

Abstract
Peripheral neuropathy is a dose-limiting and debilitating side effect of the chemotherapeutic drug, paclitaxel. Consequently, elucidating the mechanisms by which this drug alters sensory neuronal function is essential for the development of successful therapeutics for peripheral neuropathy. We previously demonstrated that chronic treatment with paclitaxel (3–5 days) reduces neuropeptide release stimulated by agonists of TRPV1. Because the activity of TRPV1 channels is modulated by conventional and novel PKC isozymes (c/nPKC), we investigated whether c/nPKC mediate the loss of neuropeptide release following chronic treatment with paclitaxel (300 nM; 3 and 5 days). Release of the neuropeptide, calcitonin gene-related peptide (CGRP), was measured as an index of neuronal sensitivity. Following paclitaxel treatment, cultured dorsal root ganglia sensory neurons were stimulated with a c/nPKC activator, phorbol 12,13-dibutyrate (PDBu), or a TRPV1 agonist, capsaicin, in the absence and presence of selective inhibitors of conventional PKCα and PKCβI/II isozymes (cPKC). Paclitaxel (300nM; 3 days and 5 days) attenuated both PDBu- and capsaicin-stimulated release in a cPKC-dependent manner. Under basal conditions, there were no changes in the protein expression, phosphorylation or membrane localization of PKC α, βI or βII, however, paclitaxel decreased cPKC activity as indicated by a reduction in the phosphorylation of cPKC substrates. Under stimulatory conditions, paclitaxel attenuated the membrane translocation of phosphorylated PKC α, βI and βII, providing a rationale for the attenuation in PDBu- and capsaicin-stimulated release. Our findings suggest that a decrease in cPKC activity and membrane localization are responsible for the reduction in stimulated peptide release following chronic treatment with paclitaxel in sensory neurons.
Keywords: paclitaxel, peripheral neuropathy, dorsal root ganglia, PKC, TRPV1, CGRP
1. Introduction
Paclitaxel is a microtubule stabilizing agent that is used to treat solid tumors including breast, ovarian and non-small cell lung cancer (Schiff and Horwitz, 1980, McGuire et al., 1989, Holmes et al., 1991, Murphy et al., 1993). A major debilitating side effect associated with paclitaxel treatment is peripheral neuropathy (Postma et al., 1995, Tamura et al., 1995). This neuropathy is characterized by sensory abnormalities indicative of both a loss and gain of sensory neuronal function (Wolfe and Trivedi, 2004) with symptoms that include numbness, loss of proprioception, tingling, burning pain and mechanical and cold hypersensitivity in the distal extremities of the hands and feet (Wiernik et al., 1987, Dougherty et al., 2004). The severity of neuropathic symptoms, which is dependent on factors including the single and cumulative dose, the number of treatment cycles and the patients’ medical history, often results in either a dose reduction or discontinuation of therapy (Postma et al., 1995, Hershman et al., 2011), and therefore compromises the successful treatment of cancer. While it is evident that paclitaxel alters sensory neuronal function, the exact mechanisms by which this occurs still remain unclear, and as a result, there are no current therapies to alleviate paclitaxel-induced peripheral neuropathy (PIPN).
Animal models of PIPN have been established in an effort to study the effects of paclitaxel on sensory neuronal activity. In a low dose model, intraperitoneal injections of paclitaxel (1 mg/kg or 2 mg/kg) on four alternate days resulted in thermal, cold and mechanical hypersensitivity when assessed using behavioral testing; however, no overt structural nerve damage or toxicity was found to explain this hypersensitivity (Polomano et al., 2001, Flatters and Bennett, 2006). In contrast, in a higher dose model, paclitaxel (cumulative doses ranging from 32 mg/kg – 80 mg/kg) induced thermal hypoalgesia, suggesting a loss, rather than gain, of function of the heat-responsive transient receptor potential vanilloid 1 (TRPV1) channel (Authier et al., 2000). Examining the effects of paclitaxel on neuronal sensitivity using a different functional end-point also showed loss of TRPV1 function following paclitaxel administration. Using vasodilation as an indirect measure of neuropeptide release, paclitaxel (cumulative dose 4 mg/kg) decreased capsaicin-evoked vasodilation in the dermal skin layer (Gracias et al., 2011). Such behavioral findings indicate that paclitaxel has dual effects on neuronal sensitivity which are dependent on the concentration and length of exposure to paclitaxel. Our laboratory has recapitulated similar effects of paclitaxel on sensory neuronal function in isolated dorsal root ganglia (DRG) sensory neurons. We found that a 5-day treatment with 10 nM paclitaxel increased capsaicin-evoked neuropeptide release whereas treatment with 300 nM decreased capsaicin-evoked release (Pittman et al., 2014). In this study, we questioned whether protein kinase C (PKC), a well-established modulator of TRPV1 channel function, was important for mediating the effects of paclitaxel on sensory neuronal function in isolated DRG cultures.
Studies show that modulating the activity of conventional and novel protein kinase C isozymes (c/nPKC) alters the sensitivity of sensory neurons. In isolated DRG sensory neurons, exposure to phorbol esters, which activate c/nPKC, enhanced the release of the neuropeptide, calcitonin gene-related peptide (Supowit et al., 1995, Barber and Vasko, 1996), whereas downregulation of c/nPKC by chronic phorbol ester treatment attenuated capsaicin-evoked neuropeptide release (Barber and Vasko, 1996). It was also shown that activation of c/nPKC enhanced heat- and capsaicin-evoked currents in isolated DRG sensory neurons (Cesare et al., 1999, Vellani et al., 2001, Zhou et al., 2001, Li et al., 2014) and increased calcium uptake via TRPV1 (Olah et al., 2002), thus c/nPKC play a large role in modulating the TRPV1 channel.
Recent studies have suggested that c/nPKC mediate enhanced neuronal sensitivity induced by paclitaxel. In rodent models, administration of PKCβII, PKCδ and PKCε inhibitory peptides reversed heat, cold and mechanical hypersensitivity induced by chronic administration of paclitaxel (Dina et al., 2001, Chen et al., 2011, He and Wang, 2015). In an acute in vitro model, c/nPKC mediate an enhanced neuropeptide release following 10–30 minute exposures to paclitaxel in isolated DRG sensory neurons (Miyano et al., 2009, He and Wang, 2015). However, the onset of symptoms of peripheral neuropathy is typically 3–6 weeks following the first infusion of paclitaxel (Forsyth et al., 1997), and it is not known whether c/nPKC contribute to the maintenance of changes in neuronal sensitivity following chronic treatment with paclitaxel in sensory neurons. As such, we examined whether c/nPKC were responsible for changes in neuronal sensitivity induced by chronic treatment with paclitaxel (1–5 days) in isolated DRG sensory neurons. We found that paclitaxel attenuated phorbol ester- and capsaicin-stimulated peptide release in a conventional PKC-dependent manner. Under basal conditions, paclitaxel attenuated the activity of PKC α, βI and βII but had no effect on the protein expression, phosphorylation status or membrane localization. However, under stimulatory conditions, paclitaxel attenuated the activity of PKC α, βI and βII, as well as phorbol ester-stimulated membrane localization of phosphorylated PKC α, βI and βII. In conclusion, we demonstrated that PKCα and PKCβI/II were critical mediators of the reduction in capsaicin-stimulated neuropeptide release following chronic treatment with paclitaxel, thus implicating these isozymes in modulating the function of TRPV1 channels following paclitaxel treatment.
2. Materials and Methods
2.1. Sensory neuronal culture
Dorsal root ganglia (DRG) were isolated and cultured as previously described (Pittman et al., 2014). Male Sprague Dawley rats (150–250 g, Envigo, Indianapolis, IN) were sacrificed by CO2 asphyxiation followed by decapitation. The DRG from all levels of the vertebral column were collected, trimmed and enzymatically digested in collagenase (0.125%; ≥ 470 collagen digestion units) for 1 hour at 37°C. Using a small diameter fire-polished glass Pasteur pipette, DRG were mechanically dissociated in F-12 media supplemented with 10% heat-inactivated horse serum, 2 mM glutamine, 50 μg/mL penicillin and streptomycin, 50 μM 5-fluoro-2-deoxyuridine, 150 μM uridine and 30 ng/mL nerve growth factor. The neuronal cell suspension was plated on 12-well plates previously coated with poly-D-lysine and laminin to an approximate density of 30,000 cells per well. Neuronal cultures were maintained at 37°C with 3% CO2 atmosphere. The media was changed every other day. For paclitaxel experiments, cultured sensory neurons were treated with 300 nM paclitaxel for 1–5 days or 1 μM paclitaxel for 1–3 days after 7 days in culture, as previously described (Pittman et al., 2014). All experiments were done in compliance with the Animal Care and Use Committee at Indiana University School of Medicine (Indianapolis, IN) and the National Institute of Health Guide for the Care and Use of Lab Animals. Prior to sacrifice, all animals were housed in group cages in a light controlled room with access to food and water ad libitum.
2.2. Drug Stocks
10 mM stocks of paclitaxel and 4-(3-Chloro-2-pyridinyl)-N-[4-(1,1-dimethylethyl)phenyl]-1-piperazinecarboxamide (BCTC), and 1 mM stocks of phorbol 12,13-dibutyrate (PDBu), 4α-PDBu, bisindolylmaleimide I (Bis I), Gö6976, LY333531 and Bis VIII were diluted in methyl-2-pyrrolidone (MPL). The stock solutions were stored at −20°C and were further diluted in media or HEPES buffer to the desired concentrations for experiments.
2.3. Calcitonin Gene-Related Peptide Release Assay
CGRP release assays were performed on days 7–12 as previously described (Hingtgen and Vasko, 1994, Pittman et al., 2014) with slight modifications as follows. Briefly, neuronal cultures were washed with HEPES buffer (25 mM HEPES, 135 mM NaCl, 3.5 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 3.3 mM D-glucose and 0.1 % bovine serum albumin, pH 7.4) followed by four sequential 10-minute incubations to assess the basal and stimulated release of immunoreactive calcitonin gene-related peptide (CGRP) under specific conditions. Neuronal cultures were maintained at 37°C with 3% CO2 atmosphere during the 10-minute intervals. For the first and second interval, neuronal cultures were incubated with HEPES buffer +/− drug to establish basal release of CGRP. For the third interval, neuronal cultures were incubated with HEPES buffer containing 10 nM PDBu or 30 nM capsaicin +/− drug to stimulate the release of CGRP. For the fourth interval, neuronal cultures were incubated with HEPES buffer to reestablish the basal release of CGRP. Neuronal cultures were incubated in 0.1N HCl for 20 minutes at room temperature to release all remaining CGRP present in the neurons. The supernatant was collected after each interval and the amount of CGRP released into the supernatant was determined using radioimmunoassay, as previously described (Hingtgen and Vasko, 1994, Pittman et al., 2014).
