

ABSTRACT
Coagulation, complement, and innate immunity are tightly interwoven and form an alliance that can be traced back to early eukaryotic evolution. Here we employed an ecoimmunological approach using Tissue Factor Pathway Inhibitor (TFPI)-1-derived peptides from the different classes of vertebrates (i.e. fish, reptile, bird, and mammals) and tested whether they can boost killing of various human bacterial pathogens in plasma. We found signs of species-specific conservation and diversification during evolution in these peptides that significantly impact their antibacterial activity. Though all peptides tested executed bactericidal activity in mammalian plasma (with the exception of rodents), no killing was observed in plasma from birds, reptiles, and fish, pointing to a crucial role for the classical pathway of the complement system. We also observed an interference of these peptides with the human intrinsic pathway of coagulation though, unlike complement activation, this mechanism appears not to be evolutionary conserved.
KEYWORDS: antimicrobial, coagulation, complement, evolution, Immunoglobulins, IgG, peptide, TFPI-1, vertebrates
Introduction
The existence of infectious organisms in nature and their damage to the invaded hosts have led to the evolution of a diverse arsenal of defense mechanisms, ranging from the creation of simple mechanical barriers to the development of complex immune responses. Blood coagulation and the complement cascade belong to such ancestral defense systems and are two major contributors to the first line of defense against infection [1]. Their multiple roles include prevention of blood loss, opsonization and immobilization of the invading pathogens, and their subsequent destruction. Notably both defense systems have in common that they react extremely rapid and can function in a non-specific manner against potentially invasive pathogenic microorganisms. In order to execute some of their immune response functions, coagulation and complement factors need to be proteolytically processed. This in turn results in the generation of small peptides with antimicrobial and host defense modulating activity. The number of identified proteins involved in coagulation and complement pathways that can release such peptides, has considerably increased during the last two decades and include proteins such as kininogens [2], fibrinogen [3, 4], thrombin [5], protein C inhibitor [6], TFPI-1 [7], TFPI-2 [8, 9], heparin cofactor [10], antithrombin [11] and complement factor 3 [12]. Because the generated fragments execute bactericidal activity they are also considered as part of the antimicrobial peptide (AMP) family. AMPs are ubiquitous and their precursors can be found among proteins with less ordered structures as exemplified above. Most AMPs have in common that their positively charged amino acids are over-represented and often clustered. This results in patches of extremely high positive net charges (from +2 to +14) that are needed to penetrate and perforate the bacterial cell wall [13–16]. From a clinical point of view, naturally generated AMPs are at the moment attracting much attention as they constitute a potential alternative to the antibiotics that are currently in use. This is not only earned by the fact that AMPs provide a rapid and broad-spectrum response towards Gram-negative and Gram-positive bacterial species [17, 18], but also because there are so far no reports about resistances to AMPs.
Tissue factor pathway inhibitor belongs to the family of Kunitz-type serine proteinase inhibitor (serpins), 1 (TFPI-1). The protein consists of a highly negatively charged N-terminus, three tandemly linked Kunitz-type domains, and a highly positively charged C-terminus. Structural and functional studies have shown that the first two Kunitz domains of TFPI-1 are involved in binding to the complex formed by tissue factor and coagulation factor VII and coagulation factor X, respectively [19, 20]. The third Kunitz-type domain located at the C-terminal region can interact with heparin, via its cationic amino acids sequence motif [21]. TFPI-1 expression is induced under inflammatory conditions by mediators such as bacterial lipopolysaccharide, IL-1, TNF-α, PDGF and heparin [22]. In human plasma TFPI-1 exists in a full-length form but also a truncated version lacking the C-terminal part can be found. The processing of TFPI-1 is achieved by proteolytic cleavage with host derived enzymes, such as plasmin [23], thrombin [24], and matrix metalloproteinase-8 [25]. However, the E. coli derived OmpT protease has been described to process full-length TFPI-1 [26]. The released C-terminal peptides can interact with plasma lipoproteins, thrombospondin-1, clearance receptors [22], lipopolysaccharide [27] and have been reported to inhibit cell growth [28] and blood coagulation [29, 30].
Previous studies show a multi-functionality role of the human TFPI-1 C-terminal cationic peptides [7]. During wound healing processes, TFPI-1 is highly upregulated and C-terminal peptides released from TFPI-1 were found to be associated with the bacteria and fibrin. Under ex vivo conditions, these fragments have broad spectrum against Gram-negative bacteria as they can cause membrane lysis. Moreover, they interfere with complement activation by boosting the formation of the membrane attack complex (MAC) and generation of antimicrobial C3a [7]. In animal models, a synthetic peptide (GGL27) derived from the C-terminal of TFPI-1 has been found to exert anticoagulant and antimicrobial activities paired with anti-inflammatory effects. Subsequent animal experiments have shown that GGL27 is effective in ameliorating E. coli and Pseudomonas sepsis and LPS-induced shock [5].
Maroney and colleagues reported in 2009 that TFPI-1 has a high degree of sequence conservation among various species including zebrafish [31], which let the authors conclude that the protein has been highly conserved throughout evolution. The present study was conducted to decipher whether the antimicrobial activity of TFPI-1 has been conserved during evolution or is the outcome of a more recent adaptation. Our findings show that TFPI-1-derived peptides from all species can modulate coagulation, but only the mammalian-derived peptides can trigger complement activation, suggesting that both systems have developed differently during evolution.
