

Abstract
RIAM is a Rap1 effector that mediates the recruitment of talin to integrins thereby supporting their activation. Here, we studied the role of RIAM in an adoptive transfer model for Type I diabetes, and report that RIAM expression in T cells is necessary for diabetes development. Loss of RIAM did not prevent lymphocyte recruitment to draining lymph nodes 24 hours after transfer, but was required for antigen-driven proliferation and cytotoxic killing. RIAM is recruited to immune synapses along with talin and LFA-1 and loss of RIAM profoundly suppresses antigen-dependent conjugate formation in primary naïve and effector T cells. These data identify the requirement of RIAM for formation of immunological synapses and in resulting T cell functions in autoimmunity. Moreover, since RIAM null mice are healthy, fertile, and display no bleeding abnormalities, our results identify RIAM and its regulators as potential targets for therapies of T cell mediated autoimmunity.
INTRODUCTION
In Type I Diabetes (T1D), autoreactive T cells are activated in draining lymph nodes, undergo clonal expansion, home to pancreatic islets, and destroy beta cells (1). Integrins are cell surface adhesion receptors that control T cell activation in the immunological synapse (IS) and govern naive and effector T cell homing (2). Integrins are proven therapeutic targets in treating autoimmune diseases (3), but agents that completely block their functions are limited by mechanism-based toxicities (4).
An alternative strategy is to inhibit integrin activation, which is dependent upon talin binding to the integrin beta subunit cytoplasmic tail (5, 6). Interference with talin or its interactions is a powerful way to disable integrin activation in vivo (7–9), but talin is ubiquitously expressed and direct blockade of talin-mediated integrin activation in all tissues is lethal (10). However, talin-dependent integrin activation is finely tuned by adaptor proteins with the potential to serve as targets to inhibit integrin activation in particular cell types and in response to specific stimuli.
Rap1-Interacting Adaptor Molecule (RIAM) is a Rap1 effector, encoded by the gene Apbb1ip, which mediates integrin activation downstream of Rap1 by targeting talin to the integrin beta subunit cytoplasmic tail (11–13). In contrast to talin deletion, germline loss of RIAM in mice does not affect development, hemostasis, or platelet integrin function (14–16). Although RIAM is abundant in leukocytes and localizes to the immunological synapse of Jurkat T cells or mouse CD4+ T cells with APC cell lines (17, 18), its value to the synapse has remained unknown.
Here we define the requirement of RIAM for the assembly of antigen-dependent immunological synapses in primary T cells and in their ensuing activation, proliferation, and effector functions. We demonstrate for the first time that loss of RIAM in mouse T cells provides protection from autoimmunity, impairs T cell-APC conjugate formation, and curbs T cell proliferation and cytotoxic effector function. These effects are ascribable to the requirement of RIAM for formation of immunological synapses of naïve and effector T cells. In contrast to the lethality of talin1 deletion, genetic loss of RIAM results in viable and fertile offspring with normal hemostasis and vascular development. Thus, these data suggest that targeting RIAM provides an avenue for cell type-specific inhibition of integrin activation to ameliorate autoimmunity.
MATERIALS AND METHODS
Mice
RIAM−/−, RIAMfl/fl, CD4-Cre, OT-1, and RIPmOVA mice are described (14, 16, 19–21). All mice were 8–20 weeks in age and from a mixed C57BL/6-Sv129 genetic background. RIAMfl/fl, OT-1, CD4-Cre+ mice were thus compared with RIAMfl/fl, OT-1 sex-matched littermates, and RIAM−/−, OT-1 mice with RIAM+/+, OT-1 sex-matched littermates. C57BL/6 mice were from Charles River. Mice were housed at the UCSD animal facility, and all experiments were approved by the Institutional Animal Care and Use Committee.