2.4. Western blotting
Cells were scraped in F-12 media and transferred to eppendorf tubes. The sample was centrifuged at 14,000 rpm for 4 minutes and the supernatant was discarded. The cell pellet was washed in ice-cold sterile phosphate-buffered saline (PBS). The sample was then centrifuged at 14,000 rpm for 4 minutes and the supernatant was discarded. Modified RIPA lysis buffer containing 0.2 μg/mL phenylmethylsulfonyl fluoride (PMSF), 1 μg/mL pepstatin A, 1 mM sodium orthovanadate, 25 mM sodium fluoride, 0.1% sodium dodecyl sulfate and 200 μL/mL protease inhibitor cocktail (Roche Diagnostics) was added to the cell pellet. Following sonication, the protein concentration was determined using a Bio-Rad protein assay, based on the Bradford protein-dye binding method (Bradford, 1976). Equivalent amounts of whole cell lysate protein were separated on NuPAGE 4–12% Bis-Tris Gels and transferred to PVDF membranes. Membranes were incubated in a 5% dry milk Tris buffered saline (TBS) solution containing 1% Tween 20 (TBS-T) for 1 hour and incubated with primary antibodies diluted in 1% TBS-T overnight at 4°C. Primary antibodies included: mouse anti-PKCα (1:1000, Santa Cruz), rabbit anti-PKCβI (1:500, Santa Cruz), rabbit anti-PKCβII (1:1000, Santa Cruz), rabbit anti-phospho(Ser) PKC substrate (1:1000, Cell Signaling Technology) and mouse anti-Actin (1:1000, Thermo Fisher Scientific). Membranes were washed in 1% TBS-T and incubated in HRP-conjugated secondary antibodies diluted in 1% TBS-T for 2 hours at room temperature. Secondary antibodies included: donkey anti-rabbit IgG-HRP (1:3000, Santa Cruz) and donkey anti-mouse IgG-HRP (1:3000, Thermo Fisher Scientific). The membranes were washed in 1% TBS-T. A Pierce ECL Western Blotting Substrate kit (Thermo Fisher Scientific) was used to detect immunoreactive bands on blue X-Ray films and the band density was quantified using KODAK 1D 3.6 (Scientific Imaging Systems, New Haven, CT).
2.5. Isolation of Cytosolic and Membrane Protein Fractions
The isolation of cytosolic and membrane protein fractions was done as previously described (Uehara et al., 2004) with slight modifications. The cell pellet was incubated in homogenization buffer (20 mM Tris-HCl pH 7.5, 330 mM sucrose, 0.5 mM EGTA, 2 mM EDTA, 2 μg/mL aprotinin, 25 μg/mL leupeptin and 1 mM PMSF, 200 μL/mL protease inhibitor cocktail), homogenized with a polytron and centrifuged at 25,000 rpm for 35 minutes at 4°C. The supernatant was collected (cytosolic fraction) and the cell pellet (membrane fraction) was re-suspended in homogenization buffer containing 1% Triton X-100 and placed on ice for 1 hour. The solution was centrifuged at 25,000 rpm for 35 minutes at 4°C. The resulting supernatant (membrane fraction) was collected. The protein concentrations of the cytosolic and membrane protein fractions were determined using the Bio-Rad protein assay and equivalent amounts of protein were separated on NuPAGE 4–12% Bis-Tris Gels. We then followed the Western blot protocol described above. In addition to the listed antibodies, primary antibodies included: mouse anti-p-PKCα (1:1000; Ser 657; Santa Cruz), rabbit anti-p-PKCβI (1:500; Thr 641; Santa Cruz), mouse anti-p-PKCβII/δ (1:1000; Ser 660; Santa Cruz) and rabbit anti-Na+/K+ ATPase α (1:1000, Cell Signaling Technology).
2.6. Immunofluorescence and Membrane Translocation
Neuronal cultures were rinsed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) for 20 minutes. The 4% PFA was aspirated and cells were rinsed with PBS and blocked in filtered blocking solution (normal donkey serum, Triton x-100 in 0.1M PBS, pH 6.0) for 1 hour. Cultures were incubated in a mixed pool of primary antibodies diluted in filtered blocking solution overnight at room temperature. Primary antibodies included: mouse anti-p-PKCα (1:500; Ser 657; Santa Cruz), rabbit anti-p-PKCβI (1:500; Thr 641; Santa Cruz) and mouse anti-p-PKCβII/δ (1:500; Ser 660; Santa Cruz). The following day, cells were rinsed with PBS and incubated with fluorescently labeled secondary antibodies diluted in filtered blocking solution for two hours at room temperature. Secondary antibodies included: Alexa Fluor 488 donkey anti-rabbit (1:200, Thermo Fisher Scientific) and Alexa Fluor 488 donkey anti-mouse (1:200, Thermo Fisher Scientific). Cultures were rinsed in PBS and 1mL PBS was added to each well of the 12-well plate. Fluorescent images were acquired using the Leica DMI6000 B inverted microscope. To measure translocation of phospho-PKC α, βI and βII, five random fields of view were acquired for each treatment group and neurons were scored as positive or negative for membrane translocation and the data expressed as % of cells with phospho-PKC α/βI/βII translocation, as previously described (Ron et al., 1995, Gray et al., 1997).
2.7. Reagents
F-12 media, heat-inactivated horse serum, glutamine, penicillin, streptomycin, 5-fluoro-2-deoxyuridine, uridine, NuPAGE 4–12% Bis-Tris Gels and PVDF membranes were obtained from Thermo Fisher Scientific (Waltham, MA). PDBu, Bis I, Gö6976 and BCTC were obtained from Tocris-BioTechne (Minneapolis, MN). The myristolyated PKCα/β peptide inhibitor (Myr-RFARKGALRQKNV) and nerve growth factor were obtained from Promega (Madison, WI) and Envigo (Indianapolis, IN), respectively. 4α-PDBu and the RIPA lysis buffer were obtained from Santa Cruz (Dallas, Texas) and EMD Millipore (Billerica, MA), respectively. Bis VIII was obtained from Cayman Chemical (Ann Arbor, Michigan). The CGRP antibody was a kind gift provided by Dr. M. Iadarola (National Institute of Health). All other reagents were purchased from Sigma Aldrich (St. Louis, MO).
2.8. Statistical Analysis
Data are represented as the mean ± standard error of mean (SEM). Data were analyzed by one-way analysis of variance (ANOVA) or two-way ANOVA and post-hoc tests were performed using either the Tukey’s or Dunnett’s multiple comparisions test using GraphPad Prism 7 software (La Jolla, CA) with p < 0.05 considered significant.
3. Results
3.1. Chronic treatment with paclitaxel decreases PDBu-stimulated CGRP release
We previously showed that long-term treatment with paclitaxel (300 nM; 3 days and 5 days) attenuated capsaicin-stimulated CGRP release in sensory neurons (Pittman et al., 2014). Because phorbol ester-induced activation of conventional and novel PKC isozymes (c/nPKC) increases TRPV1 activity (Vellani et al., 2001, Zhou et al., 2001, Numazaki et al., 2002, Bhave et al., 2003, Jeske et al., 2009), it is plausible that the attenuation in capsaicin-stimulated release upon treatment with paclitaxel is due to a loss of c/nPKC activity. To address this, we first examined the effects of treating sensory neuronal cultures with 300 nM or 1 μM paclitaxel for 1–5 days on the ability of phorbol 12, 13-dibutyrate (PDBu; 10 nM), a phorbol ester, to augment CGRP release. We expressed the data as % of total CGRP content since we have previously shown that changes in stimulated release following treatment with 300 nM paclitaxel for 3 days are not secondary to changes in peptide levels, and that treatment with 300 nM paclitaxel for 5 days does not alter CGRP content nor affect neuronal viability (Pittman et al., 2014). We found that treatment with 300 nM paclitaxel attenuated PDBu-stimulated CGRP release in a time-dependent manner to 19.0 ± 1.9 (1 day), 11.8 ± 1.0 (3 days) and 7.7 ± 0.9% of total content (5 days) compared to vehicle-treated neurons (24.0 ± 1.3% of total content; 0.003% methylpyrrolidone (MPL); Figure 1A). Similar observations were made using a higher concentration of paclitaxel at shorter time intervals in which treatment with 1 μM paclitaxel attenuated PDBu-stimulated CGRP release to 12.2 ± 1.3 (1 day), 7.7 ± 1.2 (2 days) and 3.7 ± 0.4% of total content (3 days) compared to vehicle-treated neurons (23.7 ± 0.4% of total content; 0.01% MPL; Figure 1B). These data indicate that chronic treatment with paclitaxel attenuates the release of CGRP evoked by PDBu-induced activation of c/nPKC isozymes.
Figure 1.
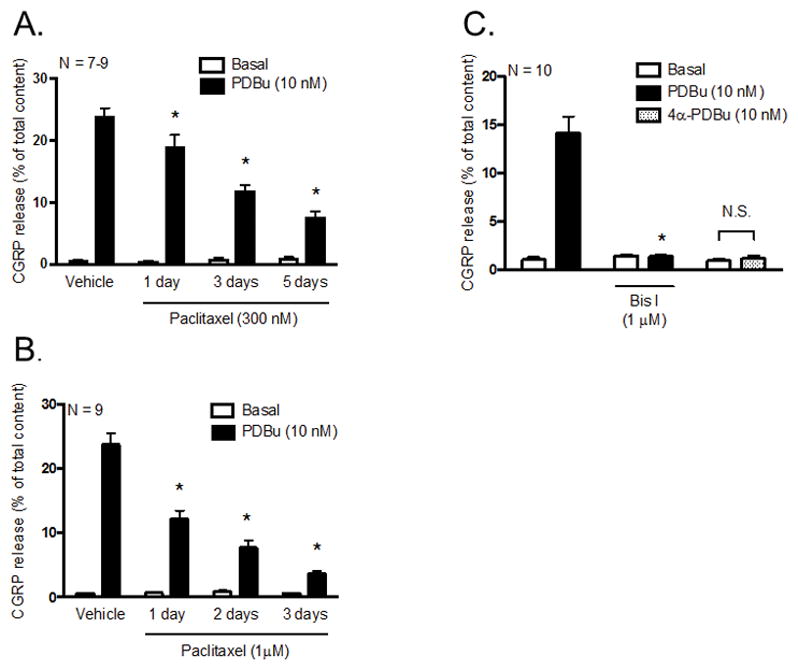
Paclitaxel decreases PDBu-stimulated CGRP release in cultured sensory neurons. Each column represents the mean ± SEM of basal (white columns) or stimulated (black columns) CGRP release expressed as % of total content. Cultures were exposed to (A) 300 nM or (B) 1 μM paclitaxel for 1, 2, 3 or 5 days prior to stimulation with PDBu (10 nM). An * indicates a significant decrease in PDBu-stimulated release in paclitaxel-treated (300 nM and 1 μM) neurons compared to vehicle-treated neurons (p < 0.05, N = 7–9). (C) Naïve cultures were pre-treated with Bis I (1 μM) for 10 minutes prior to stimulation with PDBu. As a control experiment, naïve cultures were also incubated with 10 nM 4α-PDBu (dotted columns). An * indicates a significant decrease in PDBu-stimulated release in neurons pre-treated with Bis I (p < 0.05, N = 10). Significance was determined using a two-way ANOVA with Tukey’s post-hoc test. N.S. – not significant.