Results
Phylogenetic distribution and sequence homology of the C-terminal region from TFPI-1. Using the neighbor-joining method, we constructed a phylogenetic tree from 87 different vertebrate species. This resulted in four distinct vertebrate classes referred to as mammals, birds, reptiles, and fishes (Fig. 1a). Subsequent sequence homology analysis using ClustalW multiple sequence alignment revealed that the C-terminal TFPI-1 peptide region is conserved in all vertebrates tested (Fig. 1b, Extended Data Figure 1, Supplementary Information). Even though the putative C-terminal peptide lengths can vary within the species, this region forms a positively charged core epitope, ranging from +7 to +15 (Extended Data Figs. 1–2, Table 1, Supplementary Information). As a positive net charge is a key property for many antimicrobial peptides, we speculated that the evolutionary conservation of the C-terminal region is important for an antimicrobial activity of these peptides. In human plasma TFPI-1 exists in various truncated forms and previous studies have shown that the C-terminal portion of the protein can be cleaved by plasmin and thrombin, resulting in release of GGLIKTKRKRKKQRVKIAYEEIFVKNM (27-mer, +8), RKRKKQRVKIAYEEIFVKNM (20-mer, +6), and TKRKRKKQRVKIAYEEIFVKNM (22-mer, +7) peptides, respectively [23, 24]. Indeed the three peptides have a broad antimicrobial activity and subsequent analyses have shown that this is dependent on their charge and amino acid sequence. Thus, no bacterial killing was noted when a scrambled C-terminal TFPI-1 peptide was used or positively charged amino acids were preplaced [7]. The human peptide contains two potential plasmin cleavage sites (between positions 1 and 2 and positions 8 and 9) and one site (lysine and threonine, positions 6 and 7) that is targeted by thrombin (Fig. 1b). The second plasmin cleavage motif (positions 8 and 9) is highly conserved in all species (Fig. 1b). This is in contrast to the first cleavage site (positions 1 and 2), which is found in most vertebrate species, but not in birds and reptiles (Fig. 1b). In birds, this site is often replaced by glutamic acid and arginine and in reptiles by glutamine and arginine. Likewise, the thrombin cleavage site, found in all mammals, except mice and rats, has been altered in birds and reptiles. While lysine (position 6) has remained, threonine (position 7) has changed to isoleucine in these two species (Fig. 1b). In fishes, no predicted plasmin or thrombin cleavage sites were noticed.
Figure 1.
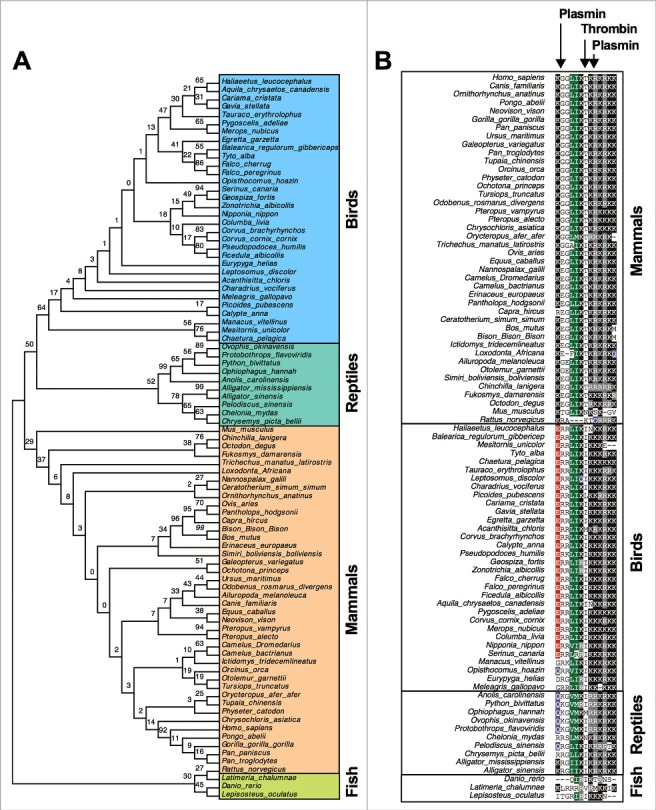
Phylogenetic tree analysis and sequence homology of TFPI-1. (a) From a total of 87 vertebrate species, a phylogenetic tree for the TFPI-1 C-terminal was constructed using Neighbour-Joining tree with 1000 bootstrap replications using the Mega 6 software. (b) Human plasmin and thrombin enzymatic cleavage sites on human TFPI-1 are indicated with arrows. ClustalW multiple sequence alignment of the TFPI-1 C-terminal highlighting amino acids with at least 70% similarity in all species.
Figure 2.