Reagents
Fluorochrome-conjugated antibodies were from Biolegend, antigen affinity-purified anti-RIAM polyclonal antibody from R&D Systems, anti-talin1 (8d4) from Novus Biologicals, and secondary antibodies from Jackson Immunoresearch. Azide-free blocking antibody against CD11a (M17/4) and control antibody (RTK2758) were from Biolegend. SIINFEKL peptide was from Anaspec, and IL-2 from the NCI preclinical repository.
Flow cytometry
Spleen, lymph node, or thymic cells were stained with fluorochrome-conjugated antibodies and analyzed by flow cytometry. Intracellular staining with anti-RIAM polyclonal antibody was performed in 0.5% saponin staining buffer after fixation with 2% formaldehyde.
T cell purification
T cells were purified using the Dynabeads Untouched Mouse T Cells kit (Life Technologies) and were routinely 93–98% CD3+ by flow cytometry. Where indicated, cells were labeled with 4 μM CFSE for 10 min at 37°C.
Type I Diabetes disease model
Diabetes transfer with OT-1 T cells into RIPmOVA recipients has been described (20). Splenic T cells (7×106) from CD4-Cre+, RIAMfl/fl, OT-1 mice or control RIAMfl/fl, OT-1 littermates were injected i.v. into RIPmOVA mice. Mice with >350 mg/dl blood glucose for 3 consecutive days were considered diabetic, sacrificed, and pancreata collected. Remaining non-diabetic mice were sacrificed on day 18 and pancreata collected. Formalin-fixed, paraffin-embedded pancreatic tissue sections (5 μm) were cleared with orange oil, dehydrated, stained with hematoxylin and eosin, and analyzed by light microscopy. A 0–3 islet infiltration scoring system was utilized by a blinded technician. 0 = intact islet with no infiltrate, 1 = peri-islet infiltrate or sparse intra-islet infiltration, 2 = 25–75% intra-islet infiltrate, and 3 ≥ 75% intra-islet infiltrate.
Homing
Splenic T cells from CD4-Cre+, RIAMfl/fl, OT-1 or control RIAMfl/fl, OT-1 mice were labeled with 0.4 μM or 4 μM CFSE and injected as a 50/50 mixture (1.5×106 of each) i.v. into RIPmOVA mice. After 3 or 24h, pancreatic lymph nodes were analyzed by flow cytometry for the ratio of RIAM-deficient (CFSElo) to control (CFSEhi) T cells.
Proliferation
For in vitro antigen-dependent proliferation, CFSE-labeled CD4-Cre+, RIAMfl/fl, OT-1 or control RIAMfl/fl, OT-1 splenocytes were stimulated with SIINFEKL or whole ovalbumin and 100 U/ml IL-2 for 3 days, and assayed by flow cytometry. The average number of times a cell divides within the total cell population (division index) or within the dividing portion (proliferation index) was calculated using FlowJo®. For in vivo expansion, CFSE-labeled splenic T cells from CD4-Cre+, RIAMfl/fl OT-1 or control RIAMfl/fl, OT-1 mice were injected (2×106) i.v. into RIPmOVA mice. After 3 days, pancreatic lymph node cells were stained for TCR Vα2 and analyzed for CFSE dilution by flow cytometry.
CD8+ expansion, differentiation to CTL, and effector function
To generate CTL, CD4-Cre+, RIAMfl/fl, OT-1 or RIAMfl/fl, OT-1 control splenocytes were stimulated with SIINFEKL and IL-2 for 6–7 days, harvested, and cultured with SIINFEKL pulsed or unpulsed target splenocytes at various effector to target ratios in the presence of anti-LAMP-1 antibodies for 2.5 h at 37°C. Effector/target cultures were harvested, stained with anti-CD8, and analyzed by flow cytometry. For target lysis in vitro, CTL were cultured overnight with a ~50/50 mixture of peptide-pulsed (CFSElo) and unpulsed (CFSEhi) splenocyte targets. Specific lysis was calculated as % decrease in the % of the peptide-pulsed peak between CTL-containing and no-CTL control cultures. For in vivo target cell lysis, differentially labeled splenocyte targets were prepared as for in vitro experiments and injected i.v. into mice that had received OT-1 CTL 24h prior. Four hours later, mice were sacrificed and splenocytes analyzed by flow cytometry for decrease in peptide-pulsed CFSE peak. Specific lysis was calculated as for in vitro experiments, comparing with mice that had not received OT-1 CTL.
