post-hoc test. B) Each column is the mean ± SEM of total CGRP content in fmol/well (N=9) from vehicle treated cultures (Veh) or paclitaxel treated cultures (Pac) as indicated.
Capsaicin stimulates the release of neuropeptides from small diameter sensory neurons expressing the TRPV1 receptor (Holzer, 1988) by increasing the permeability of the channel and inducing an inward calcium current. To determine if paclitaxel alters the function of the TRPA1 channel, we stimulated the sensory neurons with 30 μM AITC, a TRPA1 agonist, after treatment with 10 or 300 nM paclitaxel for 5 days. AITC-evoked CGRP release was significantly greater from neurons treated with 10 nM paclitaxel as compared to vehicle-treated neurons: the stimulated release was increased from 347 ± 26 fmol/well/10min in vehicle-treated neurons to 472 ± 29 fmol/well/10min in neurons treated with 10 nM paclitaxel (Figure 2A). Treatment for 5 days with 300 nM paclitaxel decreased AITC-evoked release to 117 ± 16 fmol/well/10min (Figure 2A). Total CGRP content in the neurons was not changed by paclitaxel treatment. Content levels were 1107 ± 77 fmol/well, 1343 ± 93 fmol/well, and 895 ± 85 fmol/well in the vehicle, 10 nM, and 300 nM-treated groups, respectively (Figure 2B).
Figure 2.
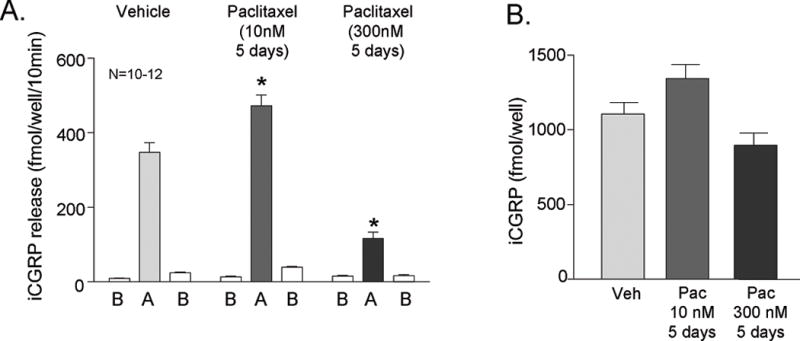
Paclitaxel alters AITC-evoked release of CGRP from sensory neurons in culture. A) Each column represents the mean ± SEM of CGRP released in fmol/well/10 min from wells treated with 10 nM or 300 nM paclitaxel for 5 days. The first open column of each group represents basal release (B), the shaded column represents release in the presence of 30 nM AITC (A), and the second open column of each group represents the recovery of basal release following stimulation (B). An asterisk indicates a significant difference in AITC-evoked release compared to release from the vehicle-treated neurons (p<0.05, N=10–12) using a two-way ANOVA with Bonferroni’s post-hoc test. B) Each column is the mean ± SEM of total CGRP content in fmol/well (N=10–12) from vehicle treated cultures (Veh) or paclitaxel treated cultures (Pac) as indicated.
To examine whether the effects of paclitaxel were dependent on altered sensitivity of TRP channels, we stimulated neuronal cultures treated with vehicle, 10 nM paclitaxel or 300 nM paclitaxel for 5 days with a general depolarizing stimulus, 50 mM potassium. In vehicle-treated cultures, high extracellular potassium increased the release of CGRP from basal levels of 25 ± 2 fmol/well/10min to 291 ± 12 fmol/well/10min (open vs shaded HK columns, Figure 3A). Exposing cultures to 10 nM paclitaxel for 5 days resulted in an increase in the potassium-stimulated release of CGRP to 353 ± 16 fmol/well/10min without altering resting basal release in a manner analogous to that observed when release was stimulated by the TRP channel agonists (Figure 3A). In contrast to our results with capsaicin- and AITC-evoked release, exposing cultures to 300 nM paclitaxel for 5 days also augmented the CGRP release evoked by potassium to 346 ± 16 fmol/well/10min (Figure 3A). Differences in potassium-evoked release were not due to an alteration of neuropeptide content, as total contents for the vehicle, 10 nM, and 300 nM treated cultures were 1219 ± 78 fmol/well, 1277 ± 87 fmol/well, and 1190 ± 63 fmol/well, respectively (Figure 3B).
Figure 3.
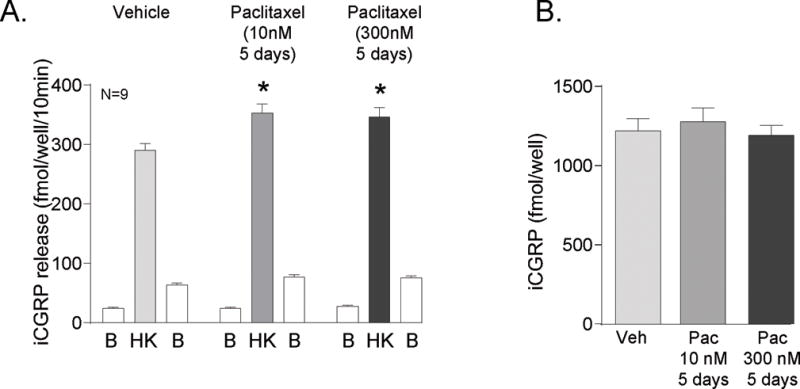
Paclitaxel augments potassium-evoked release of CGRP from sensory neurons in culture. A) Each column represents the mean ± SEM of CGRP released in fmol/well/10 min from wells treated with 10 nM or 300 nM paclitaxel for 5 days. The first open column of each group represents basal release (B), the shaded column represents release in the presence of 50 mM KCl (HK), and the second open column of each group represents the recovery of basal release following stimulation (B). An asterisk indicates a significant difference in HK-evoked release compared to release from the vehicle-treated neurons (p<0.05, N=9) using a two-way ANOVA with Bonferroni’s post-hoc test. B) Each column is the mean ± SEM of total CGRP content in fmol/well (N=9) from vehicle treated cultures (Veh) or paclitaxel treated cultures (Pac) as indicated.