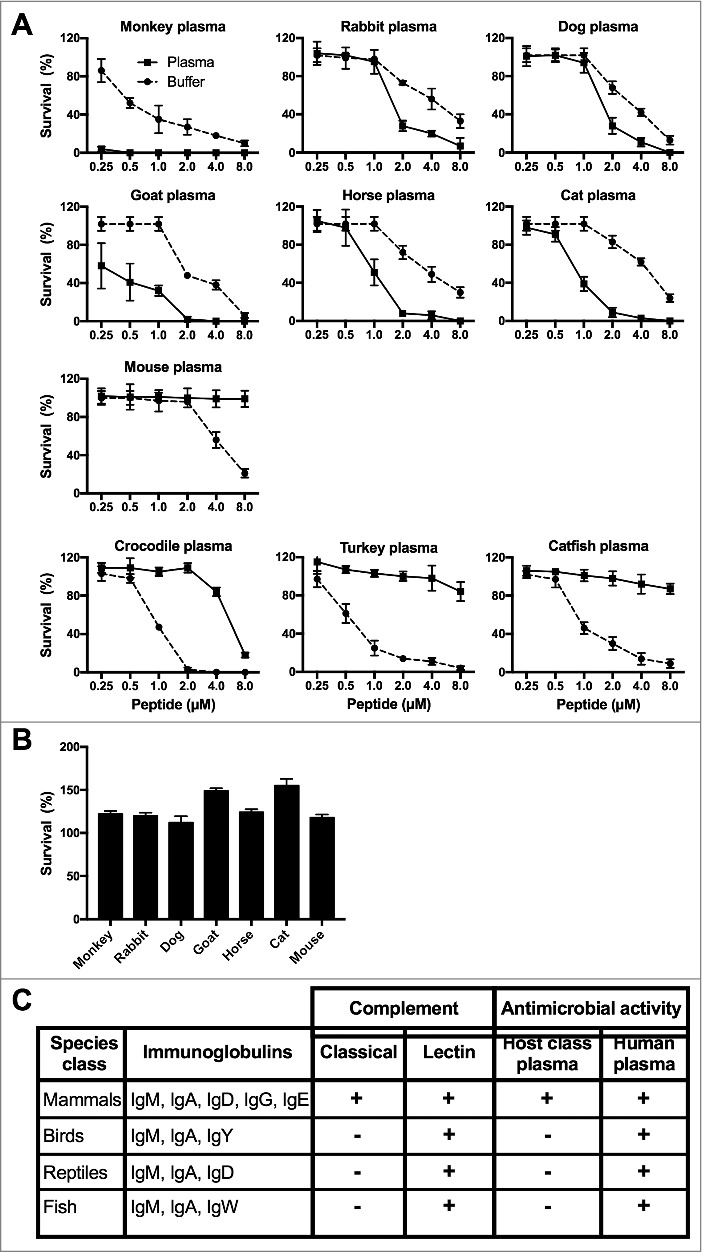
Complement mediated bacterial killing of TFPI-1 C-terminal peptides. (a-b) Bactericidal activity of TFPI-1 peptides was assessed under ex vivo conditions, using viable count analysis. E. coli bacteria were grown to mid-logarithmic phase and incubated with varying concentrations of peptides from mammals, birds, reptiles, and fish species. (a) Peptides were tested either in buffer containing 10 mM Tris, pH 7.4 alone or in presence of 20% citrated plasma from the corresponding host species. (b) Peptides (8 µM) were tested in presence of 20% citrated heat inactivated plasma from the corresponding host species. The bactericidal activity was determined by plating serial dilutions of bacteria on TH agar plates and the number of cfu was counted after overnight incubation. Data are presented as bacterial survival percentage; values are mean ± SD (n = 3) with a non-linear x-axis. (c) TFPI-1 peptides recruit the classical complement pathway.
Host defense properties of vertebrate C-terminal derived TFPI-1 peptides. Based on the phylogenetic analysis, C-terminal-derived TFPI-1 peptides from different species were synthesized (Extended Data Table 1, Supplementary Information) and their role in host defense was investigated. In a first series of experiments the antimicrobial activity of these peptides was tested in radial diffusion against the Gram-negative bacteria Escherichia coli and Pseudomonas aeruginosa. Species included were mammals: (primates (human, gorilla, orangutan, bonobo, and chimpanzee share 100% sequence identity), horse, dog, goat, cat, rabbit, and mouse), birds (turkey and penguin), reptiles (alligator and viper), and fishes (zebra fish and coelacanth). Supplementary figure 3a depicts that the human peptide had the highest antimicrobial activity, but also all other peptides were able to kill E. coli and P. aeruginosa bacteria. Many antibacterial peptides such as LL-37 lose their killing activity in buffer with physiological salt content or when added to plasma [7]. However, previous work has shown that a peptide derived from the C-terminal part of human TFPI-1 (GGL27) enhances the binding of complement factor C1q to bacterial surfaces [7]. This then displays enhanced bactericidal activity in human plasma, which in turn can boost the classical complement system and trigger the formation of the antimicrobial C3a peptide [7].
Figure 3.