In addition to activation of c/nPKC, studies have reported that PDBu binds and activates other C1 domain-containing proteins (Colon-Gonzalez and Kazanietz, 2006) that have been implicated in neurotransmitter release (Betz et al., 1998). To confirm that CGRP release was due to a specific effect of PDBu to activate c/nPKC, neurons were pre-treated with bisindolylmaleimide I (Bis I), a selective inhibitor that competitively inhibits ATP binding in c/nPKC (Toullec et al., 1991), for 10 minutes prior to stimulation with 10 nM PDBu. Pre-treatment with Bis I fully abolished PDBu-stimulated CGRP release from 14.1 ± 1.7 to 1.4 ± 0.2% of total content (Figure 1C). There were no changes in basal release of CGRP (1.5 ± 0.1% of total content) compared to untreated neurons (1.1 ± 0.3% of total content) pre-treated with Bis I. Treatment with 10 nM 4α-PDBu, an inactive analog of PDBu that is incapable of activating c/nPKC (Silinsky and Searl, 2003), did not alter the release of CGRP (1.2 ± 0.2% of total content) compared to basal release (1.0 ± 0.2% of total content). These data indicate that PDBu stimulates the release of CGRP through the specific activation of c/nPKC.
3.2. Chronic treatment with paclitaxel inhibits the activity of conventional PKC isozymes, α and βI/II, to elicit a decrease in PDBu-stimulated CGRP release
Since PDBu is a direct activator of both conventional and novel PKC isozymes, we first wanted to determine which class of PKC isozymes mediates the increase in CGRP release evoked by PDBu in the absence of paclitaxel. To differentiate between the two classes of PKC isozymes, neuronal cultures were pre-treated with Gö6976, an ATP competitive inhibitor that is selective for conventional PKC isozymes (Martiny-Baron et al., 1993), prior to stimulation with 10 nM PDBu. Pre-treatment with 100 nM Gö6976 attenuated PDBu-stimulated release from 11.3 ± 0.7 (control neurons) to 3.4 ± 0.2% of total content (Figure 2A), suggesting that conventional PKC isozymes, and not novel PKC isozymes, mediate PDBu-stimulated CGRP release. Having identified the class of PKC isozymes, we wanted to narrow down which specific isozymes mediate PDBu-stimulated release of CGRP. We investigated the specific contribution of PKCα and PKCβI/II versus PKCγ by pre-treating neuronal cultures with a myristolyated PKCα/β peptide inhibitor, prior to stimulation with 10 nM PDBu. The PKCα/β peptide, which is derived from a homologous sequence within the pseudosubstrate region of PKCα, PKCβI and PKCβII, consists of Ala25 in place of a phosphorylatable Ser/Thr and binds to the PKC substrate binding cavity to maintain the kinase in an inactive conformation (House and Kemp, 1987, Eichholtz et al., 1993). The activity of PKCγ, a conventional PKC isozyme, is unaffected by this peptide due to sequence dissimiliarities. Pre-treatment with 10 μM PKCα/β peptide inhibitor attenuated PDBu-stimulated CGRP release from 11.3 ± 0.7 (control neurons) to 3.5 ± 0.3% of total content (Figure 3A). There was no significant difference between basal (2.8 ± 0.2% of total content) and PDBu-stimulated release (3.5 ± 0.2% of total content) in the presence of the PKCα/β peptide inhibitor (Figure 2A). These data indicate that conventional PKC isozymes, α, βI and βII, mediate PDBu-stimulated release of CGRP.
Figure 2.
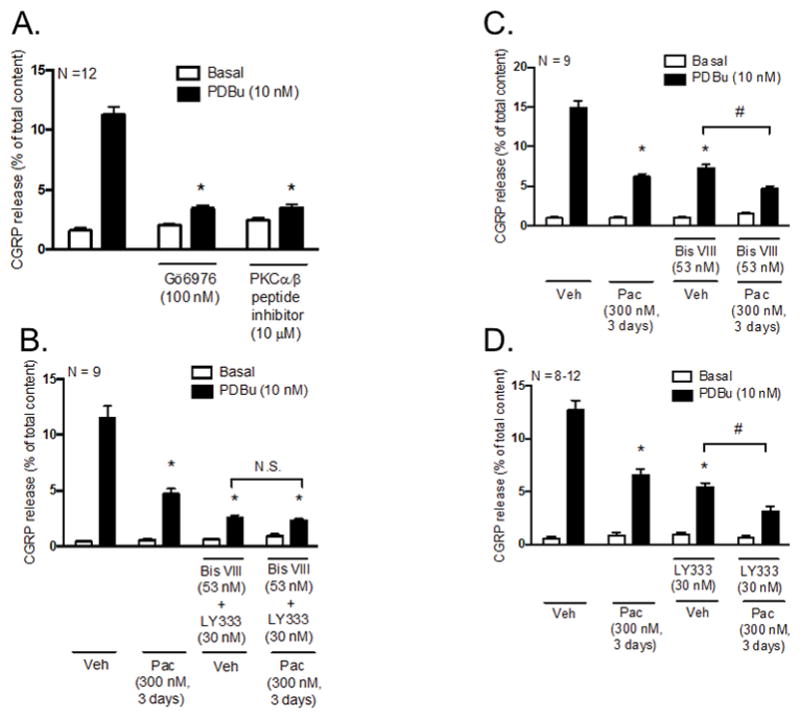
Inhibition of PKCα and PKCβI/II activity mediates the decrease in PDBu-stimulated CGRP release following chronic exposure to paclitaxel in cultured sensory neurons. Each column represents the mean ± SEM of basal (white columns) or PDBu-stimulated (black columns) CGRP release expressed as % of total content in the absence and presence of PKCα and PKCβI/II inhibitors. (A) Naïve cultures were pre-treated with Gö6976 (100 nM) or a myristoylated PKCα/β peptide inhibitor (10 μM) for 10 minutes prior to stimulation with PDBu (10 nM). An * indicates a significant decrease in PDBu-stimulated release in neurons pre-treated with Gö6976 or the myristoylated PKCα/β peptide inhibitor (p < 0.05, N = 12). Cultures were exposed to 300 nM paclitaxel for 3 days prior to stimulation with PDBu (10 nM) in the presence and absence of the (B) PKCα inhibitor (Bis VIII; 53 nM) + PKCβI/II inhibitor (LY333531; 30 nM), (C) Bis VIII or (D) LY333531. An * indicates a significant decrease in PDBu-stimulated release in paclitaxel-only treated neurons, vehicle- and paclitaxel-treated neurons pre-treated with (B) Bis VIII + LY333531, (C) Bis VIII or (D) LY333531 compared to vehicle-only treated neurons and a # indicates a significant decrease in PDBu-stimulated release in paclitaxel-treated neurons compared to vehicle-treated neurons pre-treated with (C) Bis VIII or (D) LY333531. Significance was determined using a two-way ANOVA with Tukey’s post-hoc test (p < 0.05, N = 8–12). Veh - Vehicle; Pac - Paclitaxel; LY333 – LY333531; N.S. – not significant.
Figure 3.
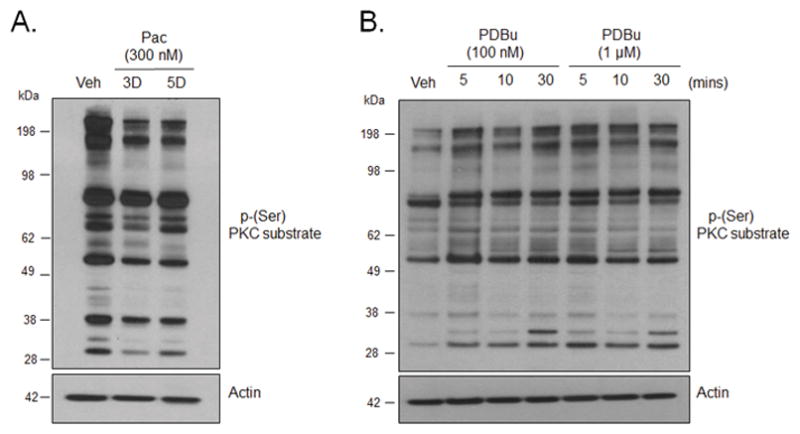
Chronic treatment with paclitaxel decreases the basal phosphorylation of PKC α/β substrates in cultured sensory neurons. (A) Representative western blot image showing immunoreactivity for phosphorylation of PKC α, βI and βII substrates in whole cell lysates in neurons treated with vehicle and paclitaxel (300 nM, 3 and 5 days). (B) Representative western blot image showing immunoreactivity for phosphorylation of PKC α, βI and βII substrates in neurons treated with PDBu (100 nM and 1 μM; 5, 10 or 30 minutes). Veh - Vehicle; Pac – Paclitaxel; 3D – 3 days; 5D – 5 days.
We next examined the relative contribution of PKCα and PKCβI/II in PDBu-stimulated release in the absence and presence of paclitaxel using small molecule inhibitors of these kinases. We chose to use selective small molecule inhibitors of PKCα and PKCβI/II, as opposed to the PKCα/β peptide inhibitor, as this would allow us to later discern the effects of each individual kinase. Cultured sensory neurons were treated with 300 nM paclitaxel for 3 days and pre-treated with a combination of PKCα (Bis VIII) and PKCβI/II (LY333531) inhibitors prior to stimulation with 10 nM PDBu; inhibitor concentrations used were based on reported values in the literature (Wilkinson et al., 1993, Jirousek et al., 1996, Zhou et al., 2006, Gray et al., 2013). In vehicle-treated neurons, combined pre-treatment with 53 nM Bis VIII and 30 nM LY333531 significantly attenuated PDBu-stimulated release from 11.5 ± 1.1 (vehicle-only) to 2.5 ± 0.2 % of total content (Figure 2B), confirming our earlier findings that PKC isozymes, α, βI and βII, mediate PDBu-stimulated CGRP release (see Figure 2A). In the presence of combined inhibition of PKCα and PKCβI/II, there was no further decrease in PDBu-stimulated release in paclitaxel-treated neurons (2.3 ± 0.2% of total content; Figure 2B) compared to vehicle-treated neurons (2.5 ± 0.2% of total content), suggesting that PKCα and PKCβI/II signal through common pathways. These data demonstrate that inhibition of PKCα and PKCβI/II activity is responsible for the reduction in CGRP release induced by paclitaxel.