The effects of paclitaxel on neuronal viability
Previous reports demonstrate that paclitaxel treatment can cause significant neuronal death in DRG cultures derived from embryonic day 16 (E16) rats and that this death is mediated through necrosis (Scuteri et al., 2006). To determine whether paclitaxel’s alteration in transmitter release could be secondary to a change in sensory neuronal viability in adult DRG cultures, we stained neuronal cultures with propidium iodide (PI) to differentiate between live and dead cells and with annexin V to determine if cells were in the initial stages of apoptosis. In cultures treated with vehicle for 5 days, the percentage of neurons that stained positive for annexin V or PI was 8 ± 2 % and 8 ± 2% of the total number of neurons counted, respectively (Figure 4). In cultures exposed to 300 nM paclitaxel for 5 days, the percentage of neurons that stained positive for annexin V or PI were 10 ± 2% and 10 ± 3 % of the total number of neurons counted. Five days of exposure to 1 μM paclitaxel also did not significantly affect cell viability (Figure 4). As a positive control for decreases in cell viability, neuronal cultures were exposed to 300 μM H2O2 for 1 hour at 37°C, which has previously been shown to produce significant loss in cell viability (Vasko et al., 2005). As seen in Figure 4, H2O2 treatment significantly increased the percentage of annexin V and PI positive cells to 63 ± 14% and 69 ± 18% of the total number of neurons, respectively.
Figure 4.
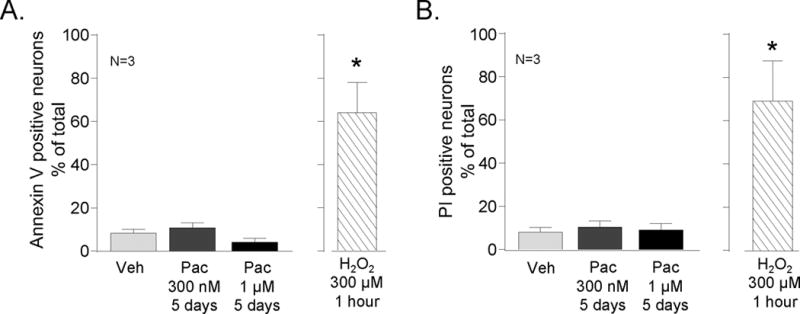
Paclitaxel does not decrease the survival of sensory neurons in culture. Sensory neuronal cultures were treated with vehicle (open columns, Veh) or with 10 nM or 300 nM paclitaxel for 5 days (Pac; shaded columns) and then cell viability was measured. As a positive control, vehicle-treated cultures were exposed to H2O2 (300 μM) for 1 hour 24 hours prior to analysis of cell survival (shaded columns). The number of annexin V positive cells (A) and propidium iodine (PI) positive cells (B) were counted in a minimum of 5 fields from 3 different harvests and normalized to the total number of neurons in the field. Each column is the mean ± SEM of % of positively stained neurons. An asterisk indicates a significant difference from untreated controls (p<0.05, N=3) using a one way-ANOVA and Bonferroni’s post-hoc test.
The time-course of paclitaxel-induced changes in basal and stimulated CGRP release
To investigate if paclitaxel-induced alterations in capsaicin-evoked CGRP release were dependent on the duration of paclitaxel exposure, we treated sensory neuronal cultures with 10 nM or 300 nM paclitaxel for 1, 3, or 5 days or with vehicle for 5 days. We then measured the basal release of CGRP and release upon stimulation with 30 nM capsaicin. The treatment with vehicle (.003% MPL), 10 nM or 300 nM paclitaxel did not alter the resting basal release of CGRP (B columns, Figures 5A and 6A) over the course of treatment. The capsaicin-evoked CGRP release from neurons treated with 10 nM paclitaxel for 1 day did not differ from the vehicle controls; the release was 339 ± 11 and 353 ± 10 fmol/well/10min in the vehicle and treated neurons, respectively (C columns, Figure 5A). However, the capsaicin-evoked CGRP release from the 3 day and 5 day 10 nM paclitaxel-treated wells significantly increased to 380 ± 14 and 395 ± 17 fmol/well/10min, respectively. The increase in capsaicin-evoked release of iCGRP was not secondary to an altered content of CGRP in the neurons since treatments with 10 nM paclitaxel did not alter the content of the peptide in the cultures (Figure 5B).
Figure 5.