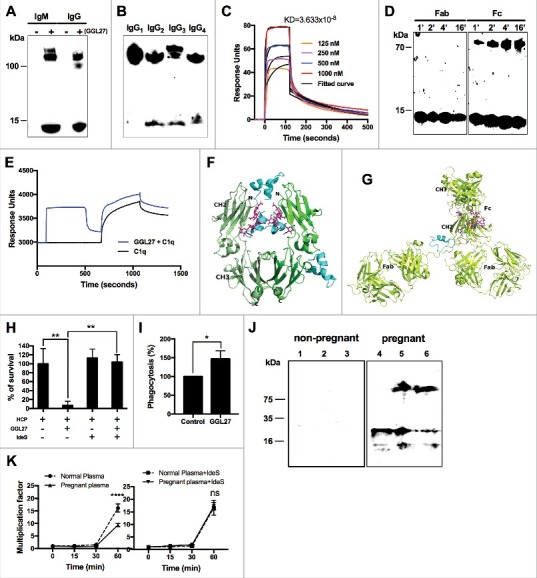
The enhanced bacterial killing by TFPI-1 endogenous C-terminal peptides in human plasma is dependent on immunoglobulins. (a-b, d) 4 µg GGL27 were incubated with 20 µg of human IgM, IgG, and IgG subclasses. In another experiment, 2 µg GGL27 were incubated with 50 µg of human IgG Fab and Fc fragments at indicated time points. GGL27 binding was determined by using rabbit anti-human TFPI-1 C-terminal monoclonal primary antibody and followed by secondary goat anti-rabbit polyclonal antibody (a) binding of GGL27 to IgM and IgG as indicated by arrows (b) binding of GGL27 to different human IgG subclasses (d) binding of GGL27 to Fc fragment at indicated time points. (c) Surface plasmon resonance (SPR) sensorgrams illustrating interactions between GGL27 (analyte) and immobilized IgG (ligand). The curves were obtained after injection of different concentrations of the peptide (125, 25, 500 and 1000 µM respectively) and analysis shows binding incidence with association and dissociation curves between GGL27 and IgG. (e) Biacore graph shows the association and dissociation curve for GGL27 and C1q binding to immobilized IgG. (f-g) ClusPro 2.0 results for the GGL27 peptide (cyan cartoon) docking to the human IgG1. (f) Docking to the homodimeric Fc domain (green and light green cartoon): The top most pose in the figure is at the FcγR site and the middle one in the glycan binding site. The glycan is shown as magenta sticks. The pose on the right hand side is at the hydrophobic patch of the CH2-CH3 domain interface. (g) Docking to the complete IgG1 (lemon green cartoon). The red color regions in the Fc CH2 domain denote the major binding site of C1q globular head. The blue regions denote the amino acids that somewhat restrict the C1q access to the binding site [64]. (h) The importance of GGL27 binding to IgG and mediating complement killing activity was tested via viable count assay. E. coli was incubated for 1 hour with 1 μM GGL27 comparing two different plasma conditions; in normal human citrated plasma or in human citrated plasma pre-treated with IdeS (0.2 µg/µl). Serial dilutions of bacteria were plated on TH agar plates and incubated overnight at 37°C and number of colonies was counted next day. Percentage of E. coli survival when incubating the GGL27 peptide in human citrated plasma alone or in citrated plasma pretreated with IdeS. (i) GGL27 (10 µM) peptide was incubated with fluorescence-labeled E. coli bio-particles for 1 hour in 20% human citrated plasma followed by incubation with murine macrophages (RAW 264.7) for 1 hour at 37°C. Percentage of phagocytosis activity of macrophages was measured by detection of fluorescence using a microtiter plate reader. (j) Plasma samples from three non-pregnant (numbers 1-3) and pregnant (numbers 4-6) women were analyzed by immunoblotting using TFPI-1 C-terminal monoclonal primary antibodies. (k) Endogenously generated TFPI-1 c-terminal peptides inhibit E. coli bacterial growth in pregnant women plasma (left) and this activity was completely abolished when IgG was inactivated with IdeS (right). Plasma from non-pregnant women was used as a control (control n=3, pregnant women n=3).
To investigate whether this feature has been conserved during evolution, the TFPI-1-derived peptides were incubated with E. coli in presence of physiological buffer containing 20% of citrated plasma. The peptide corresponding to the primate sequence was incubated in cynomolgus monkey plasma, whereas the rabbit, dog, goat, horse, cat, mouse, and turkey peptides were incubated with plasma from their corresponding endogenous species. The alligator peptide was incubated in crocodile plasma and the zebra fish peptide in sturgeon plasma. Fig. 2a shows that all mammalian peptides, with the exception of the mouse peptide, triggered increased complement-mediated bacterial killing in the presence of plasma. When plasma was heat-treated in order to inactivate the complement system, the antimicrobial ability of the mammalian peptides was abolished (Fig. 2b). These results further strengthen our findings that bacterial killing is complement-mediated. However, the antimicrobial activity was reduced or lost in the presence of plasma incubated with bird, reptile, or fish peptides. These findings indicate that only TFPI-1 derived peptides from higher organisms have the ability to interact with the complement system (Fig. 2c, Extended Data Figure 3b, Supplementary Information).
Endogenous peptides from the C-terminal region of TFPI-1 interact with immunoglobulins. The next experiments were set up to test whether complement-mediated killing is linked to the interaction of GGL27 with immunoglobulins, and whether this can trigger an augmentation of classical complement pathway. To this end we performed a panel of peptide-protein interaction assays. Fig. 3A shows that the binding of GGL27 to human IgM or IgG leads to the formation of clusters as verified by Western blot analysis probing with an antibody against GGL27 (Fig. 3a). Notably, GGL27 was also found to interact with immunoglobulins from all other species tested (Extended Data Figure 3c, Supplementary Information). The binding of GGL27 to human IgG was further confirmed by surface plasmon resonance (SPR) analysis (Fig. 3c). When performing additional Western blot analysis no difference in the specificity of the peptide for the different human subclasses (IgG1, IgG2, IgG3, and IgG4, respectively) was noted (Fig. 3b). To map the interaction site on IgG, GGL27 was incubated with human IgG Fab and Fc fragments at different incubation times. Fig. 3d depicts that GGL27 binds only to the Fc region, but not to the Fab fragment. Finally we wished to determine if GGL27 and C1q share same or different binding sites on IgG. To address this experimentally we performed SPR analysis by running GGL27 as a first analyte on an IgG coated chip, followed by an incubation with C1q. These data reveal that GGL27 and C1q have two distinct binding sites on IgG (Fig. 3e), since the sequential binding leads to an additive increase in the mass bound to the sensor chip. Together these findings support the notion that the interaction of GGL27 with IgG can boost the activation of the classical pathway of complement.