Microscopy of immunological conjugates
Dendritic cells (DC) were purified from spleens of C57/BL6 mice injected i.p. with 1×106 Flt3L-expressing B16 cells (gift of G. Dranoff) 10 days earlier. DC were labeled with CellTrace Violet (Life Technologies), pulsed with SIINFEKL peptide, and cultured with naïve OT-1 TCR transgenic CD8+ T cells at a 1:2 ratio for 45 min at 37°C on coverslips coated with poly-L-lysine (Sigma), before fixation with 2% formaldehyde. Cytotoxic immunological conjugates were studied using CellTrace Violet-labeled splenocytes as targets with OT-1 TCR transgenic CTL in a 1:1 ratio. Staining for talin, CD11a and RIAM was done on coverslips in permeabilization buffer at RT for 1h, as was subsequent incubation with secondary antibodies. Coverslips mounted on slides were imaged microscopically, and post-acquisition processing was limited to adjusting contrast or correcting registration with Volocity (PerkinElmer) and ImageJ (NIH).
Conjugate formation
eFluor 670-labeled DCs (5×104) were pulsed with SIINFEKL peptide for 1h at 37°C. Where indicated, T cells were pre-incubated with blocking anti-CD11a or control antibody for 15 min at 37°C. CFSE-labeled naïve OT-1 TCR transgenic CD8+ T cells (105) were cultured with DC for 45 min to allow conjugate formation. Conjugates were vortexed at 1300 rpm for 30 sec, fixed in 2% formaldehyde, analyzed by flow cytometry, and quantified as the % of CFSE-labeled T cells bound to eFluor 670 DC. Formation of cytotoxic immunological synapses was analyzed as for naïve T:APC synapses, using 105 splenocytes as targets with 105 OT-1 TCR transgenic CTL.
Statistical analysis
Results are mean ± SEM. Statistical significance was assayed by two-tailed t-test for single comparisons or ANOVA for multiple comparisons with Bonferroni post-hoc test. Chi-square test was used for diabetes incidence. A P value <0.05 was considered significant. *p<0.05, **p<0.01, ***p<0.001; ns, not significant.
RESULTS AND DISCUSSION
Genetic deletion of RIAM protects from experimental Type I Diabetes
We found, as has been previously reported (14–16) that RIAM−/− mice are viable and healthy with stable hemostasis, with reduced marginal zone B cells, increased follicular B cells, and increased dendritic cells (DC) (Supplemental Fig. 1A). To specifically study the role(s) of RIAM in T cell-mediated autoimmunity, we crossed a RIAMfl/fl strain (14, 16) with mice that express Cre recombinase in mature CD4 and CD8 T cells (21), resulting in loss of RIAM in >95% of T cells (Fig. 1A), but largely normal myeloid and lymphoid cell abundance in spleen (Supplemental Fig. 1B). T cell thymic development was grossly normal, although selection efficiency was somewhat reduced (Supplemental Fig. 1C–H). These data indicate that the CD4-Cre+, RIAMfl/fl mouse is suitable for studies of RIAM function in T cells during autoimmune disease.
FIGURE 1. Loss of T cell RIAM protects mice from experimental diabetes.