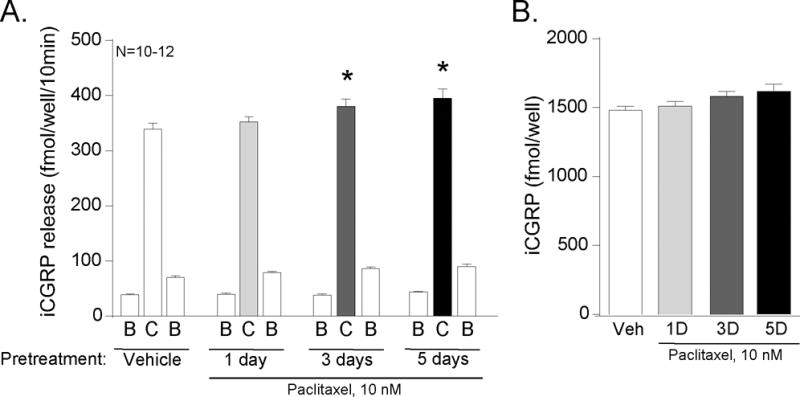
Paclitaxel (10 nM) increases the release of CGRP from sensory neurons in culture in a time-dependent manner. A) Each column represents the mean ± SEM of CGRP released in fmol/well/10 min from wells treated with vehicle or 10 nM paclitaxel for 1, 3, or 5 days as indicated. The first open column of each group represents basal release (B), the shaded column represents release in the presence of 30 nM capsaicin (C), and the second open column of each group represents the recovery of basal release following stimulation (B). An asterisk indicates a significant difference in capsaicin-evoked release compared to release from the vehicle-treated neurons (p<0.05, N=10–12) using a two-way ANOVA with Bonferroni’s post-hoc test. B) Each column is the mean ± SEM of total CGRP content in fmol/well (N=10–12) from cultures treated with vehicle (Veh) or paclitaxel (Pac) for the duration of time as indicated.
Figure 6.
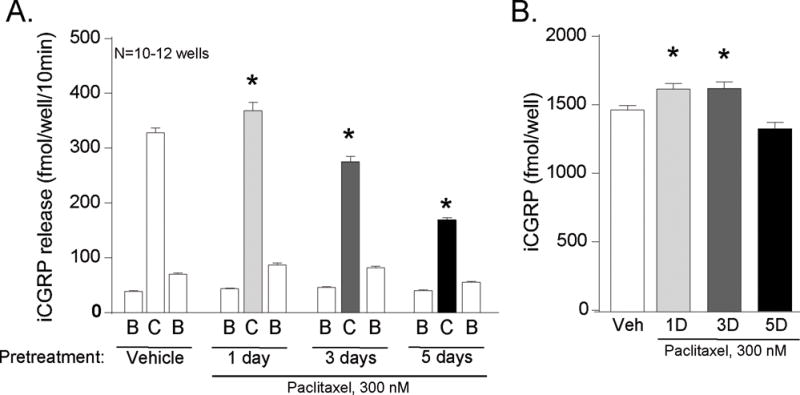
Time-dependent effects of 300 nM paclitaxel on the release of CGRP from sensory neurons in culture. A) Each column represents the mean ± SEM of CGRP released in fmol/well/10 min from wells treated with vehicle or 300 nM paclitaxel for 1, 3, or 5 days as indicated. The first open column of each group represents basal release (B), the shaded column represents release in the presence of 30 nM capsaicin (C), and the second open column of each group represents the recovery of basal release following stimulation (B). An asterisk indicates a significant difference in capsaicin-evoked release compared to release from the vehicle-treated neurons (p<0.05, N=10–12) using a two-way ANOVA with Bonferroni’s post-hoc test. B) Each column is the mean ± SEM of total CGRP content in fmol/well (N=10–12) from cultures treated with vehicle (Veh) or paclitaxel (Pac) for the duration of time as indicated. An asterisk indicates a significant difference in the total content of CGRP compared to the vehicle-treated cultures (p<0.05, N=10–12) using a one way-ANOVA and Bonferroni’s post-hoc test.
We also observed a small, but significant augmentation in the capsaicin-evoked release of CGRP following 1 day of exposure of neuronal cultures to 300 nM paclitaxel. The release of CGRP in vehicle- and paclitaxel-treated neurons was 328 ± 9 and 369 ± 15 fmol/well/10min, respectively (C columns, Figure 6A). This increase in CGRP release, however, was likely because of an increase in the content of CGRP in the cultures, since 300 nM paclitaxel increased the peptide content levels from 1470 ± 30 to 1623 ± 40 (Figure 6B). Calculating the capsaicin-stimulated release as the % of total peptide content, release from vehicle-treated neurons was 22%, and it was 23% from cultures treated with 300 nM paclitaxel for 1 day. Following 3 or 5 days of treatment with paclitaxel (300 nM), the capsaicin-evoked release was significantly reduced to 275 ± 10 and 169 ± 5 fmol/well/10min, respectively. The total content of CGRP in the neurons following 3 and 5 days of treatment was 1626 ± 49 and 1332 ± 47 fmol/well (Figure 6B). Thus the capsaicin-stimulated release at 3 and 5 days of paclitaxel exposure represents 17% and 13% of the total content of CGRP, demonstrating that the reduced release was not secondary to changes in peptide levels.
Discussion
Research examining the effects of paclitaxel on sensory neuronal function has focused largely on in vivo studies using a well-characterized animal model of paclitaxel-induced neuropathic pain, in which integrated nociceptive withdrawal behaviors are utilized as indices of neuropathy (Bennett et al., 2011; Cata et al., 2008; Deng et al., 2012; Pascual et al., 2005; Polomano et al., 2001). Using this approach, investigators have examined the efficacy of antidepressants, gabapentin, COX inhibitors, antioxidants, mitochondrial protective agents, and immune suppressing drugs to reverse paclitaxel-induced mechanical hypersensitivity and cold allodynia (Boyette-Davis et al., 2011; Flatters et al., 2006; Ito et al., 2012; Jin et al., 2008; Kim et al., 2010; Xiao et al., 2007). Although these putative therapeutics are moderately successful in the animal model, they fall short in translation into the clinical setting (Hershman et al., 2012; Kautio et al., 2009; Kottschade et al., 2011; Rao et al., 2008; Rao et al., 2007; Smith et al., 2013). These strategies largely inhibit the basal and pathological function of primary afferent neurons, thus the fundamental questions of how paclitaxel augments or reduces sensory neuronal function and whether it has dissimilar effects on the different subpopulations of sensory neurons have remained unanswered. Although Dougherty and co-workers suggest that a predominant clinical effect of the taxanes is on discrimination of sensations conducted by large diameter fibers (Dougherty et al., 2004), a role for the small diameter sensory neurons has been intimated based upon nociceptive endpoints (Campana et al., 1998; Polomano et al., 2001). The current findings demonstrate that paclitaxel can either augment or reduce TRP channel-evoked transmitter release from peptidergic small diameter sensory neurons dependent upon the concentration of the drug and the duration of exposure. A relatively low concentration of paclitaxel augments transmitter release, whereas a high concentration can reduce transmitter release. Our results are analogous to animal studies using chronic systemic injection of paclitaxel, which suggest that both sensitizing and desensitizing mechanisms may contribute to the clinical symptoms of neurotoxicity, dependent on the dosing and on the experimental endpoints measured.