Molecular modeling of the IgG/GGL27 complex and its implication for structural modifications. To predict the structure of GGL27 when released from TFPI-1, the C-terminal peptide was analyzed by web-based PSIPRED and JPred4 software (secondary structure prediction tools). The PSIPRED results for the peptide alone suggest that there are two helical sequence stretches (residues 4-12 and 14-25) in the peptide. This result was also confirmed by the JPred4 tool that predicted in addition that the two helical sequences (predicted at residues 5-10 and 20-24) are linked by a short beta strand (residues 15-19). However, within TFPI-1 the fold of the GGL27 sequence is altered and predicted to be either almost fully helical (residues 4-25; PSIPRED) or mainly coiled with a short beta strand formed by residues 22-25 (JPred4). Similarly to the secondary structure model of the alpha helical regions, also the 3D structure of the GGL27 peptide consists of two alpha helical portions linked by a coil region according to the PEPFOLD 3 program prediction as shown in Supplementary figure 4a. Comparing the five best models from PEPFOLD 3, we found that the length of the helical portions can vary a little bit, but all five models predict that the consensus helical sequences include the peptide residues 5-11 and 19-23. Finally the peptide was subjected to a 20-ns molecular dynamics (MD) simulation. Based on this analysis it can be concluded that the secondary structures of the peptide did not change during the simulation. Moreover the two helical sequence motifs seem to have kept their length and principally remained at their expected positions at residues 5-10 and 19-26 (initially residues 6-11 and 19-26). In the next series of structural studies, the GGL27 peptide was virtually docked to the Fc domain of human IgG and the whole IgG. The peptide was found to interact with the hydrophobic patch at the CH2-CH3 junction (Fig. 3f) or with the Fc-Fab linker region (Fig. 3g). Our analysis further suggests that the binding is mediated by van der Waals (hydrophobic) forces and also involves hydrogen bonds at the hinge region including the residues Met252, Ile253, Met428, Asn434 and Tyr436, respectively (Supplementary Figure 4b). Moreover, the predicted binding sites for the C-terminal GGL27 peptide are not overlapping with the putative C1q binding site at CH2 domain (Fig. 3f-g). As the CH2-CH3 hinge and the Fc-Fab linker region are flexible, our model also suggests that the binding of GGL27 can induce a conformational change in the IgG molecule that may facilitate its interaction with the globular head of C1q. Interestingly, also bacterial immune-modulating virulence factors such as the B1 domain of staphylococcal protein A and the C domain of streptococcal protein G are utilizing the CH2-CH3 hinge as a binding site [32], which could be a virulence mechanism to dampen the immuno-stimulatory effect of GGL27.
Figure 4.
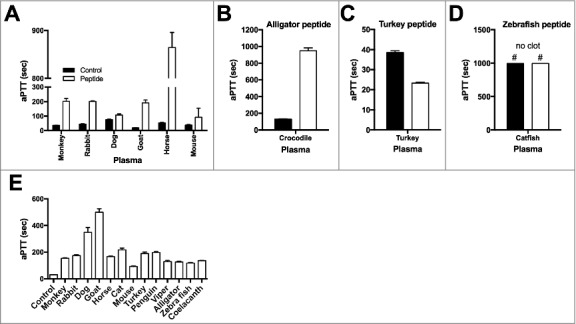
Anticoagulant activities of TFPI-1 C-terminal peptides. Anticoagulant activities of vertebrate TFPI-1 derived peptides (50 µM) in citrated plasma were determined using activated partial thromboplastin time (aPTT). Mammalian peptides were tested in their corresponding host species plasma (a), alligator peptide in crocodile plasma (b), bird peptides in turkey plasma (c) and fish peptides in catfish plasma (d). Note that clotting times > 1000 sec are considered as “no clot”. Data are presented as clotting time in seconds; values are mean ± SD (n = 3). (e) Anticoagulant effects of different vertebrate TFPI-1 peptides were tested in human plasma. Data are presented as clotting time in seconds; values are mean ± SD (n = 3).
IgG is crucial for GGL27-mediated killing through complement. Based on these findings we concluded that the antimicrobial activity of GGL27 is dependent on its interaction with IgG and the subsequent activation of the classical pathway of complement. To prove this concept, bacterial killing assays were performed by incubating E. coli bacteria with GGL27 (1 µM) for 1 hour at 37°C in the presence of normal human citrated plasma. In a second set of killing assays the plasma was first treated for 3 h with IdeS enzyme (0.2 µg/µl) to remove all IgG [33] and then mixed with GGL27 and E. coli bacteria. Fig. 3h demonstrates that almost 100% killing is seen when bacteria and GGL27 were incubated with plasma. However, when plasma was pretreated with IdeS the antimicrobial activity was completely lost (Fig. 3h). In addition to direct complement mediated killing, IgG opsonization of microorganisms also triggers a cellular immune response as the priming increases the ability of macrophages to phagocytize. Using fluorescein-labeled E. coli (K-12) bio-particles and mouse macrophages (RAW 264.7), particle uptake was significantly increased in the presence of GGL27 (Fig. 3i). It can therefore be concluded that the binding of GGL27 to IgG is important to mount classical complement activation and increase phagocytosis.