To differentiate between the individual contributions of PKCα and PKCβI/II, cultured sensory neurons were treated with 300 nM paclitaxel for 3 days and pre-treated with either Bis VIII or LY333531 prior to stimulation with 10 nM PDBu. In vehicle-treated neurons, we found that individual pre-treatment with either Bis VIII or LY333531 partially attenuated PDBu-stimulated release to similar extents (Figures 2C and 2D), but did not completely block the increase in release as observed following combined pre-treatment with both inhibitors (see Figure 2B). This suggests that both PKCα and PKCβI/II are necessary to elicit an increase in CGRP release induced by PDBu; the single activity of either kinase is not sufficient to reach the given threshold. In the presence of paclitaxel, however, individual pre-treatment with Bis VIII (Figure 2C) and LY333531 (Figure 2D) resulted in a further significant reduction of PDBu-stimulated release. Based on the findings in Figure 2B, these results suggest that the additional reduction in release from paclitaxel-treated neurons following inhibition of PKCα is due to the residual activity of PKCβI/II (Figure 2C); likewise, the additional reduction in release following inhibition of PKCβI/II is due to the residual activity of PKCα (Figure 2D). Overall, these sets of data suggest that inhibition of both PKCα and PKCβI/II activity, as oppsosed to inhibition of a single kinase, is responsible for reduction in CGRP release induced by paclitaxel. As such, the two kinases share a common signaling pathway to elicit changes in sensory neuronal function following chronic treatment with paclitaxel.
3.3. Chronic treatment with paclitaxel decreases the basal activity of conventional PKC isozymes, α and βI/II, as indicated by a reduction in the phosphorylation of cPKC substrates
Researchers measure PKC activity by examining the phosphorylation of downstream PKC substrates (Numazaki et al., 2002, Zhang et al., 2002, Bhave et al., 2003, Ferreira et al., 2005, Mandadi et al., 2006, Jeske et al., 2009). To determine whether paclitaxel altered the basal activity of PKC, neuronal cultures were treated with 300 nM paclitaxel for 3 or 5 days and the phosphorylation of PKC substrates was determined using an anti-phospho (Ser) PKC substrate antibody. This antibody recognizes PKC substrates when phosphorylated at serine residues surrounded by arginine or lysine at positions −2 and +2 and a hydrophobic residue at +1. Studies show that PKC isozymes can be distinguished from one another based on their substrate sequence motif (Nishikawa et al., 1997). All PKC isozymes select for substrates with a hydrophobic residue (i.e. Phe) at +1 and basic residues at −6, −4, −2, however, conventional PKC isozymes (α, βI, βII, and γ) and the novel PKC isozyme (η) have been shown to have higher selectivity for substrates with basic residues (i.e. arginine and lysine) at positions +2 and +3 (Nishikawa et al., 1997). We found that treatment with 300 nM paclitaxel for 3 and 5 days decreased the phosphorylation of cPKC substrates (Figure 3A). These data strongly indicate that paclitaxel decreases the activity of cPKC with subsequent decreases in the phosphorylation of downstream cPKC substrates. As a positive control, neuronal cultures were treated with the c/nPKC activator, PDBu (100 nM and 1 μM), for 5, 10 or 30 minutes and the phosphorylation of cPKC substrates was determined using the anti-phospho (Ser) PKC antibody. As expected, treatment with PDBu, at all tested concentrations and time-points, increased phosphorylation of cPKC substrates (Figure 3B), confirming that phorbol ester-induced activation of PKC enhances the phosphorylation of cPKC substrates in this assay.
3.4. Chronic treatment with paclitaxel does not decrease the basal protein expression of conventional PKC isozymes, α, βI or βII
To ascertain whether the decrease in CGRP release induced by paclitaxel is due to a reduction in the basal protein expression of PKC α, βI and/or βII, cultured sensory neurons were treated with 300 nM paclitaxel for 3 days and the protein expression in whole cell lysates was determined using Western blots. For our studies, the term “basal” encapsulates the cellular environment of cells treated with paclitaxel in the absence of a stimulatory agent. Treatment with 300 nM paclitaxel for 3 days did not attenuate the expression of PKCα (1.0 ± 0.1 arbitrary units; Figures 4A and 4B), PKCβI (1.6 ± 0.4 a.u.; Figures 4C and 4D) or PKCβII (1.1 ± 0.1 a.u.; Figures 4E and 4F) compared to vehicle (1.0 ± 0.0 a.u.). These findings suggest that changes in neuronal sensitivity induced by paclitaxel cannot be explained by a reduction in protein levels of the conventional PKC isozymes, α, βI and βII.
Figure 4.
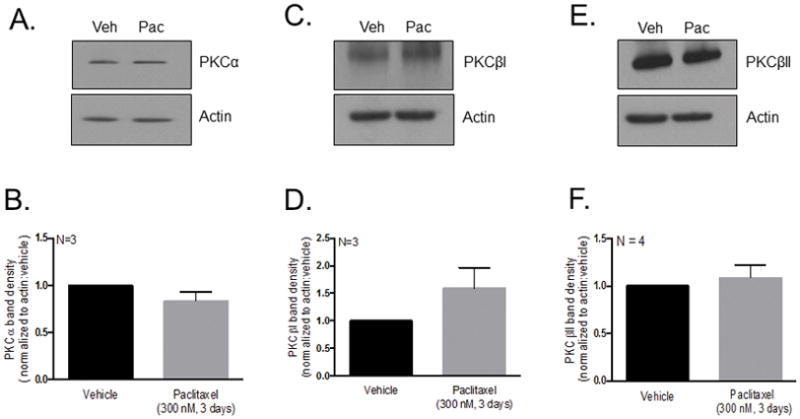
Chronic treatment with paclitaxel does not reduce basal PKC α, βI or βII, immunoreactivity in cultured sensory neurons. (A, C, E) Representative western blot images showing immunoreactivity for PKC α, βI and βII in whole cell lysates in neurons treated with vehicle and paclitaxel (300 nM, 3 days). (B, D, F) Densitometry analysis showing no significant differences in PKC immunoreactivity normalized to actin in paclitaxel-treated (300 nM, 3 days) neurons compared to vehicle-treated neurons using a t-test (N = 3–4, a.u. ± SEM). Veh - Vehicle; Pac -Paclitaxel.
3.5. Chronic treatment with paclitaxel does not attenuate the basal phosphorylation or membrane localization of conventional PKC isozymes, α, βI or βII
The phosphorylation status and localization of conventional PKC isozymes (cPKC) are important determinants of cPKC activity. Lack of phosphorylation at three conserved phosphorylation sites in the catalytic domain predisposes PKC for degradation (Gould and Newton, 2008) and membrane translocation upon cPKC activation serves to position cPKC in close proximity to their downstream substrates (Mochly-Rosen and Gordon, 1998, Jaken and Parker, 2000). To determine whether loss of phosphorylation or membrane localization under basal conditions accounted for the reduction of PDBu-stimulated CGRP release induced by paclitaxel, neuronal cultures were treated with 300 nM paclitaxel for 3 days and differential centrifugation assays were performed to isolate the cytosolic and membrane protein fractions. Actin and Na+/K+ ATPase α were used as loading controls for cytosolic and membrane protein fractions, respectively. Treatment with 300 nM paclitaxel for 3 days did not significantly alter the levels of phosphorylated PKCα in the cytosol (1.04 ± 0.1 a.u.) or membrane (1.48 ± 0.7 a.u.) compared to vehicle-treated neurons (1.00 ± 0.0 a.u.; Figures 5A and 5B). There also were no significant differences in total PKCα in the cytosol (1.07 ± a.u.) or membrane (0.98 ± a.u.) following treatment with paclitaxel compared to vehicle-treated neurons (1.00 ± 0.0 a.u.; Figures 5A and 5B). Similarly, treatment with 300 nM paclitaxel for 3 days did not significantly alter the levels of phosphorylated PKCβI in the cytosol (1.24 ± 0.0 a.u.) or membrane (1.71 ± 0.4 a.u.) or total PKCβI in the cytosol (1.15 ± 0.1 a.u.) or membrane (1.41 ± 0.3 a.u.) following treatment with paclitaxel compared to vehicle-treated neurons (1.00 ± 0.0 a.u.; Figures 5C and 5D). There also were no significant differences in phosphorylated PKCβII in the cytosol (1.15 ± 0.2 a.u.) or membrane (0.96 ± 0.1 a.u.) or total PKCβII in the cytosol (1.16 ± 0.1 a.u.) or membrane (0.89 ± 0.2 a.u.) following treatment with paclitaxel (300 nM, 3 days) compared to vehicle-treated neurons (1.00 ± 0.0 a.u.; Figures 5E and 5F). These data indicate that under basal conditions, chronic treatment with paclitaxel does not alter the phosphorylation status of PKC α, βI, or βII demonstrating that these isozymes are stable and catalytically competent. We also show that paclitaxel does not alter the membrane localization of PKC α, βI, or βII under basal conditions.
Figure 5.
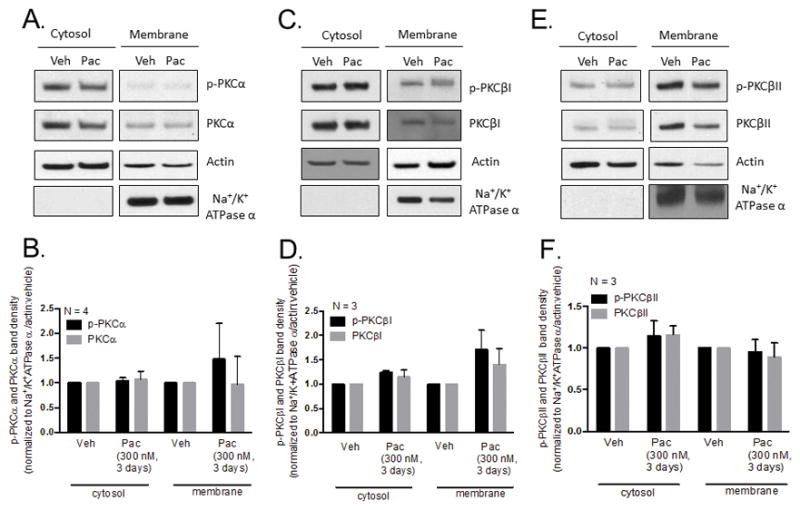
Chronic treatment with paclitaxel does not attenuate the basal phosphorylation level or membrane localization of PKC α, βI or βII in cultured sensory neurons. (A, C, E) Representative western blot images showing immunoreactivity for PKC α, βI and βII in cytosolic and membrane fractions of neurons treated with vehicle and paclitaxel (300 nM, 3 days). (B, D, F) Densitometry analysis showing no significant differences in PKC immunoreactivity in paclitaxel-treated (300 nM, 3 days) neurons compared to vehicle-treated neurons in cytosolic and membrane fractions when normalized to actin and Na+/K+ ATPase α, respectively. Significance was determined using a two-way ANOVA with Tukey’s post-hoc test (N = 3–4, a.u. ± SEM). Veh - Vehicle; Pac -Paclitaxel.