Paclitaxel-induced increases in high extracellular potassium-stimulated release suggest that a gain of function is present within the small diameter peptidergic neurons and is independent of both TRPV1 and TRPA1. This finding also argues against a possible microtubule-induced defect in the synthesis and/or release of CGRP, or paclitaxel-induced neuronal death as potential mechanisms by which paclitaxel induces desensitization. Our findings, that paclitaxel treatment of sensory neuron cultures derived from adult rats does not cause neuronal death, contrast with findings from Scuteri and colleagues (2006), who demonstrate 50–75% mortality of neurons within cultures derived from E16 rats following just 48 hours of treatment with paclitaxel. Differences in paclitaxel-induced neurotoxicity between sensory neurons derived from embryonic and adult DRG are not surprising, as injury to axons of embryonic sensory neurons induces neuronal death quicker than injury to axons of adult sensory neurons following axotomy (Koliatsos and Price, 1996; Whiteside et al., 1998). The data observed from cultures of adult sensory neurons more closely mimics the effects of paclitaxel in an animal model of paclitaxel-induced neuropathy, where neuronal death is seldom observed (Polomano et al., 2001).
The mechanisms for paclitaxel-induced increases in transmitter release remain unknown, but the observed gain in function is supported by in vivo studies showing an augmentation in thermal and mechanical sensitivity. The major proposed mechanism of action for the oncolytic effects of paclitaxel, to bind to and alter microtubule dynamics (Manfredi et al., 1982; Schiff et al., 1979), could result in subsequent changes in axonal trafficking (LaPointe et al., 2013), localization of ion channels and receptors on the plasma membrane (Gambino et al., 2007), and altered mitochondrial function (Kidd et al., 2002), all of which could contribute to enhanced neuronal sensitivity. Alternate hypotheses are that paclitaxel binds to other targets; as both neuronal calcium sensor-1 and Bcl-2 bind to paclitaxel and subsequently disrupt neuronal calcium homeostasis and/or the function of mitochondria, respectively (Boehmerle et al., 2006; Ferlini et al., 2009; Voehringer, 1999). Recent work from Hara and co-workers indicates that systemic administration of paclitaxel in the rat resulted in an increase in TRPV1 mRNA and protein in the dorsal root ganglia (2013). It is possible that an increase in TRPV1 expression may underlie the augmentation of release in our neuronal cultures; however, increases in TRPV1 expression would not explain paclitaxel-induced sensitization of CGRP release evoked with high extracellular potassium or AITC. The fact that we see a reduction in capsaicin-evoked release suggests either a decrease in expression or a functional inhibition of TRPV1 after exposure to high concentrations of paclitaxel. There is a vast literature on desensitization of the TRPV1 channels, and our future studies will examine whether desensitization mechanisms, established in the literature, are involved in the loss of function of TRPV1 induced by paclitaxel treatment.
The observation that higher concentrations of paclitaxel inhibit TRPV1-mediated activation of sensory neurons corroborates our previous in vivo work which showed a loss of capsaicin-evoked bloodflow in the dermal layers of the rat hindpaw skin after chronic administration of paclitaxel (Gracias et al., 2011). Furthermore, the decrease in capsaicin-evoked release could be a mechanism to explain why thermal hyponociception is observed in some studies following high-dose administration of paclitaxel, while thermal hyperalgesia is observed in studies following low-dose treatment of the animals. For example, injecting 16 mg/kg of paclitaxel per week for 5 weeks produced nerve degeneration with concomitant mechanical hyperalgesia, but thermal hypoalgesia (Authier et al., 2000). In a similar manner, administering 1.2 mg/kg paclitaxel 5 times per week for three weeks or a cumulative dose of 50 mg/kg over 10 days resulted in thermal hypoalgesia in the absence of gross nerve degeneration (Campana et al., 1998). In contrast, administering 1mg/kg/day or 2 mg/kg/day on alternating days until four doses are given produced mechanical and cold allodynia and thermal hyperalgesia (Polomano et al., 2001). Thus, it is possible that a reduction of thermal nociceptive responses following high dose treatment with paclitaxel is mediated through losses of TRPV1 function, whereas the mechanical and cold hypersensitivity are not.