(A) RIAM deletion in T cells. RIAM protein level was assessed in T cells by intracellular staining and flow cytometry. (B) Diabetes Incidence. T cells were injected i.v. into RIPmOVA mice and blood glucose measured on indicated days; n=12 (CD4-Cre+) and n=9 (no Cre) recipients pooled from two experiments. (C–D) Islet infiltration. Pancreatic sections were stained with H&E, and islet infiltration graded using a 0–3 scale. (C) Cumulative data are plotted as mean of the % distribution of infiltration scores. (D) Representative infiltration stages. Scale bar, 100 μm. (E) T cell draining LN homing. CFSE-labeled T cells were enumerated by flow cytometry 24h after injection into RIPmOVA mice; n=3 per group; performed twice. (F) Competitive T cell draining LN homing. The ratio of CD4-Cre+, RIAMfl/fl, OT-1 (CFSElo) to control RIAMfl/fl, OT-1 (CFSEhi) T cells was determined by flow cytometry from pancreatic lymph nodes after injection; n=3 per group; performed twice.
We crossed the CD4-Cre+, RIAMfl/fl strain with OT-1 TCR transgenic mice (19) before transfer into RIPmOVA mice expressing the cognate antigen on islet β cells (20). This T1D model compresses the time for disease development and permits analysis of β cell-specific T cell priming, clonal expansion, and homing (20, 22). Transfer of purified RIAMfl/fl, OT-1 control T cells resulted in the expected rapid onset of T1D in the majority of recipient mice (Fig. 1B), whereas transfer of CD4-Cre+, RIAMfl/fl, OT-1 T cells did not cause disease in any recipients tested. Likewise, histological analyses revealed heavy infiltration of control RIAMfl/fl, OT-1 T cells into pancreatic islets and little infiltration after transfer of RIAM-deficient OT-1 T cells (Fig. 1C–D).
Because RIAM deficiency impairs homeostatic trafficking of lymphocytes to lymph nodes (16), we measured their ability to home to pancreatic-draining lymph nodes after intravenous injection into RIPmOVA recipient mice. To our surprise, we found similar numbers of CD4-Cre+, RIAMfl/fl, OT-1 or control RIAMfl/fl, OT-1 T cells at 24h after transfer (Fig. 1E). We next performed competitive homing assays to more easily detect any subtle defect in homing of the RIAM null cells. At 3h after transfer, control RIAMfl/fl, OT-1 T cells could be found in 3-fold higher abundance than CD4-Cre+, RIAMfl/fl, OT-1 T cells in pancreatic-draining lymph nodes of RIPmOVA mice (Fig. 1F), confirming previous findings that initial trafficking from the blood to secondary lymphoid organs is RIAM-dependent (16). Importantly, by 24h, a time point critical for T cell priming in pancreatic lymph nodes (20), we did not observe significant differences in the numbers of RIAM-deficient and control T cells (Fig. 1F and S2A). These results indicate that protection from experimental T1D seen with RIAM-deficient T cells cannot be explained solely by defects in lymphocyte homing to peripheral lymph nodes.
RIAM is necessary for optimal antigen-driven T cell activation and rapid clonal expansion
To address alternative mechanism(s) whereby RIAM contributes to pathogenesis of T1D, we tested activation of naïve RIAM-null OT-1 T cells in response to antigen (ovalbumin protein) and antigen-presenting cells (APC) in a cross-priming assay. CD4-Cre+, RIAMfl/fl, OT-1 T cells displayed reduced upregulation of the early activation markers CD69 and CD98 (Fig. 2A–B), suggesting that RIAM loss in T cells impairs their ability to respond to antigenic stimulation.
FIGURE 2. RIAM is necessary for Ag-driven T cell activation and rapid proliferation.