3.6. Chronic treatment with paclitaxel inhibits PDBu-stimulated membrane localization of conventional PKC isozymes, α, βI or βII
We demonstrated that chronic treatment with paclitaxel does not alter the membrane localization of phosphorylated PKC α, βI, or βII under basal conditions. However, activation of PKC and subsequent membrane translocation and localization at cellular membranes is a short-lived transient response (Cesare et al., 1999, Gould and Newton, 2008). Although we did not detect paclitaxel-induced changes in membrane localization under basal conditions, the sensitivity of the translocation assay is too low to detect minor shifts in PKC localization. It was therefore important for us to ascertain whether chronic treatment with paclitaxel altered membrane translocation of phosphorylated PKC α, βI and βII following acute PDBu-induced activation of PKC. Cultured neurons were treated with 300 nM paclitaxel for 3 days or 5 days, and stimulated with 300 nM PDBu for 2 minutes. The changes in phosphorylated PKC α, βI and βII localization at the plasma membrane were determined using immunofluorescence in which neurons where stained with a pool of fluorescently labeled PKC α, βI, and βII antibodies. Studies have previously shown that PDBu-induced activation of c/nPKC increases membrane translocation of c/nPKC (Tsutsumi et al., 1993, Cesare et al., 1999, Zhu et al., 2007). Similarly, we demonstrate that treatment with 300 nM PDBu for 2 minutes increased the number of neuronal cell bodies which exhibit membrane localization of phosphorylated PKC α, βI, and βII from 0.0 ± 0.0 to 31.6 ± 3.0% (Figure 6A and 6B). However, treatment with 300 nM paclitaxel for 3 days and 5 days decreased the percentage of cells with membrane localized phosphorylated PKC α, βI and βII to 13.7 ± 5.0 and 16.8 ± 8.0%, respectively (Figure 6A and 6B). Neither 3 day nor 5 day treatment with paclitaxel altered the percentage of cells with membrane localized phosphorylated PKC α/βI/βII under basal conditions (Figure 6A and 6B), confirming our findings in Figures 5A–E. These data indicate that paclitaxel attenuates the membrane localization of phosphorylated PKC α, βI and βII under stimulatory conditions and demonstrates that the loss of functional PKC effects could be due to misdirected cPKC localization.
Figure 6.
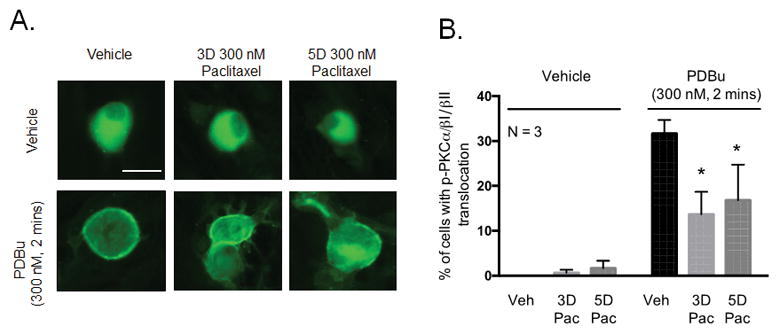
Chronic treatment with paclitaxel decreases PDBu-stimulated membrane localization of PKC α, βI and βII in cultured sensory neurons. (A) Representative fluorescent images showing membrane localization of immunoreactive phosphorylated PKC α, βI and βII in neurons treated with paclitaxel (300 nM, 3 days or 5 days) following stimulation with PDBu (300 nM, 2 minutes). (B) Graphical analysis showing the number of neuronal cell bodies demonstrating translocation of phosphorylated PKC α, βI and βII in paclitaxel-treated neurons (300 nM, 3 and 5 days) following stimulation with PDBu (300 nM, 2 minutes) expressed as % of phospho- PKC α/βI/II translocation. An * indicates a significant decrease in the number of cells with PKC α, βI and βII translocation. Significance was determined using a two-way ANOVA with Dunnett’s post-hoc test (p < 0.05, N = 3). Scale bar = 20 μm. Veh - Vehicle; Pac –Paclitaxel, 3D – 3 days, 5D – 5 days.
3.7. Chronic treatment with paclitaxel inhibits the activity of conventional PKC isozymes, α and βI/II, to elicit a decrease in capsaicin-stimulated CGRP release
It is well-established that the function of TRPV1 channels is modulated by conventional and novel PKC isozymes (c/nPKC). Studies have shown that phorbol-ester induced activation of c/nPKC enhanced the phosphorylation of TRPV1 channels (Jeske et al., 2009) and augmented capsaicin-induced currents in sensory neurons (Vellani et al., 2001, Zhou et al., 2001), whereas downregulation of c/nPKC by chronic phorbol ester treatment attenuated capsaicin-stimulated CGRP release (Barber and Vasko, 1996). Furthermore, there is strong evidence to support that PDBu-induced CGRP release is mediated by TRPV1 activation in spinal cord tissue (Mogg et al., 2013). In these studies, it was shown that PDBu- and capsaicin-stimulated CGRP release was attenuated following pre-treatment with multiple TRPV1 antagonists (BCTC, AMG517, A425619, A784168, AMG 49a, AMG9810, JNJ 17203212, SB705498, Neurogen) and was also blocked in spinal cord tissue from TRPV1 null mutant mice (Mogg et al., 2013).
To assess whether PDBu-stimulated release was dependent on TRPV1 channels in cultured sensory neurons, cultured sensory neurons were pre-treated with increasing concentrations of the TRPV1 antagonist, 4-(3-Chloro-2-pyridinyl)-N-[4-(1,1 dimethylethyl)phenyl]-1-piperazinecarboxamide (BCTC), for 10 minutes prior to stimulation with 10 nM PDBu. Pre-treatment with 1 nM BCTC significantly attenuated PDBu-stimulated release from 9.2 ± 1.2 to 2.3 ± 0.5% of total content (Figure 7A); neither 0.01 nM nor 0.1 nM BCTC altered PDBu-stimulated release. The basal release following pre-treatment with 0.01 nM (0.8 ± 0.1), 0.1 nM (1.2 ± 0.1) and 1 nM BCTC (0.9 ± 0.1% of total content) was not significantly different when compared to control (0.7 ± 0.1% of total content; Figure 7A). These findings indicate that PDBu-stimulated release is mediated by activation of TRPV1 channels.
Figure 7.
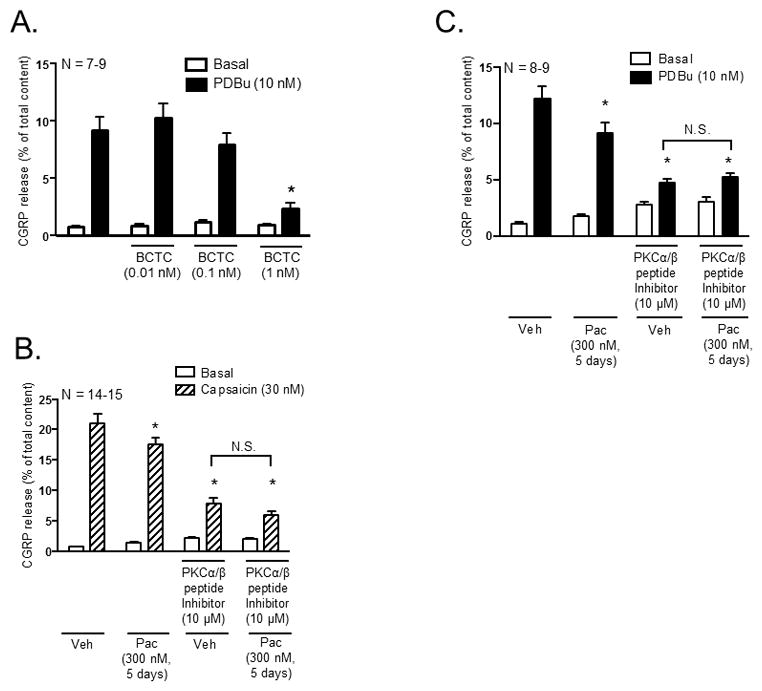
Inhibition of PKCα and PKCβI/II activity mediates the decrease in capsaicin-stimulated CGRP release following chronic exposure to paclitaxel in cultured sensory neurons. Each column represents the mean ± SEM of basal (white columns), capsaicin-stimulated (striped columns) or PDBu-stimulated (black columns) CGRP release expressed as % of total content. (A) Naïve cultures were pre-treated with the TRPV1 antagonist, BCTC, for 10 minutes prior to stimulation with capsaicin (30 nM). An * indicates a significant decrease in capsaicin-stimulated release in neurons pre-treated with BCTC compared to control neurons (p < 0.05, N = 7–9). Cultures were exposed to 300 nM paclitaxel for 3 days and pre-treated with a myristolyated PKCα/β peptide inhibitor (10 μM) for 10 minutes prior to stimulation with (B) 30 nM capsaicin or (C) 10 nM PDBu. An * indicates a decrease in (B) capsaicin-stimulated and (C) PDBu-stimulated release in paclitaxel-only treated neurons, vehicle- and paclitaxel treated neurons pre-treated with the myristolyated PKCα/β peptide inhibitor compared to vehicle-only treated neurons. Significance was determined using a two-way ANOVA with Tukey’s post-hoc test (p < 0.05, N = 8–15). N.S. – not significant.
Since our previous studies demonstrated that chronic treatment with paclitaxel (300 nM; 3 and 5 days) attenuates capsaicin-stimulated peptide release (Pittman et al., 2014), we wanted to ascertain whether conventional PKCα and PKCβI/II were responsible for the changes in capsaicin-stimulated CGRP release following chronic treatment with paclitaxel. Cultured sensory neurons were treated with 300 nM paclitaxel for 5 days and pre-treated with the myristolyated PKCα/β peptide inhbitior (10 μM) for 10 minutes prior to stimulation with 30 nM capsaicin. Treatment with paclitaxel significantly attenuated capsaicin-stimulated release from 21.1 ± 1.5 (vehicle-only) to 17.6 ± 1.0% of total content (Figure 7B), confirming our previous findings (Pittman et al., 2014). Pre-treatment with the PKCα/β peptide inhibitor in vehicle-treated neurons significantly attenuated capsaicin-stimulated release to 7.9 ± 1.0% of total content compared to vehicle-only treated neurons, suggesting that a major component of capsaicin-stimulated release is due to PKC α, βI and βII. However, pre-treatment with the PKCα/β peptide inhibitor did not significantly alter capsaicin-stimulated release in paclitaxel-treated neurons (6.0 ± 0.7% of total content) compared to vehicle-treated neurons (7.9 ± 1.0% of total content; Figure 7B), suggesting that paclitaxel inhibits that activity of PKCα and PKCβI/II to elicit a decrease in capsaicin-stimulated peptide release.