Activation of the TRPA1 channel is thought to be induced by a wide variety of stimuli, including noxious mechanical, noxious cold and environmental chemicals (see review by Kwan et al., 2006), although sensitivity of the channel to some of these stimulating modalities is disputed (Dunham et al., 2010). Several investigators have examined the role of TRPA1 in mediating paclitaxel-induced mechanical and cold allodynia by systemically administering a TRPA1 antagonist or by using TRPA1-deficient animals and examining the effects of these manipulations in animals treated with paclitaxel (Chen et al., 2011; Materazzi et al., 2012). Their findings suggest that TRPA1, in conjunction with another TRP channel, TRPV4, mediates mechanical allodynia, whereas TRPA1 alone mediates the cold allodynia induced by paclitaxel. These in vivo findings, demonstrating enhanced or sustained sensitivity of the TRPA1 channel following paclitaxel administration, contrast with our in vitro findings that treatment with a high concentration of paclitaxel inhibits TRPA1-stimulated neuropeptide release. A recent series of manuscripts demonstrated that the TRPA1 agonist, AITC, also can activate the TRPV1 channel (Alpizar et al., 2013; Everaerts et al., 2011; Gees et al., 2013), suggesting that the paclitaxel-induced inhibition of AITC-evoked CGRP release might be mediated by the TRPV1 channel. However, evidence against a role for AITC in evoking CGRP release via the TRPV1 channel has been demonstrated by Kunkler et al., who determined that AITC-evoked release of CGRP from sensory neurons in culture was not inhibited by the TRPV1 antagonist, capsazepine (2011). Interestingly, a recent study suggests that functional TRPA1 channels, defined as those which mediate AITC-induced increases in intracellular calcium in cultures derived from the dorsal root ganglion, are expressed in both peptidergic and nonpeptidergic sensory neurons (Barabas et al., 2012). Thus, while we have demonstrated paclitaxel-induced inhibition of the function of TRPA1 channels in peptidergic neurons, persistent or enhanced activation of the TRPA1 channels expressed within nonpeptidergic neurons could explain why desensitization to cold or mechanical stimulation is not observed in vivo. Further studies examining whether paclitaxel alters TRP-mediated increases in intracellular calcium and the release of glutamate will be critical to appreciate the effects of paclitaxel on TRPA1 function in the population of sensory neurons which does not contain CGRP.
Use of an in vitro model of paclitaxel-induced changes in neuronal sensitivity could serve as an alternative approach to examine mechanisms by which paclitaxel alters the function of sensory neurons. Although it is difficult to equate the concentrations of paclitaxel used in isolated cells with concentrations achieved in tissues in patients, pharmacokinetic studies show that dosing patients with 125–225 mg/m2 of paclitaxel results in maximal plasma concentrations between 1 and 10 μM (Henningsson et al., 2001; Rowinsky et al., 1999), with a reduction in concentration to between 0.01 μM and 1 μM by 20 hours. Furthermore, when paclitaxel concentrations were measured in laboratory animals 24 hours after the last of a number of doses, levels were significantly higher in the dorsal root ganglia and in sensory nerve bundles than in the plasma (Cavaletti et al., 2000; Xiao et al., 2011). Based on the pharmacokinetic studies in humans and the animal data suggesting that the drug concentrates in tissues, the amounts of paclitaxel we used in the current experiments are within the range that is achieved clinically.
The data in this manuscript validate the release of CGRP as a functional endpoint of sensory neuron activity to observe the effects of paclitaxel on neuronal function. We can use this in vitro model to perform interventional studies to identify and substantiate putative inhibitors of paclitaxel-induced PN. Further studies will elucidate different hypothesized signaling pathways to determine whether they contribute to the illustrated alterations in neuropeptide release. Paclitaxel-induced increases in neuropeptide release could contribute to a ‘gain of function’ of primary sensory neurons, experienced by patients as mechanical allodynia and tingling (Dougherty et al., 2004; Forsyth et al., 1997; Lipton et al., 1989; Wiernik et al., 1987). Alternatively, the decreased release of neuropeptides could mediate a ‘loss of function’ of primary sensory neurons. A loss of sensory neuron activity could produce numbness (Dougherty et al., 2004) and alter the sensitivity to cold temperatures, either through neuronal crosstalk (McCoy et al., 2013) or via disruption of peripheral bloodflow in patients (Gracias et al., 2011), and therefore mediate cold-induced burning pain in patients (Dougherty et al., 2004; Lipton et al., 1989). Further studies are clearly needed to substantiate these proposed relationships between in vitro and in vivo observations. Only after we understand the signaling mechanisms can we subsequently develop therapeutics to specifically prevent the development or maintenance of paclitaxel-induced neurotoxicity without compromising the oncolytic activity of the drug.
Acknowledgments
This publication was supported by a Project Development Team within the ICTSI NIH/NCRR grant number RR025761 to JCF and MRV and by NIH RO1CA 121168 to MRV.