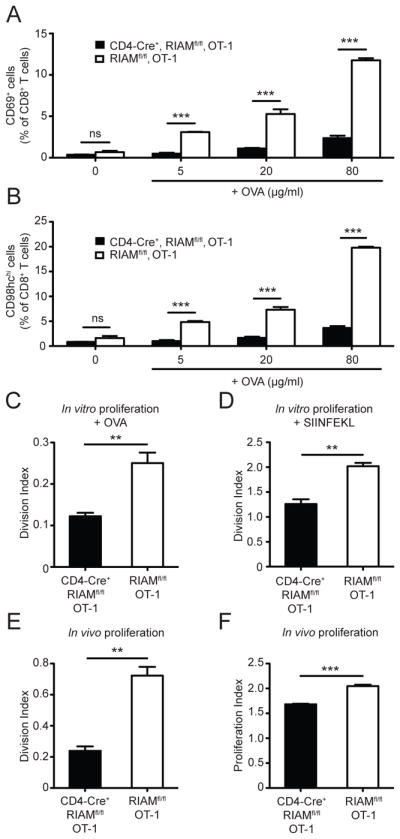
(A–B) Activation. Splenocytes were stimulated as indicated, and CD69 (A) and CD98hc (B) measured by flow cytometry; n=3 per group; performed twice. (C–D) In vitro proliferation. Splenocytes were stimulated as indicated and analyzed by flow cytometry; performed twice. (E–F) In vivo clonal expansion. T cells from CD4-Cre+, RIAMfl/fl, OT-1 (eFluor670-labeled) and control RIAMfl/fl, OT-1 littermates (CFSE-labeled) were mixed equally, transferred into RIPmOVA mice, and assessed by flow cytometry from pancreatic lymph nodes 3 days later; n=3 mice per group; performed twice.
To assess the role of RIAM in Ag-induced T cell proliferation in vitro, CFSE-labeled splenocytes from CD4-Cre+, RIAMfl/fl, OT-1 or control RIAMfl/fl, OT-1 mice were cultured with ovalbumin- or SIINFEKL-loaded APC. T cell division, quantified as division index, was significantly decreased upon loss of RIAM (Fig. 2C–D). We next tested whether the proliferation defect in RIAM-null T cells occurred in vivo. T cells purified from CD4-Cre+, RIAMfl/fl, OT-1 or control RIAMfl/fl, OT-1 mice were labeled with eFluor 670 and CFSE dyes, respectively, mixed in equal proportion, and transferred into RIPmOVA recipients. RIAM-deficient OT-1 T cells from pancreatic-draining lymph nodes of recipient mice showed reduced division compared to control OT-1 T cells (Fig. 2E). The proliferation index, a measure of how many doublings a dividing cell undergoes, was only minimally affected in the RIAM null T cells (Fig. 2F), suggesting that lack of RIAM likely impaired the initiation of proliferation rather than the speed or extent of cell division. Thus, RIAM is important for Ag-driven T cell activation and proliferation.
RIAM is required to form stable conjugates with APC
The interaction between T cells and APC resulting in formation of the immunological synapse is a central event in the activation of T cells (23, 24). The affinity and avidity of lymphocyte-associated antigen 1 (LFA-1, or αLβ2 integrin) for ICAM-1 are naturally regulated during participation in T cell-APC immunological synapses (25). TCR or chemokine receptor engagement generates signals that increase affinity of LFA-1 for extracellular ligand (integrin activation) (26, 27). This occurs through the binding of the cytoskeletal adaptor talin to the β2 cytoplasmic tail of LFA-1 (28). RIAM localizes to the immunological synapse of human T cell lines or mouse CD4+ T cells with transformed APC (17, 18). We found that endogenous RIAM localized to LFA-1 and talin-containing contacts formed between primary naive OT1 T cells and SIINFEKL-pulsed dendritic cells (DC) (Fig. 3A). Furthermore, RIAM-deficient T cells manifested profound impairment of Ag-dependent conjugation to DC (Fig. 3B–C). Blocking LFA-1 abolished T cell adhesion to DC, confirming the integrin dependence of these conjugates (Fig. 3D). RIAM-deficient T cells did not display major differences in expression level of LFA-1 surface protein (Fig. 3E), in agreement with reports of T cells from germline RIAM−/− mice (14, 16), excluding a possible decrease in LFA-1 protein as an explanation for loss of synapse formation. Our findings thus support the hypothesis that RIAM recruits talin to LFA-1-dependent immunological contacts for formation of stable T cell-APC conjugates.