We next verified whether inhibition of PKCα and PKCβI/II activity mediates loss of PDBu-stimulated release following treatment with 300 nM paclitaxel for 5 days, as demonstrated using the 3-day treatment paradigm. Treatment with 300 nM paclitaxel for 5 days significantly attenuated PDBu-stimulated release from 12.2 ± 1.1 (vehicle-only) to 9.1 ± 0.9% of total content (Figure 7C), in a manner analogous to that observed with the 3 day treatment (see Figures 2B–D). As expected, pre-treatment with the 10 μM myristolyated PKCα/β peptide inhibitor completely abolished PDBu-stimulated release; PDBu-stimulated release decreased from 12.2 ± 1.1 (vehicle-only) to 4.7 ± 0.4% of total content (Figure 7C). Pre-treatment with the PKCα/β peptide inhibitor did not significantly alter PDBu-stimulated release in paclitaxel-treated neurons (5.2 ± 0.4% of total content) compared to vehicle-treated neurons (4.7 ± 0.4% of total content; Figure 7C). These findings are analgous to that observed following combined pre-treatment with the PKCα and PKCβI/II inhibitors, Bis VIII and LY333531, following treatment with paclitaxel for 3 days (see Figure 2B). Similar to the 3-day paclitaxel treatment paradigm, a more prolonged exposure to paclitaxel decreases the activity of PKCα and PKCβI/II to elicit an attenuation in the stimulated release of CGRP.
4. Discussion
Our work indicates that PKCα and PKCβI/II are critical mediators of changes in neuronal sensitivity in TRPV1 positive sensory neurons following chronic exposure to paclitaxel. Using isolated sensory neurons, we show that chronic treatment with paclitaxel inhibits the activity of PKCα and PKCβI/II and the membrane localization of phosphorylated PKCα and PKCβI/II to elicit a reduction in the stimulated release of neuropeptide from capsaicin-sensitive neurons. These findings provide a mechanistic understanding of the effects of chronic exposure to paclitaxel on sensory neuronal function.
To determine the effects of paclitaxel on PKC-modulation of sensory neurons, our model relied upon measuring changes in neuropeptide release following stimulation with the phorbol ester, PDBu, in cultured sensory neurons. PDBu is a potent stimulator of CGRP release from sensory neurons, however it is evident from our experiments that the levels of release are variable among different release assay experiments. These differences in release can be explained by very slight differences in cell density. There is an approximate density of 30,000 cells per well, however, slight differences in the cell density will affect the total content of CGRP per well. To control for these differences, each individual experiment consisted of vehicle-treated (control) neurons. Furthermore, the data was normalized to the total content of CGRP per well.
PKC isozymes are differentially expressed in various cell types, exhibit differential sensitivity to calcium, and interact with different substrates (Way et al., 2000). It was imperative for us to first ascertain which class of PKC isozymes was responsible for the actions of PDBu on neuropeptide release. Pharmacological manipulation is limited by narrow ranges of concentration at which many of the small molecule inhibitors demonstrate selectivity for a given PKC isozyme. Therefore, in addition to using multiple small molecule inhibitors, we also used a myristoylated PKCα/β peptide inhibitor to discern the contribution of specific isozymes. We did not employ a genetic approach to modulate PKC expression due to the propensity for PKC isozyme compensation and PKC cross-modulation of sub-cellular localization of other isozymes (Collazos et al., 2006). Pre-treatment with 100 nM Gö6976, a selective inhibitor of conventional PKC isozymes (cPKC) significantly reduced PDBu-stimulated release. Studies have shown that concentrations up to 3 μM selectively inhibit cPKC while having no effect on the activity of novel PKC isozymes (Martiny-Baron et al., 1993), therefore our findings indicate that novel PKC isozymes are not involved in mediating PDBu-stimulated neuropeptide release from isolated sensory neurons. Using the PKCα/β peptide inhibitor, which consists of a sequence derived from homologous regions within the pseudosubstrate of PKC isozymes α, βI and βII (House and Kemp, 1987, Eichholtz et al., 1993), we were able to further isolate and examine the contributions of specific conventional PKC isozymes. The PKCα/β peptide inhibitor does not alter the activity of PKCγ due to differences in amino acid sequences, therefore allowing us to specifically examine the contributions of PKCα and PKCβI/II. Pre-treatment with the PKCα/β peptide inhibitor fully abolished PDBu-stimulated release indicating that PKCα and PKCβI/II were responsible for mediating the actions of PDBu. These findings were substantiated using a combined pre-treatment with selective small molecule inhibitors of PKCα (Bis VIII) and PKCβI/II (LY333531). Importantly, in paclitaxel-treated neurons, combined pre-treatment with PKCα (Bis VIII) and PKCβI/II (LY333531) inhibitors and pre-treatment with the PKCα/β peptide inhibitor, did not further alter PDBu-stimulated release, suggesting that loss of PKCα and PKCβI/II activity was responsible for the loss of neuronal sensitivity induced by paclitaxel. In addition to the use of selective PKCα and PKCβI/II inhiibitors, we demonstrated that paclitaxel has a direct effect on the activity of cPKC using an antibody that recognizes the phosphorylated levels of cPKC substrates. We showed that paclitaxel decreased the phosphorylated levels of cPKC substrates, therefore indicating loss of cPKC activity. Our data shows that PKCα and PKCβI/II are the major components of PDBu-evoked neuropeptide release, and that paclitaxel inhibits the activity of these kinases to elicit a reduction in neuronal sensitivity.
In addition to modulating the activity of cPKC, it is plausible that paclitaxel alters the basal expression, phosphorylation status and/or localization of cPKC. The term “basal” is used to describe the cellular environment of cells that were treated with paclitaxel for days, but were not exposed to a stimulatory agent. We first investigated whether paclitaxel modulated total cPKC protein levels under basal conditions. A reduction in cPKC protein levels could provide an explanation for the loss in neuropeptide release induced by chronic treatment with paclitaxel since PDBu-stimulated release is dependent on PKC activity, however, we observed no changes in the total protein levels of either PKC α, βI or βII. While total cPKC levels remained unchanged, it was plausible that paclitaxel attenuated the phosphorylated levels of cPKC and/or inhibited the localization of phosphorylated cPKC at cellular membranes under basal conditions. Studies have shown that phosphorylation of residues in the kinase domain, turn motif and hydrophobic motif, are required for catalytic competence and stability of PKC (Shirai and Saito, 2002, Gould and Newton, 2008) whereas a lack of phosphorylation predisposes the protein to undergo degradation via the ubiquitin-proteasome pathway (Lu et al., 1998, Gould and Newton, 2008). Following activation, phosphorylated cPKC translocate from the cytosol to cellular membranes positioning them in close proximity to membrane-bound substrates involved in altering neuronal sensitivity. We found that paclitaxel did not decrease the degree of phosphorylation of either PKC α, βI or βII in the cytosolic or membrane fractions under basal conditions. Studies examining the acute (10 minutes –1 hour) effects of paclitaxel have demonstrated enhanced membrane localization of conventional PKC isozymes in cultured DRG sensory neurons (He and Wang, 2015). Another investigator found that treatment with paclitaxel for 2–10 minutes also enhanced translocation of PKCβ, using a pan PKCβ antibody, to the membrane fraction while having no effect on PKCα localization (Miyano et al., 2009). It is apparent that acute exposure to paclitaxel enhances the translocation of PKCβII to cellular membranes, however, membrane localization of PKCβII is not maintained following chronic exposure to paclitaxel under basal conditions. Given the physiological nature of PKC membrane localization, it is somewhat unsurprising that we did not observe any effects on cPKC translocation following chronic treatment with paclitaxel under basal conditions. PKC localization at cellular membranes upon activation is a short-lived response (Cesare et al., 1999, Gould and Newton, 2008). In fact, prolonged localization of activated PKC at cellular membranes results in downregulation of PKC (Gould and Newton, 2008). Having determined that cPKC was not downregulated, it became apparent that our experimental design examining changes in phosphorylated cPKC localization under basal conditions was not sensitive enough to detect small changes in PKC localization, given the temporal nature of PKC activation and the potential short time intervals of membrane localization. Therefore, to better understand the effects of chronic paclitaxel treatment on membrane localization of phosphorylated cPKC, neurons treated with paclitaxel were stimulated acutely (2 minutes) with PDBu and the percentage of cells that exhibited cPKC membrane localization was determined. We demonstrated that chronic treatment with paclitaxel decreased phorbol ester-stimulated membrane localization of phosphorylated PKC α, βI and βII. The observation that chronic treatment with paclitaxel (under basal conditions) decreased cPKC substrate phosphorylation while having no effects on phosphorylated cPKC membrane localization highlights the temporal nature of PKC signaling. We believe that the effects of paclitaxel on cPKC substrate phosphorylation is a composite of phosphorylation events over time. On the other hand, because of the short-lived nature of cPKC membrane localization, changes in membrane translocation of phosphorylated PKC after chronic treatment with paclitaxel would be undetectable in the absence of an acute stimulus to activate PKC.
There is precedence in the literature to support that the activation of PKC differentially alters membrane ion channel function via phosphorylation. Studies have found that N- and L-type calcium currents are enhanced following activation of PKC whereas sodium currents are attenuated (Yang and Tsien, 1993, Hall et al., 1995, Chen et al., 2005). Notably, the activity of the nociceptive ligand-gated TRPV1 ion channel (Caterina et al., 1997) is also modulated by PKC phosphorylation. Under physiological conditions, PKC dependent phosphorylation of TRPV1 occurs downstream of Gq coupled receptor activation by inflammatory substances such as bradykinin (Cesare et al., 1999, Sugiura et al., 2002). Studies have shown that enhancing PKC activity, either via phorbol ester activation or via overexpression of a constitutively active PKCε construct, potentiates capsaicin- and heat-activated TRPV1 currents (Cesare et al., 1999, Numazaki et al., 2002, Bhave et al., 2003) whereas downregulation of PKCα following chronic treatment with PDBu attenuates PDBu-stimulated calcium uptake in NIH 3T3 cells expressing TRPV1 (Olah et al., 2002). More recently, it was found that PKCβII phosphorylation of TRPV1 increased the sensitivity of the channel (Li et al., 2014). Unlike PKCε, a novel PKC isozyme which is believed to be positioned in close proximity to TRPV1 through A-kinase anchoring protein 150 (Jeske et al., 2009), PKCβII directly binds to TRPV1 to alter its function (Li et al., 2014).