Abbreviations
- Pac
paclitaxel
- PN
peripheral neuropathy
- TRP
transient receptor potential
- CGRP
calcitonin gene-related peptide
- AITC
allyl isothiocyanate
References
- Alpizar YA, Boonen B, Gees M, Sanchez A, Nilius B, Voets T, Talavera K. Allyl isothiocyanate sensitizes TRPV1 to heat stimulation. Pflugers Archiv : European journal of physiology. 2013 doi: 10.1007/s00424-013-1334-9. [DOI] [PubMed] [Google Scholar]
- Authier N, Gillet JP, Fialip J, Eschalier A, Coudore F. Description of a short-term Taxol-induced nociceptive neuropathy in rats. Brain Res. 2000;887:239–249. doi: 10.1016/s0006-8993(00)02910-3. [DOI] [PubMed] [Google Scholar]
- Barabas ME, Kossyreva EA, Stucky CL. TRPA1 is functionally expressed primarily by IB4-binding, non-peptidergic mouse and rat sensory neurons. PLoS One. 2012;7:e47988. doi: 10.1371/journal.pone.0047988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Liu GK, Xiao WH, Jin HW, Siau C. Terminal arbor degeneration–a novel lesion produced by the antineoplastic agent paclitaxel. Eur J Neurosci. 2011;33:1667–1676. doi: 10.1111/j.1460-9568.2011.07652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmerle W, Splittgerber U, Lazarus MB, McKenzie KM, Johnston DG, Austin DJ, Ehrlich BE. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc Natl Acad Sci U S A. 2006;103:18356–18361. doi: 10.1073/pnas.0607240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis J, Xin W, Zhang H, Dougherty PM. Intraepidermal nerve fiber loss corresponds to the development of Taxol-induced hyperalgesia and can be prevented by treatment with minocycline. Pain. 2011;152:308–313. doi: 10.1016/j.pain.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana WM, Eskeland N, Calcutt NA, Misasi R, Myers RR, O’Brien JS. Prosaptide prevents paclitaxel neurotoxicity. Neurotoxicology. 1998;19:237–244. [PubMed] [Google Scholar]
- Capri G, Tarenzi E, Fulfaro F, Gianni L. The role of taxanes in the treatment of breast cancer. Semin Oncol. 1996;23:68–75. [PubMed] [Google Scholar]
- Cata JP, Weng HR, Dougherty PM. The effects of thalidomide and minocycline on taxol-induced hyperalgesia in rats. Brain Res. 2008;1229:100–110. doi: 10.1016/j.brainres.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaletti G, Cavalletti E, Montaguti P, Oggioni N, De Negri O, Tredici G. Effect on the peripheral nervous system of the short-term intravenous administration of paclitaxel in the rat. Neurotoxicology. 1997;18:137–145. [PubMed] [Google Scholar]
- Cavaletti G, Cavalletti E, Oggioni N, Sottani C, Minoia C, D’Incalci M, Zucchetti M, Marmiroli P, Tredici G. Distribution of paclitaxel within the nervous system of the rat after repeated intravenous administration. Neurotoxicology. 2000;21:389–393. [PubMed] [Google Scholar]
- Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience. 2011;193:440–451. doi: 10.1016/j.neuroscience.2011.06.085. [DOI] [PubMed] [Google Scholar]
- Connelly E, Markman M, Kennedy A, Webster K, Kulp B, Peterson G, Belinson J. Paclitaxel delivered as a 3-hr infusion with cisplatin in patients with gynecologic cancers: unexpected incidence of neurotoxicity. Gynecol Oncol. 1996;62:166–168. doi: 10.1006/gyno.1996.0210. [DOI] [PubMed] [Google Scholar]
- Deng L, Guindon J, Vemuri VK, Thakur GA, White FA, Makriyannis A, Hohmann AG. The maintenance of cisplatin- and paclitaxel-induced mechanical and cold allodynia is suppressed by cannabinoid CB(2) receptor activation and independent of CXCR4 signaling in models of chemotherapy-induced peripheral neuropathy. Molecular pain. 2012;8:71. doi: 10.1186/1744-8069-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty PM, Cata JP, Cordella JV, Burton A, Weng HR. Taxol-induced sensory disturbance is characterized by preferential impairment of myelinated fiber function in cancer patients. Pain. 2004;109:132–142. doi: 10.1016/j.pain.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Dunham JP, Leith JL, Lumb BM, Donaldson LF. Transient receptor potential channel A1 and noxious cold responses in rat cutaneous nociceptors. Neuroscience. 2010;165:1412–1419. doi: 10.1016/j.neuroscience.2009.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everaerts W, Gees M, Alpizar YA, Farre R, Leten C, Apetrei A, Dewachter I, van Leuven F, Vennekens R, De Ridder D, Nilius B, Voets T, Talavera K. The capsaicin receptor TRPV1 is a crucial mediator of the noxious effects of mustard oil. Current biology: CB. 2011;21:316–321. doi: 10.1016/j.cub.2011.01.031. [DOI] [PubMed] [Google Scholar]
- Ferlini C, Cicchillitti L, Raspaglio G, Bartollino S, Cimitan S, Bertucci C, Mozzetti S, Gallo D, Persico M, Fattorusso C, Campiani G, Scambia G. Paclitaxel Directly Binds to Bcl-2 and Functionally Mimics Activity of Nur77. Cancer Research. 2009;69:6906–6914. doi: 10.1158/0008-5472.CAN-09-0540. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122:245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatters SJ, Xiao WH, Bennett GJ. Acetyl-L-carnitine prevents and reduces paclitaxel-induced painful peripheral neuropathy. Neurosci Lett. 2006;397:219–223. doi: 10.1016/j.neulet.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth PA, Balmaceda C, Peterson K, Seidman AD, Brasher P, DeAngelis LM. Prospective study of paclitaxel-induced peripheral neuropathy with quantitative sensory testing. J Neurooncol. 1997;35:47–53. doi: 10.1023/a:1005805907311. [DOI] [PubMed] [Google Scholar]