FIGURE 3. RIAM is required for naive T cell-APC conjugate formation.

(A) RIAM localization. Naïve OT-1 T cells were cultured with SIINFEKL-pulsed dendritic cells (DC), fixed, stained with indicated antibodies, and visualized by confocal microscopy. Scale bar = 5 μm. (B–C) Conjugate formation. Naïve OT-1 T cells were labeled with CFSE and incubated with eFluor 670-labeled DCs. The % of T cells forming conjugates with APC was determined by flow cytometry; representative dot plots (B) and cumulative data (C) are shown; performed 4 times. (D) Integrin-dependent conjugates. Conjugate formation was assayed as in (C) with either anti-CD11a blocking antibody or control isotype antibody; performed twice. (E) T cell integrin expression. Representative histograms show surface levels of integrin CD11a (αL) and CD18 (β2) on CD3+ splenocytes.
RIAM is necessary for T cell cytotoxic effector function
T cell effector functions, including cytotoxicity, are important in T cell-dependent autoimmunity (29, 30). Since RIAM-deficient T cells are capable of modest proliferation (Fig. 2C–F), we wondered whether they could generate functional CTL. To test whether RIAM is required for CD8+ T cell cytotoxicity per se, we generated CTL from splenocytes of CD4-Cre+, RIAMfl/fl, OT-1 or control RIAMfl/fl, OT-1 mice and confirmed that RIAM was absent in >93% of differentiated CTL (Fig. 4A). The capacity of RIAM-null CTL to kill specific targets in vitro was reduced compared to equivalent numbers of control CTL (Fig. 4B). To determine if this defect also occurs in vivo, we adoptively transferred CD4-Cre+, RIAMfl/fl, OT-1 or control RIAMfl/fl, OT-1 CTL into recipient C57BL/6 mice. One day later, we injected SIINFEKL-pulsed (CFSElo) or control unpulsed (CFSEhi) targets and measured specific lysis. RIAM-null CTLs exhibited dramatically reduced cytotoxic activity (Fig. 4C). This defective killing could be ascribed to the failure of RIAM-deficient CTL to degranulate, as measured by decreased LAMP-1 exposure on the outer membrane during exposure to SIINFEKL-bearing targets (Fig. 4D–E). Thus, RIAM is required for CD8 T cell cytotoxic effector functions. Defective degranulation and target lysis could be an indirect result of inadequate activation of naïve RIAM-deficient T cells leading to poorly differentiated CTL, or it could be a result of RIAM loss directly hindering cytotoxic functions.
FIGURE 4. RIAM mediates CTL function.

OT-1 splenocytes were differentiated to CTL by culture with SIINFEKL peptide and IL-2. (A) RIAM deletion in CTL. RIAM expression in OT-1 CTL was measured by intracellular staining and flow cytometry. (B) In vitro target lysis. CTL were cultured overnight with a mixture of SIINFEKL-pulsed (CFSElo) and unpulsed (CFSEhi) target splenocytes; lysis was detected by flow cytometry; performed twice. (C) In vivo target lysis. CTL were injected i.v. into recipient C57BL/6 mice, which received a mixture of SIINFEKL-pulsed (CFSElo) and unpulsed (CFSEhi) target splenocytes 24h later. Recipient splenocytes were analyzed 4h later by flow cytometry for decrease in the % of pulsed targets; n=4 mice per group; performed twice. (D–E) In vitro degranulation. CTL were cultured with SIINFEKL-pulsed target splenocytes in the presence of anti-CD107a (LAMP-1), followed by staining for CD8 and flow cytometric analysis; performed twice. (F) RIAM localization to CTL immune synapses. CTL were allowed to form conjugates with SIINFEKL-pulsed splenocyte targets, fixed, stained with indicated antibodies, and visualized by confocal microscopy. Scale bar = 5 μm. (G) CTL-Target conjugate formation. CTL were labeled with CFSE and incubated with eFluor 670-labeled target SIINFEKL-pulsed splenocyte targets. The % conjugation was determined by flow cytometry; performed twice.