Our previous studies have shown that paclitaxel has concentration- and time-dependent effects on TRPV1 function in cultured sensory neurons. Treatment with 300 nM paclitaxel for 5 days decreased capsaicin-stimulated release, whereas treatment with lower concentrations (10 nM) increased capsaicin-stimulated release (Pittman et al., 2014). Using our in vitro culture system, we recapitulated the gain and loss of TRPV1 function that is observed in animal models of paclitaxel-induced peripheral neuropathy (Campana et al., 1998, Authier et al., 2000, Chen et al., 2011, Gracias et al., 2011, Hara et al., 2013, Li et al., 2015). In this manuscript, we chose to focus on the importance of PKC in the loss of TRPV1 function in neurons treated with 300 nM paclitaxel for 3 or 5 days. However, we do speculate that PKC has a role in the gain of TRPV1 function upon chronic treatment with paclitaxel since treatment with 10 nM paclitaxel for 3 days increased PDBu-stimulated peptide release in cultured sensory neurons (data not shown). Given the numerous studies implicating PKC in modulating TRPV1 function, it was unsurprising that we found a significant attenuation of capsaicin-evoked release upon pre-treatment with the myristolyated PKCα/β peptide inhibitor. These findings strongly demonstrate that neuropeptide release upon TRPV1 activation is largely mediated by PKCα and PKCβI/II in isolated sensory neurons. Our additional studies in paclitaxel-treated neurons show that the reduction in capsaicin-stimulated release is due to loss of PKCα and PKCβI/II activity. We speculate that paclitaxel-induced inhibition of PKCα and PKCβI/II activity causes a decrease in TRPV1 phosphorylation resulting in a subsequent reduction of neuropeptide release. Such reasoning could provide a plausible explanation for the loss of TRPV1 function observed following chronic exposure to paclitaxel. Another possible explanation for the reduction in capsaicin-stimulated release is the desensitization of TRPV1 channels. Various signaling proteins have been implicated in regulating the desensitization of TRPV1 channels. Activation of protein kinase A (PKA) and inhibition of the phosphatase, calcineurin, have both been shown to reduce capsaicin-induced TRPV1 desensitization (Mohapatra and Nau, 2003, 2005, Por et al., 2010, Sanz-Salvador et al., 2012). Unlike with PKA and calcineurin, there has been conflicting data regarding the role of PKC in TRPV1 desensitization. It was found that phorbol ester-induced activation of PKC reversed TRPV1 desensitization in a calcium-dependent manner, whereas inhibition of PKC, using an inhibitory peptide, prevented the recovery of TRPV1 sensitization (Mandadi et al., 2004, Mandadi et al., 2006). While these findings oppose the observations of others who found no involvement of PKC in the reversal of TRPV1 desensitization (Mohapatra and Nau, 2003, 2005), these studies suggest the decrease in capsaicin-stimulated release following chronic exposure to paclitaxel could be attributable to a loss in the recovery of desensitized TRPV1 channels via decreased activity of PKC. Although not within the scope of this work, future studies will examine the mechanism by which loss of PKC activity alters TRPV1 function.
The observed loss of TRPV1 function in isolated sensory neurons is analogous to findings in in vivo models of paclitaxel-induced peripheral neuropathy. It was shown that systemic injections of paclitaxel (cumulative doses ranging between 18 and 80 mg/kg) induced thermal hypoalgesia (Campana et al., 1998, Authier et al., 2000) and (4 mg/kg cumulative dose) attenuated capsaicin-evoked blood flow in the rat hindpaw (Gracias et al., 2011). These in vivo studies demonstrate that paclitaxel decreased the function of small diameter TRPV1-expressing neurons. Interestingly, a recent study found that the loss of peptidergic TRPV1-expressing sensory neurons unmasked cold hypersensitivity (McCoy et al., 2013), a major symptom of paclitaxel-induced peripheral neuropathy (Dougherty et al., 2004). Therefore, it is plausible that the observed loss of TRPV1 function following chronic treatment with paclitaxel enhances neuronal sensitivity to cold temperatures. In conjunction with our studies, the aforementioned findings highlight a possible physiological role for PKC α, βI and βII as critical regulators of hot, cold and mechanical sensitivity following chronic exposure to paclitaxel in patients.
In conclusion, our studies show that the conventional PKC isozymes, α, βI and βII, are critical mediators of changes in neuronal sensitivity following chronic treatment with paclitaxel. Chronic treatment with paclitaxel attenuated the activity and membrane localization of PKC α, βI and βII to elicit a reduction in stimulated peptide release from capsaicin-sensitive neurons.
Highlights.
Stimulated CGRP release was measured as an index of neuronal sensitivity.
Paclitaxel attenuates phorbol ester- and TRPV1-stimulated release.
Paclitaxel inhibits cPKC activity to elicit changes in stimulated release.
Paclitaxel attenuates phorbol ester-stimulated cPKC membrane translocation.
Paclitaxel does not affect basal cPKC expression, phosphorylation or localization.
Acknowledgments
We would like to thank Dr. Michael Vasko and Dr. Travis Jerde for their invaluable critical review of this manuscript. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for profit sectors.
Abbreviations
- Pac
paclitaxel
- c/nPKC
conventional and novel PKC isozymes
- cPKC
conventional PKC isozymes
- DRG
dorsal root ganglia
- TRPV1
Transient Receptor Potential Vanilloid 1
- CGRP
calcitonin gene-related peptide
- PDBu
phorbol 12,13-dibutyrate
- BCTC
4-(3-Chloro-2-pyridinyl)-N-[4-(1,1-dimethylethyl)phenyl]-1-piperazinecarboxamide
- MPL
methylpyrrolidone
References
- Authier N, Gillet JP, Fialip J, Eschalier A, Coudore F. Description of a short-term Taxol-induced nociceptive neuropathy in rats. Brain research. 2000;887:239–249. doi: 10.1016/s0006-8993(00)02910-3. [DOI] [PubMed] [Google Scholar]
- Barber LA, Vasko MR. Activation of protein kinase C augments peptide release from rat sensory neurons. Journal of neurochemistry. 1996;67:72–80. doi: 10.1046/j.1471-4159.1996.67010072.x. [DOI] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RWt. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci U S A. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Campana WM, Eskeland N, Calcutt NA, Misasi R, Myers RR, O’Brien JS. Prosaptide prevents paclitaxel neurotoxicity. Neurotoxicology. 1998;19:237–244. [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cantrell AR, Messing RO, Scheuer T, Catterall WA. Specific modulation of Na+ channels in hippocampal neurons by protein kinase C epsilon. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:507–513. doi: 10.1523/JNEUROSCI.4089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience. 2011;193:440–451. doi: 10.1016/j.neuroscience.2011.06.085. [DOI] [PubMed] [Google Scholar]
- Collazos A, Diouf B, Guerineau NC, Quittau-Prevostel C, Peter M, Coudane F, Hollande F, Joubert D. A spatiotemporally coordinated cascade of protein kinase C activation controls isoform-selective translocation. Molecular and cellular biology. 2006;26:2247–2261. doi: 10.1128/MCB.26.6.2247-2261.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Gonzalez F, Kazanietz MG. C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochimica et biophysica acta. 2006;1761:827–837. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Dina OA, Chen X, Reichling D, Levine JD. Role of protein kinase Cepsilon and protein kinase A in a model of paclitaxel-induced painful peripheral neuropathy in the rat. Neuroscience. 2001;108:507–515. doi: 10.1016/s0306-4522(01)00425-0. [DOI] [PubMed] [Google Scholar]
- Dougherty PM, Cata JP, Cordella JV, Burton A, Weng HR. Taxol-induced sensory disturbance is characterized by preferential impairment of myelinated fiber function in cancer patients. Pain. 2004;109:132–142. doi: 10.1016/j.pain.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Eichholtz T, de Bont DB, de Widt J, Liskamp RM, Ploegh HL. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. The Journal of biological chemistry. 1993;268:1982–1986. [PubMed] [Google Scholar]
- Ferreira J, Triches KM, Medeiros R, Calixto JB. Mechanisms involved in the nociception produced by peripheral protein kinase c activation in mice. Pain. 2005;117:171–181. doi: 10.1016/j.pain.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122:245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth PA, Balmaceda C, Peterson K, Seidman AD, Brasher P, DeAngelis LM. Prospective study of paclitaxel-induced peripheral neuropathy with quantitative sensory testing. J Neurooncol. 1997;35:47–53. doi: 10.1023/a:1005805907311. [DOI] [PubMed] [Google Scholar]
- Gould CM, Newton AC. The life and death of protein kinase C. Current drug targets. 2008;9:614–625. doi: 10.2174/138945008785132411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracias NG, Cummins TR, Kelley MR, Basile DP, Iqbal T, Vasko MR. Vasodilatation in the rat dorsal hindpaw induced by activation of sensory neurons is reduced by paclitaxel. Neurotoxicology. 2011;32:140–149. doi: 10.1016/j.neuro.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. The Journal of biological chemistry. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- Gray RD, Lucas CD, Mackellar A, Li F, Hiersemenzel K, Haslett C, Davidson DJ, Rossi AG. Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. Journal of inflammation (London, England) 2013;10:12. doi: 10.1186/1476-9255-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KE, Browning MD, Dudek EM, Macdonald RL. Enhancement of high threshold calcium currents in rat primary afferent neurons by constitutively active protein kinase C. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1995;15:6069–6076. doi: 10.1523/JNEUROSCI.15-09-06069.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Chiba T, Abe K, Makabe A, Ikeno S, Kawakami K, Utsunomiya I, Hama T, Taguchi K. Effect of paclitaxel on transient receptor potential vanilloid 1 in rat dorsal root ganglion. Pain. 2013;154:882–889. doi: 10.1016/j.pain.2013.02.023. [DOI] [PubMed] [Google Scholar]
- He Y, Wang ZJ. Nociceptor Beta II, Delta, and Epsilon Isoforms of PKC Differentially Mediate Paclitaxel-Induced Spontaneous and Evoked Pain. J Neurosci. 2015;35:4614–4625. doi: 10.1523/JNEUROSCI.1580-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershman DL, Weimer LH, Wang A, Kranwinkel G, Brafman L, Fuentes D, Awad D, Crew KD. Association between patient reported outcomes and quantitative sensory tests for measuring long-term neurotoxicity in breast cancer survivors treated with adjuvant paclitaxel chemotherapy. Breast cancer research and treatment. 2011;125:767–774. doi: 10.1007/s10549-010-1278-0. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Vasko MR. Prostacyclin enhances the evoked-release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain research. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- Holmes FA, Walters RS, Theriault RL, Forman AD, Newton LK, Raber MN, Buzdar AU, Frye DK, Hortobagyi GN. Phase II trial of taxol, an active drug in the treatment of metastatic breast cancer. Journal of the National Cancer Institute. 1991;83:1797–1805. doi: 10.1093/jnci/83.24.1797-a. [DOI] [PubMed] [Google Scholar]