- Gambino F, Pavlowsky A, Begle A, Dupont JL, Bahi N, Courjaret R, Gardette R, Hadjkacem H, Skala H, Poulain B, Chelly J, Vitale N, Humeau Y. IL1-receptor accessory protein-like 1 (IL1RAPL1), a protein involved in cognitive functions, regulates N-type Ca2+-channel and neurite elongation. Proc Natl Acad Sci U S A. 2007;104:9063–9068. doi: 10.1073/pnas.0701133104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gees M, Alpizar YA, Boonen B, Sanchez A, Everaerts W, Segal A, Xue F, Janssens A, Owsianik G, Nilius B, Voets T, Talavera K. Mechanisms of transient receptor potential vanilloid 1 activation and sensitization by allyl isothiocyanate. Molecular pharmacology. 2013;84:325–334. doi: 10.1124/mol.113.085548. [DOI] [PubMed] [Google Scholar]
- Gracias NG, Cummins TR, Kelley MR, Basile DP, Iqbal T, Vasko MR. Vasodilatation in the rat dorsal hindpaw induced by activation of sensory neurons is reduced by paclitaxel. Neurotoxicology. 2011;32:140–149. doi: 10.1016/j.neuro.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Chiba T, Abe K, Makabe A, Ikeno S, Kawakami K, Utsunomiya I, Hama T, Taguchi K. Effect of paclitaxel on transient receptor potential vanilloid 1 in rat dorsal root ganglion. Pain. 2013;154:882–889. doi: 10.1016/j.pain.2013.02.023. [DOI] [PubMed] [Google Scholar]
- Henningsson A, Karlsson MO, Vigano L, Gianni L, Verweij J, Sparreboom A. Mechanism-based pharmacokinetic model for paclitaxel. J Clin Oncol. 2001;19:4065–4073. doi: 10.1200/JCO.2001.19.20.4065. [DOI] [PubMed] [Google Scholar]
- Hershman DL, Unger JM, Crew KD, Moinpour C, Minasian LM, Hansen L, Lew D, OKane P, Wade JL, Wong SF, Hortobagyi GN, Meyskens FL, Albain KS. SWOG S0715: Randomized placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy during adjuvant breast cancer therapy. ASCO Meeting Abstracts. 2012;30:9018. [Google Scholar]
- Hingtgen CM, Vasko MR. Prostacyclin enhances the evoked-release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain Res. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- Holzer P. Local effector functions of capsaicin-sensitive sensory nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience. 1988;24:739–768. doi: 10.1016/0306-4522(88)90064-4. [DOI] [PubMed] [Google Scholar]
- Ito S, Tajima K, Nogawa M, Inoue N, Kyoi T, Takahashi Y, Sasagawa T, Nakamura A, Kotera T, Ueda M, Yamashita Y, Banno K. Etodolac, a cyclooxygenase-2 inhibitor, attenuates paclitaxel-induced peripheral neuropathy in a mouse model of mechanical allodynia. J Pharmacol Exp Ther. 2012;342:53–60. doi: 10.1124/jpet.111.187401. [DOI] [PubMed] [Google Scholar]
- Jin HW, Flatters SJ, Xiao WH, Mulhern HL, Bennett GJ. Prevention of paclitaxel-evoked painful peripheral neuropathy by acetyl-L-carnitine: effects on axonal mitochondria, sensory nerve fiber terminal arbors, and cutaneous Langerhans cells. Exp Neurol. 2008;210:229–237. doi: 10.1016/j.expneurol.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautio AL, Haanpaa M, Leminen A, Kalso E, Kautiainen H, Saarto T. Amitriptyline in the prevention of chemotherapy-induced neuropathic symptoms. Anticancer Res. 2009;29:2601–2606. [PubMed] [Google Scholar]
- Kawakami K, Chiba T, Katagiri N, Saduka M, Abe K, Utsunomiya I, Hama T, Taguchi K. Paclitaxel Increases High Voltage-Dependent Calcium Channel Current in Dorsal Root Ganglion Neurons of the Rat. Journal of pharmacological sciences. 2012 doi: 10.1254/jphs.12123fp. [DOI] [PubMed] [Google Scholar]
- Kidd JF, Pilkington MF, Schell MJ, Fogarty KE, Skepper JN, Taylor CW, Thorn P. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J Biol Chem. 2002;277:6504–6510. doi: 10.1074/jbc.M106802200. [DOI] [PubMed] [Google Scholar]
- Kim HK, Zhang YP, Gwak YS, Abdi S. Phenyl N-tert-butylnitrone, a free radical scavenger, reduces mechanical allodynia in chemotherapy-induced neuropathic pain in rats. Anesthesiology. 2010;112:432–439. doi: 10.1097/ALN.0b013e3181ca31bd. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Price DL. Axotomy as an Experimental Model of Neuronal Injury and Cell Death. Brain Pathology. 1996;6:447–465. doi: 10.1111/j.1750-3639.1996.tb00875.x. [DOI] [PubMed] [Google Scholar]
- Kottschade LA, Sloan JA, Mazurczak MA, Johnson DB, Murphy BP, Rowland KM, Smith DA, Berg AR, Stella PJ, Loprinzi CL. The use of vitamin E for the prevention of chemotherapy-induced peripheral neuropathy: results of a randomized phase III clinical trial. Support Care Cancer. 2011;19:1769–1777. doi: 10.1007/s00520-010-1018-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkler PE, Ballard CJ, Oxford GS, Hurley JH. TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. Pain. 2011;152:38–44. doi: 10.1016/j.pain.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Brady ST, Feinstein SC, Wilson L, Jordan MA. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology. 2013;37:231–239. doi: 10.1016/j.neuro.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton RB, Apfel SC, Dutcher JP, Rosenberg R, Kaplan J, Berger A, Einzig AI, Wiernik P, Schaumburg HH. Taxol produces a predominantly sensory neuropathy. Neurology. 1989;39:368–373. doi: 10.1212/wnl.39.3.368. [DOI] [PubMed] [Google Scholar]