We previously found that naïve RIAM-null T cells form few stable conjugates with APC (Fig. 3B–D), and RIAM-null CTL exhibit defective cytotoxicity (Fig. 4A–C). In addition, we observed that RIAM polarized to the contact regions between CTL and target cells (Fig. 4F). Therefore, we hypothesized that RIAM might also be required for assembly of cytotoxic immunological synapses. We tested this possibility and found that, as seen with naïve T cells, RIAM loss in CTL strongly impaired their ability to form conjugates with SIINFEKL-loaded targets (Fig. 4G). Thus, RIAM participates in the formation of conjugates between both naïve CD8 T cells and APC, and of effector CD8 T cells with Ag-specific targets, and is therefore important in both naïve T cell activation/proliferation and efficient cytotoxicity. To rule out possible Cre-dependent artifact, we repeated our core experiments using T cells from germline RIAM knock-out (RIAM−/−) OT-1 mice and found similar results (Supplemental S2B–D). RIAM regulates actin dynamics in addition to promoting integrin activation through talin (11, 31). Examining the relative contribution of each of these RIAM functions to conjugate formation will be a potential avenue for future investigation.
In sum, we have shown that RIAM is required for T cell mediated immunological synapses and autoimmunity. Our work identifies this cell type-specific integrin regulator as a plausible locus for interrupting T cell driven pathologies since complete loss of RIAM in all tissues results in relatively healthy mice (14–16).
Supplementary Material
Acknowledgments
The authors thank Marina Slepak, Audrey Mays, and Quynhanh Nguyen for technical assistance.
This work was supported in part by NIH grants (K01DK090416, P30DK063491, P01HL-078784 and R01HL-117807), a Melanoma Research Alliance grant (MRA 346628), the Ludwig Center for Molecular Oncology, and the Neuroscience Microscopy Shared Facility (P30 NS047101, University of California San Diego).
References
- 1.Roep BO. The role of T-cells in the pathogenesis of Type 1 diabetes: from cause to cure. Diabetologia. 2003;46:305–321. doi: 10.1007/s00125-003-1089-5. [DOI] [PubMed] [Google Scholar]
- 2.Evans R, Patzak I, Svensson L, De Filippo K, Jones K, McDowall A, Hogg N. Integrins in immunity. Journal of cell science. 2009;122:215–225. doi: 10.1242/jcs.019117. [DOI] [PubMed] [Google Scholar]
- 3.Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nature reviews Drug discovery. 2010;9:804–820. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 4.Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annual review of medicine. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- 5.Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 6.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nature reviews Molecular cell biology. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haling JR, Monkley SJ, Critchley DR, Petrich BG. Talin-dependent integrin activation is required for fibrin clot retraction by platelets. Blood. 2011;117:1719–1722. doi: 10.1182/blood-2010-09-305433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrich BG, Fogelstrand P, Partridge AW, Yousefi N, Ablooglu AJ, Shattil SJ, Ginsberg MH. The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. The Journal of clinical investigation. 2007;117:2250–2259. doi: 10.1172/JCI31024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yago T, Petrich BG, Zhang N, Liu Z, Shao B, Ginsberg MH, McEver RP. Blocking neutrophil integrin activation prevents ischemia-reperfusion injury. The Journal of experimental medicine. 2015;212:1267–1281. doi: 10.1084/jem.20142358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monkley SJ, Zhou XH, Kinston SJ, Giblett SM, Hemmings L, Priddle H, Brown JE, Pritchard CA, Critchley DR, Fassler R. Disruption of the talin gene arrests mouse development at the gastrulation stage. Developmental dynamics: an official publication of the American Association of Anatomists. 2000;219:560–574. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1079>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 11.Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon-McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, Ginsberg MH. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Current biology: CB. 2006;16:1796–1806. doi: 10.1016/j.cub.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 12.Lagarrigue F, Kim C, Ginsberg MH. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 2016 [Google Scholar]