TFPI-1 and plasmin are highly upregulated in women during pregnancy [19, 34]. Consequently increased levels of GGL27 might be present in plasma samples from pregnant women and this should contribute to a more efficient clearance of bacteria. Thus we examined six plasma samples including three specimens from pregnant women by Western blot analysis using an anti-GGL27 antibody. Fig. 3j shows that the antibody detected both intact TFPI-1 protein and C-terminal fragments that are bound to immunoglobulins, as apparent by a shift in molecular weight of peptide/immunoglobulin complexes. Next we incubated E. coli bacteria with plasma samples from pregnant and non-pregnant women and monitored bacterial survival. Increased bacterial killing was observed in plasma samples from pregnant women compared to samples from non-pregnant women. When the plasma samples were pretreated with IdeS, no differences in bacterial growth were noted, suggesting that TFPI-1 mediated complement activation is dependent on IgG antibodies and is more efficient during pregnancy Fig. 3k.
Effect of TFPI-1 derived peptides on blood coagulation/intrinsic pathway. Having shown the physiological relevance of TFPI-1 to ameliorate complement efficiency, we next tested the interspecies activity of the TFPI-1 peptide collection used in this study. To this end, peptides from all species were incubated with 20% human citrated plasma. This was then followed by measurements of complement-mediated killing of E. coli bacteria. Supplementary figure 4 shows that without any exception, all peptides from fish to human boosted the complement system and enhanced bactericidal activity in human plasma. The coagulation as well as the complement system are both serine proteinase driven amplification cascades. Indeed evolutionary studies have shown that the vertebrate clotting cascade has originated from the complement system at a time when duplication of primitive complement proteins (also referred to as advanced deuterostome complement) already had occurred [1]. In human, the coagulation cascade can be divided into two branches, i.e. the extrinsic and intrinsic pathways of which the intrinsic pathway has evolved after the extrinsic pathway [35]. While predecessors of the intrinsic pathway are found early in evolution such as in amphibians and platypus, they are not always seen in birds and fishes suggesting that the genes have been lost in some species [35]. Based on these findings we tested the effect of the TFPI-1 peptides on the intrinsic pathway of coagulation by measuring the activated partial thromboplastin time. Fig. 4a shows that the addition of the mammalian TFPI-1 peptides to their respective plasma impaired normal clotting as clotting times were prolonged and similar findings were recorded when the alligator peptide was added to plasma from crocodile, a closely related species (Fig. 4b). However, no clotting was seen when the aPTT reagent was incubated with catfish plasma for 1000 sec, suggesting lack of an intrinsic pathway. The plasma remained unclottable when the zebrafish peptide was co-incubated with the aPTT reagent. Unexpectedly, the intrinsic pathway was activated in turkey plasma and was further amplified in the presence of the turkey TFPI-1 peptide as seen by a shortened clotting time. We next tested whether the treatment of human plasma with the TFPI-1-derived peptides impairs activation of the intrinsic pathway of coagulation. As shown in fig. 4E peptides from all species, even those lacking a functioning intrinsic pathway of coagulation, were able to prolong clotting in human plasma. These findings suggest that the function of the C-terminal part of TFPI-1 has rather evolved to eradicate invading pathogens than to modulate the intrinsic pathway of coagulation which is in line by the conclusions from Krem and Di Cera that that vertebrate blood clotting emerged as a by-product of innate immunity [1]. In conclusion, here we show that the C-terminal part of TFPI-1 is evolutionary conserved and plays an important role in the host defense to infection.
Discussion
The coagulation and complement systems belong to the blood-borne serine proteinase cascades with common evolutionary pathways that can be traced back to arthropods [36]. To prevent systemic clotting and pathologic immune responses both systems have to be tightly regulated. This task is to a great extent achieved by serine proteinases inhibitors. Among those, Kunitz type serine protease inhibitors play an important role [37]. Likewise serine proteinases, also proteins with Kunitz type motifs have been conserved during evolution and can be found in almost all organisms ranging from microbes to mammals [37]. This applies also for TFPI-1 which plays a central role in down-regulating the extrinsic pathway of coagulation [38], but also in other serine proteinase driven cascades such as the lectin pathway of complement [39]. Arachnids such as ticks, for instance, produce TFPI-1-like proteins in their salivary glands to avoid clotting during blood feeding [40]. In addition, TFPI-1-like proteins are also found in the venom of the vampire snail Colubraria reticulate [41] or in the midgut and salivary glands of Culicoides sonorensis [42].