- Manfredi JJ, Parness J, Horwitz SB. Taxol binds to cellular microtubules. The Journal of cell biology. 1982;94:688–696. doi: 10.1083/jcb.94.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materazzi S, Fusi C, Benemei S, Pedretti P, Patacchini R, Nilius B, Prenen J, Creminon C, Geppetti P, Nassini R. TRPA1 and TRPV4 mediate paclitaxel-induced peripheral neuropathy in mice via a glutathione-sensitive mechanism. Pflugers Archiv : European journal of physiology. 2012;463:561–569. doi: 10.1007/s00424-011-1071-x. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Inoue M, Hald A, Xie W, Ueda H. Inhibition of paclitaxel-induced A-fiber hypersensitization by gabapentin. J Pharmacol Exp Ther. 2006;318:735–740. doi: 10.1124/jpet.106.103614. [DOI] [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Street SE, Pribisko AL, Zheng J, Zylka MJ. Peptidergic CGRPalpha primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron. 2013;78:138–151. doi: 10.1016/j.neuron.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melli G, Taiana M, Camozzi F, Triolo D, Podini P, Quattrini A, Taroni F, Lauria G. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp Neurol. 2008;214:276–284. doi: 10.1016/j.expneurol.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Miyano K, Tang HB, Nakamura Y, Morioka N, Inoue A, Nakata Y. Paclitaxel and vinorelbine, evoked the release of substance P from cultured rat dorsal root ganglion cells through different PKC isoform-sensitive ion channels. Neuropharmacology. 2009;57:25–32. doi: 10.1016/j.neuropharm.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Mo M, Erdelyi I, Szigeti-Buck K, Benbow JH, Ehrlich BE. Prevention of paclitaxel-induced peripheral neuropathy by lithium pretreatment. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26:4696–4709. doi: 10.1096/fj.12-214643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KA, Fehrenbacher JC, Thompson EL, Duarte DB, Hingtgen CM, Vasko MR. Signaling pathways that mediate nerve growth factor-induced increase in expression and release of calcitonin gene-related peptide from sensory neurons. Neuroscience. 2010;171:910–923. doi: 10.1016/j.neuroscience.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual D, Goicoechea C, Suardiaz M, Martin MI. A cannabinoid agonist, WIN 55,212-2, reduces neuropathic nociception induced by paclitaxel in rats. Pain. 2005;118:23–34. doi: 10.1016/j.pain.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- Rao RD, Flynn PJ, Sloan JA, Wong GY, Novotny P, Johnson DB, Gross HM, Renno SI, Nashawaty M, Loprinzi CL. Efficacy of lamotrigine in the management of chemotherapy-induced peripheral neuropathy: a phase 3 randomized, double-blind, placebo-controlled trial, N01C3. Cancer. 2008;112:2802–2808. doi: 10.1002/cncr.23482. [DOI] [PubMed] [Google Scholar]
- Rao RD, Michalak JC, Sloan JA, Loprinzi CL, Soori GS, Nikcevich DA, Warner DO, Novotny P, Kutteh LA, Wong GY. Efficacy of gabapentin in the management of chemotherapy-induced peripheral neuropathy: a phase 3 randomized, double-blind, placebo-controlled, crossover trial (N00C3) Cancer. 2007;110:2110–2118. doi: 10.1002/cncr.23008. [DOI] [PubMed] [Google Scholar]
- Rowinsky EK, Gilbert MR, McGuire WP, Noe DA, Grochow LB, Forastiere AA, Ettinger DS, Lubejko BG, Clark B, Sartorius SE, et al. Sequences of taxol and cisplatin: a phase I and pharmacologic study. J Clin Oncol. 1991;9:1692–1703. doi: 10.1200/JCO.1991.9.9.1692. [DOI] [PubMed] [Google Scholar]
- Rowinsky EK, Jiroutek M, Bonomi P, Johnson D, Baker SD. Paclitaxel steady-state plasma concentration as a determinant of disease outcome and toxicity in lung cancer patients treated with paclitaxel and cisplatin. Clin Cancer Res. 1999;5:767–774. [PubMed] [Google Scholar]
- Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- Scuteri A, Nicolini G, Miloso M, Bossi M, Cavaletti G, Windebank AJ, Tredici G. Paclitaxel toxicity in post-mitotic dorsal root ganglion (DRG) cells. Anticancer Res. 2006;26:1065–1070. [PubMed] [Google Scholar]
- Smith EM, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, Bressler LR, Fadul CE, Knox C, Le-Lindqwister N, Gilman PB, Shapiro CL. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA : the journal of the American Medical Association. 2013;309:1359–1367. doi: 10.1001/jama.2013.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA repair. 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Voehringer DW. BCL-2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Radical Biology and Medicine. 1999;27:945–950. doi: 10.1016/s0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- Whiteside G, Doyle CA, Hunt SP, Munglani R. Differential time course of neuronal and glial apoptosis in neonatal rat dorsal root ganglia after sciatic nerve axotomy. European Journal of Neuroscience. 1998;10:3400–3408. doi: 10.1046/j.1460-9568.1998.00346.x. [DOI] [PubMed] [Google Scholar]
- Wiernik PH, Schwartz EL, Strauman JJ, Dutcher JP, Lipton RB, Paietta E. Phase I clinical and pharmacokinetic study of taxol. Cancer Res. 1987;47:2486–2493. [PubMed] [Google Scholar]
- Xiao W, Boroujerdi A, Bennett GJ, Luo ZD. Chemotherapy-evoked painful peripheral neuropathy: analgesic effects of gabapentin and effects on expression of the alpha-2-delta type-1 calcium channel subunit. Neuroscience. 2007;144:714–720. doi: 10.1016/j.neuroscience.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao WH, Zheng H, Zheng FY, Nuydens R, Meert TF, Bennett GJ. Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience. 2011;199:461–469. doi: 10.1016/j.neuroscience.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang IH, Siddique R, Hosmane S, Thakor N, Hoke A. Compartmentalized microfluidic culture platform to study mechanism of paclitaxel-induced axonal degeneration. Exp Neurol. 2009;218:124–128. doi: 10.1016/j.expneurol.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, Dougherty PM. Induction of Monocyte Chemoattractant Protein-1 (MCP-1) and Its Receptor CCR2 in Primary Sensory Neurons Contributes to Paclitaxel-Induced Peripheral Neuropathy. J Pain. 2013 doi: 10.1016/j.jpain.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]