- House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science (New York, NY) 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- Jaken S, Parker PJ. Protein kinase C binding partners. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22:245–254. doi: 10.1002/(SICI)1521-1878(200003)22:3<245::AID-BIES6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Jeske NA, Patwardhan AM, Ruparel NB, Akopian AN, Shapiro MS, Henry MA. A-kinase anchoring protein 150 controls protein kinase C-mediated phosphorylation and sensitization of TRPV1. Pain. 2009;146:301–307. doi: 10.1016/j.pain.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirousek MR, Gillig JR, Gonzalez CM, Heath WF, McDonald JH, 3rd, Neel DA, Rito CJ, Singh U, Stramm LE, Melikian-Badalian A, Baevsky M, Ballas LM, Hall SE, Winneroski LL, Faul MM. (S)-13-[(dimethylamino)methyl]-10,11,14,15-tetrahydro-4,9:16, 21-dimetheno-1H, 13H-dibenzo[e,k]pyrrolo[3,4-h][1,4,13]oxadiazacyclohexadecene-1,3(2H)-d ione (LY333531) and related analogues: isozyme selective inhibitors of protein kinase C beta. Journal of medicinal chemistry. 1996;39:2664–2671. doi: 10.1021/jm950588y. [DOI] [PubMed] [Google Scholar]
- Li L, Hasan R, Zhang X. The basal thermal sensitivity of the TRPV1 ion channel is determined by PKCbetaII. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:8246–8258. doi: 10.1523/JNEUROSCI.0278-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Adamek P, Zhang H, Tatsui CE, Rhines LD, Mrozkova P, Li Q, Kosturakis AK, Cassidy RM, Harrison DS, Cata JP, Sapire K, Zhang H, Kennamer-Chapman RM, Jawad AB, Ghetti A, Yan J, Palecek J, Dougherty PM. The Cancer Chemotherapeutic Paclitaxel Increases Human and Rodent Sensory Neuron Responses to TRPV1 by Activation of TLR4. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:13487–13500. doi: 10.1523/JNEUROSCI.1956-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Liu D, Hornia A, Devonish W, Pagano M, Foster DA. Activation of protein kinase C triggers its ubiquitination and degradation. Molecular and cellular biology. 1998;18:839–845. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandadi S, Numazaki M, Tominaga M, Bhat MB, Armati PJ, Roufogalis BD. Activation of protein kinase C reverses capsaicin-induced calcium-dependent desensitization of TRPV1 ion channels. Cell calcium. 2004;35:471–478. doi: 10.1016/j.ceca.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Mandadi S, Tominaga T, Numazaki M, Murayama N, Saito N, Armati PJ, Roufogalis BD, Tominaga M. Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCepsilon-mediated phosphorylation at S800. Pain. 2006;123:106–116. doi: 10.1016/j.pain.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. The Journal of biological chemistry. 1993;268:9194–9197. [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Street SE, Pribisko AL, Zheng J, Zylka MJ. Peptidergic CGRPalpha primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron. 2013;78:138–151. doi: 10.1016/j.neuron.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire WP, Rowinsky EK, Rosenshein NB, Grumbine FC, Ettinger DS, Armstrong DK, Donehower RC. Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Annals of internal medicine. 1989;111:273–279. doi: 10.7326/0003-4819-111-4-273. [DOI] [PubMed] [Google Scholar]
- Miyano K, Tang HB, Nakamura Y, Morioka N, Inoue A, Nakata Y. Paclitaxel and vinorelbine, evoked the release of substance P from cultured rat dorsal root ganglion cells through different PKC isoform-sensitive ion channels. Neuropharmacology. 2009;57:25–32. doi: 10.1016/j.neuropharm.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D, Gordon AS. Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1998;12:35–42. [PubMed] [Google Scholar]
- Mogg AJ, Mill CE, Folly EA, Beattie RE, Blanco MJ, Beck JP, Broad LM. Altered pharmacology of native rodent spinal cord TRPV1 after phosphorylation. British journal of pharmacology. 2013;168:1015–1029. doi: 10.1111/bph.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. The Journal of biological chemistry. 2003;278:50080–50090. doi: 10.1074/jbc.M306619200. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. The Journal of biological chemistry. 2005;280:13424–13432. doi: 10.1074/jbc.M410917200. [DOI] [PubMed] [Google Scholar]
- Murphy WK, Fossella FV, Winn RJ, Shin DM, Hynes HE, Gross HM, Davilla E, Leimert J, Dhingra H, Raber MN, et al. Phase II study of taxol in patients with untreated advanced non-small-cell lung cancer. Journal of the National Cancer Institute. 1993;85:384–388. doi: 10.1093/jnci/85.5.384. [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. The Journal of biological chemistry. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. The Journal of biological chemistry. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Olah Z, Karai L, Iadarola MJ. Protein kinase C(alpha) is required for vanilloid receptor 1 activation. Evidence for multiple signaling pathways. The Journal of biological chemistry. 2002;277:35752–35759. doi: 10.1074/jbc.M201551200. [DOI] [PubMed] [Google Scholar]
- Pittman SK, Gracias NG, Vasko MR, Fehrenbacher JC. Paclitaxel alters the evoked release of calcitonin gene-related peptide from rat sensory neurons in culture. Experimental neurology. 2014;253:146–153. doi: 10.1016/j.expneurol.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- Por ED, Samelson BK, Belugin S, Akopian AN, Scott JD, Jeske NA. PP2B/calcineurin-mediated desensitization of TRPV1 does not require AKAP150. The Biochemical journal. 2010;432:549–556. doi: 10.1042/BJ20100936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma TJ, Vermorken JB, Liefting AJ, Pinedo HM, Heimans JJ. Paclitaxel-induced neuropathy. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 1995;6:489–494. doi: 10.1093/oxfordjournals.annonc.a059220. [DOI] [PubMed] [Google Scholar]
- Ron D, Luo J, Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of beta protein kinase C in vivo. The Journal of biological chemistry. 1995;270:24180–24187. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- Sanz-Salvador L, Andres-Borderia A, Ferrer-Montiel A, Planells-Cases R. Agonist- and Ca2+-dependent desensitization of TRPV1 channel targets the receptor to lysosomes for degradation. The Journal of biological chemistry. 2012;287:19462–19471. doi: 10.1074/jbc.M111.289751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. Journal of biochemistry. 2002;132:663–668. doi: 10.1093/oxfordjournals.jbchem.a003271. [DOI] [PubMed] [Google Scholar]
- Silinsky EM, Searl TJ. Phorbol esters and neurotransmitter release: more than just protein kinase C? British journal of pharmacology. 2003;138:1191–1201. doi: 10.1038/sj.bjp.0705213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Tominaga M, Katsuya H, Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. Journal of neurophysiology. 2002;88:544–548. doi: 10.1152/jn.2002.88.1.544. [DOI] [PubMed] [Google Scholar]
- Supowit SC, Christensen MD, Westlund KN, Hallman DM, DiPette DJ. Dexamethasone and activators of the protein kinase A and C signal transduction pathways regulate neuronal calcitonin gene-related peptide expression and release. Brain research. 1995;686:77–86. doi: 10.1016/0006-8993(95)00461-x. [DOI] [PubMed] [Google Scholar]
- Tamura T, Sasaki Y, Nishiwaki Y, Saijo N. Phase I study of paclitaxel by three-hour infusion: hypotension just after infusion is one of the major dose-limiting toxicities. Japanese journal of cancer research : Gann. 1995;86:1203–1209. doi: 10.1111/j.1349-7006.1995.tb03316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. The Journal of biological chemistry. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Tsutsumi A, Kubo M, Fujii H, Freire-Moar J, Turck CW, Ransom JT. Regulation of protein kinase C isoform proteins in phorbol ester-stimulated Jurkat T lymphoma cells. Journal of immunology (Baltimore, Md : 1950) 1993;150:1746–1754. [PubMed] [Google Scholar]
- Uehara K, Yamagishi S, Otsuki S, Chin S, Yagihashi S. Effects of polyol pathway hyperactivity on protein kinase C activity, nociceptive peptide expression, and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes. 2004;53:3239–3247. doi: 10.2337/diabetes.53.12.3239. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. The Journal of physiology. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends in pharmacological sciences. 2000;21:181–187. doi: 10.1016/s0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
- Wiernik PH, Schwartz EL, Einzig A, Strauman JJ, Lipton RB, Dutcher JP. Phase I trial of taxol given as a 24-hour infusion every 21 days: responses observed in metastatic melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1987;5:1232–1239. doi: 10.1200/JCO.1987.5.8.1232. [DOI] [PubMed] [Google Scholar]
- Wilkinson SE, Parker PJ, Nixon JS. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. The Biochemical journal. 1993;294( Pt 2):335–337. doi: 10.1042/bj2940335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe GI, Trivedi JR. Painful peripheral neuropathy and its nonsurgical treatment. Muscle & nerve. 2004;30:3–19. doi: 10.1002/mus.20057. [DOI] [PubMed] [Google Scholar]
- Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zha X, Tan Y, Hornbeck PV, Mastrangelo AJ, Alessi DR, Polakiewicz RD, Comb MJ. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. The Journal of biological chemistry. 2002;277:39379–39387. doi: 10.1074/jbc.M206399200. [DOI] [PubMed] [Google Scholar]
- Zhou W, Wang XL, Lamping KG, Lee HC. Inhibition of protein kinase Cbeta protects against diabetes-induced impairment in arachidonic acid dilation of small coronary arteries. The Journal of pharmacology and experimental therapeutics. 2006;319:199–207. doi: 10.1124/jpet.106.106666. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou ZS, Zhao ZQ. PKC regulates capsaicin-induced currents of dorsal root ganglion neurons in rats. Neuropharmacology. 2001;41:601–608. doi: 10.1016/s0028-3908(01)00106-x. [DOI] [PubMed] [Google Scholar]
- Zhu W, Xu P, Cuascut FX, Hall AK, Oxford GS. Activin acutely sensitizes dorsal root ganglion neurons and induces hyperalgesia via PKC-mediated potentiation of transient receptor potential vanilloid I. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:13770–13780. doi: 10.1523/JNEUROSCI.3822-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]