- 13.Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. The Journal of biological chemistry. 2009;284:5119–5127. doi: 10.1074/jbc.M807117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klapproth S, Sperandio M, Pinheiro EM, Prunster M, Soehnlein O, Gertler FB, Fassler R, Moser M. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired beta2 integrin function in mice. Blood. 2015;126:2704–2712. doi: 10.1182/blood-2015-05-647453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stritt S, Wolf K, Lorenz V, Vogtle T, Gupta S, Bosl MR, Nieswandt B. Rap1-GTP-interacting adaptor molecule (RIAM) is dispensable for platelet integrin activation and function in mice. Blood. 2015;125:219–222. doi: 10.1182/blood-2014-08-597542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su W, Wynne J, Pinheiro EM, Strazza M, Mor A, Montenont E, Berger J, Paul DS, Bergmeier W, Gertler FB, Philips MR. Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood. 2015;126:2695–2703. doi: 10.1182/blood-2015-05-644104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menasche G, Kliche S, Chen EJ, Stradal TE, Schraven B, Koretzky G. RIAM links the ADAP/SKAP-55 signaling module to Rap1, facilitating T-cell-receptor-mediated integrin activation. Molecular and cellular biology. 2007;27:4070–4081. doi: 10.1128/MCB.02011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wernimont SA, Wiemer AJ, Bennin DA, Monkley SJ, Ludwig T, Critchley DR, Huttenlocher A. Contact-dependent T cell activation and T cell stopping require talin1. Journal of immunology. 2011;187:6256–6267. doi: 10.4049/jimmunol.1102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 20.Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. The Journal of experimental medicine. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 22.Cantor J, Slepak M, Ege N, Chang JT, Ginsberg MH. Loss of T cell CD98 H chain specifically ablates T cell clonal expansion and protects from autoimmunity. Journal of immunology. 2011;187:851–860. doi: 10.4049/jimmunol.1100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dustin ML. The immunological synapse. Cancer immunology research. 2014;2:1023–1033. doi: 10.1158/2326-6066.CIR-14-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 25.Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Current opinion in cell biology. 2012;24:107–115. doi: 10.1016/j.ceb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature. 1989;341:619–624. doi: 10.1038/341619a0. [DOI] [PubMed] [Google Scholar]
- 27.Hogg N, Patzak I, Willenbrock F. The insider’s guide to leukocyte integrin signalling and function. Nature reviews Immunology. 2011;11:416–426. doi: 10.1038/nri2986. [DOI] [PubMed] [Google Scholar]
- 28.Simonson WT, Franco SJ, Huttenlocher A. Talin1 regulates TCR-mediated LFA-1 function. Journal of immunology. 2006;177:7707–7714. doi: 10.4049/jimmunol.177.11.7707. [DOI] [PubMed] [Google Scholar]
- 29.Peakman M. Immunological pathways to beta-cell damage in Type 1 diabetes. Diabetic medicine: a journal of the British Diabetic Association. 2013;30:147–154. doi: 10.1111/dme.12085. [DOI] [PubMed] [Google Scholar]
- 30.Wallberg M, Cooke A. Immune mechanisms in type 1 diabetes. Trends in immunology. 2013;34:583–591. doi: 10.1016/j.it.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Lafuente EM, van Puijenbroek AA, Krause M, Carman CV, Freeman GJ, Berezovskaya A, Constantine E, Springer TA, Gertler FB, Boussiotis VA. RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Developmental cell. 2004;7:585–595. doi: 10.1016/j.devcel.2004.07.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.