In the present study we show that the C-terminal part of TFPI-1 is highly conserved among vertebrates. Notably, domains of proteins with biological activities have often been more preserved during evolution than those lacking a function. This said, previous work has shown that the C-terminal domain of human TFPI-1 is both anticoagulant and antimicrobial [7]. To investigate whether these features are unique to the human coagulation inhibitor or have ancestral origin, we synthesized a panel of TFPI-1-derived peptides from species representing birds, reptiles, mammals and fishes. Employing functional assays, we found that all 13 peptides from different species have a direct antimicrobial activity against Gram-negative pathogens. These results are not completely unexpected as the amino acid sequences and in particular the net charges do not differ significantly between the different species. However, only the mammalian TFPI-1 peptides expose an increased antimicrobial activity, when incubated in plasma instead of buffer. In humans this effect is caused by the ability of GGL27 (the human TFPI-1-derived peptide) to boost classical pathway of complement via its interaction with complement factor C3 [7].
It has been estimated that the first primitive form of the complement system was developed about 1,300 million years ago and was entirely dependent on the alternative pathway [43–45]. About 400 million years later the lectin pathways became part of the complement cascade, while it took another 300 million years for the classical pathway to appear [43]. The latter event correlates with occurrence of the adaptive immune system and explains why the classical pathway was not developed at an earlier stage, since its activation requires IgG or IgM opsonized targets.
IgG is the most abundant antibody isotype in human plasma accounting for 70–85% of all immunoglobulins. This probably also applies for the majority of all placental mammals and considering the mechanism described in this study the appearance of IgG could have been evolutionary of great help to strengthen the immune system of an unborn offspring. However, lower vertebrates such as birds, reptiles, and amphibians lack IgG. They instead express IgY as principal immunoglobulin, which is an ortholog of mammalian IgG and IgE [46] that like IgG and IgM binds to GGL27. Thus boosting complement activation by an interaction with IgGs can occur in mammals, but not in lower vertebrates. This hypothesis is further supported by our molecular modeling data suggesting that, the binding of GGL27 to IgG may trigger a conformational change that in turn can facilitate the interaction of IgG with C1q to boost complement activation. It therefore makes perfect sense that that only TFPI-1 from mammals, but not from birds, fishes, or reptiles, is susceptible to cleavage by thrombin or plasmin. This modulation will result in the release of immuno-modulatory peptides from the C-terminal part of the protein, which could not have been occurred when employing TFPI-1 from non-mammalian species (Fig. 1). Taken together, our data show that activation of the complement system is restricted to TFPI-peptides derived from mammals, but not from other species. This is in contrast to the intrinsic pathway of coagulation, which is impaired by the addition of TFPI-1 peptides from all species to human plasma. Evolutionary studies suggest that inclusion of the factors of the intrinsic pathway of coagulation (plasma kallikrein, factor XI, and factor XII, respectively) to the vertebrate clotting system occurred several hundred million years ago and started with the appearance of the respective predecessors in amphibians [35]. However, cetaceans as well as some birds are deficient in factor XII, though they can express the remaining factors of the intrinsic pathway. Obviously, different evolutionary paths have been involved in these processes. In whales, for instance, the production of the coagulation factor is prevented by introducing two nonsense mutation making stop codons and a single nucleotide insertion which about 60 million years ago rendered the gene into a pseudogene [47]. The gene in birds like chickens is completely missing as it has been removed about 300 million ago [35]. The evolutionary development is also reflected in the clotting time of the intrinsic pathway of coagulation which can be blocked when the reptile TFPI-1 peptide is added to reptile plasma, but not when the fish or bird TFPI-1 peptides were given to their respective plasma samples. Taken together these findings indicate that despite the removal of the factor XII gene in some birds and cetaceans, their TFPI-1 derived peptides have not lost their activity to block the intrinsic pathway of coagulation.
Materials and methods
Peptides. Peptides (Supplementary table 1) were synthesized by Ontores Biotechnologies (Shanghai, China). The purity (>98%) of these peptides was confirmed by mass spectral analysis (MALDI-ToF Voyager).
Microorganisms. The bacterial isolates E. coli ATCC 25922 and P. aeruginosa ATCC 27853 were obtained from the American Type Culture Collection (Manassas, VA, USA).
Ethical statement. All human samples were collected in accordance with the ethically approved guidelines by Lund University and informed consent was obtained from all subjects (Dnr 2016/178). Animal use protocols were approved by the Institutional Animal Care and Use Committee of Malmö/Lund (M185-14).
Plasma. Fresh human citrated plasma was collected from non-pregnant and pregnant women. Citrated plasma from cynomolgus monkey, rabbit, dog, goat, horse, cat, mouse, turkey, goose, and sturgeon was purchased from Seralab (West Sussex, UK). Crocodile (Crocodylus siamensis) citrated plasma was a kind gift from Yosapong Temsiripong (Sriracha Moda Co., Ltd, Chon Buri, Thailand).
Radial diffusion assay (RDA). Bacteria were grown to mid-logarithmic phase in 10 ml of full-strength (3% w/v) trypticase soy broth (TSB) (Becton-Dickinson, Cockeysville, MD) as described earlier [48]. Bacteria were then washed once with 10 mM Tris, pH 7.4 and 4 x 106 bacterial colony forming units (cfu) were added to 15 ml of the underlay agarose gel, consisting of 0.03% (w/v) TSB, 1% (w/v) low electroendosmosis type (EEO) agarose (Sigma, St Louis, MO) and 0.02% (v/v) Tween 20 (Sigma). The underlay was poured into a Ø 144 mm petri dish. After agarose solidification, 4 mm-diameter wells were punched and 6 μl of test sample was added to each well. Plates were incubated at 37°C for 3 hours to allow diffusion of the peptides. The underlay gel was then covered with 15 ml of molten overlay (6% TSB and 1% Low-EEO agarose in distilled H2O). Antimicrobial activity of a peptide was visualized as a zone of clearing around each well after 18-24 hours of incubation at 37°C.
Viable count analysis (VCA). As previously described [48] E. coli bacteria were grown to mid-exponential phase in Todd-Hewitt (TH) broth. Bacteria were washed and diluted in 10 mM Tris, pH 7.4 either alone or with 20% citrated plasma or with 20% citrated heat inactivated plasma. Bacteria (2 x 106 cfu/ml) were incubated in 50 μl, at 37°C for 1 h with the TFPI-1 C-terminal peptides at the indicated concentrations. In another experiment 2 x 108 cfu/ml bacteria were incubated at 37°C for 0, 15, 30 or 60 min with 100% plasma from non-pregnant or pregnant women. Serial dilutions of the incubation mixture were plated on TH agar plates, followed by incubation at 37°C overnight and cfu determination.
In another parallel experiment, E. coli (2 x 108 cfu/ml) was incubated for 60 min at 37°C with 1 μM GGL27 in 100% human plasma in the presence or absence of IdeS (0.2 µg/µl) treated human plasma for 3 h. Serial dilutions of bacteria were plated on TH agar plates and incubated at 37°C overnight followed by cfu determination.
Hemolysis assay. EDTA-blood was centrifuged at 800 x g for 10 min in order to remove the plasma and white blood cells. The remaining erythrocytes were washed three times, resuspended in PBS, pH 7.4 and adjusted to a 5% suspension. The cells were then incubated with end-over-end rotation for 60 min at 37°C in the presence of peptides (60 µM). Triton X-100 (Sigma) 2% served as positive control. This was followed by a centrifugation step (800 x g for 10 min) and the supernatant was transferred to a 96 well microtiter plate. The absorbance of hemoglobin release was measured at 540 nm and expressed as percentage of Triton X-100 induced hemolysis.
Phylogenetic and sequence homology analyses. Various vertebrate TFPI-1 amino acid sequences were retrieved from NCBI, Ensembl and UniProt databases. These sequences were aligned using Blosum 69 protein weight matrix settings using BioEdit software (Ibis Biosciences, Carlsbad, CA). Internal adjustments were made taking the structural alignment into account utilizing the ClustalW interface. Neighbor-joining method was used for phylogenetic tree construction, and the reliability of each branch was assessed using 1000 bootstrap replications using Mega6 software [49].
Clotting Assays. All clotting times were analyzed using a coagulometer (Amelung, Lemgo, Germany). For determination of the activated partial thromboplastin time (aPTT), 100 µl of a kaolin-containing solution (Technoclone, Vienna, Austria) were added to 100 µl of citrated plasma or citrated plasma peptide mix and incubated at 37°C for 200 s before clot formation was initiated by adding 100 µl of 30 mM fresh CaCl2 solution.
SDS-PAGE & immunoblotting
In a total volume of 20 µl, 4 µg of GGL27 peptide were incubated over night with 20 µg of antibody (IgM, IgG, IgG1, IgG2, IgG3, IgG4 or IgY) or with 20% citrated plasma. Alternatively, GGL27 (2 µg) was incubated with human IgG Fab and Fc fragments (50 µg) for 1, 2, 4 and 16 h. Samples were then separated on SDS-PAGE under non-reducing conditions (Any kD Gel, Bio-Rad, Hercules, CA) and proteins and peptides were transferred onto PVDF membrane (Merck Millipore). Membranes were blocked with 3% (w/v) skimmed milk (Bio-Rad) overnight, washed, and incubated for 1 h with a monoclonal rabbit antibody against the human TFPI-1 C-terminal peptide (GGL27) (1:1000, Abcam ab134151). The membranes were washed three times with PBS-Tween for 10 min. An HRP conjugated secondary antibody against goat immunoglobulins (1:1000) (Bio-Rad) was added for 45 min followed by another washing step to remove unbound antibody. Specific bands were visualized by an enhanced chemiluminescent substrate (ThermoFisher), and a developing ChemiDoc system (Bio-Rad).
Surface plasmon resonance (SPR)
Binding characteristics between human IgG, the peptide and C1q protein investigated were carried out using a BIAcore X-100 instrument (GE Healthcare, Uppsala, Sweden). 3000 the resonance units (RU) human IgG was immobilized on a CM5 sensor chip (GE Healthcare) by standard amine coupling and RU were calculated based on the analyte molecular weight. In parallel, one flow cell was incubated with buffer alone (i.e. without IgG), serving as control. Interaction experiments were performed with injections of 125, 250, 500, 1000 nM of peptides in running buffer (10 mM HEPES, pH 7.5, 150 mM NaCl, 0.005% surfactant P20, and 3.4 mM EDTA) at a flow rate of 30 μl/min. After the end of each injection, dissociation was performed with 50 mM NaOH, followed by a washing procedure. The BIA evaluation 3.1 analysis software (GE Healthcare) was used to determine equilibrium dissociation constants (KD) from the processed data sets by fitting to a 1:1 molecular binding model with drifting baseline. In another experiment C1q protein was injected over the immobilized IgG or immediately after the peptide was injected over the surface.
Molecular modeling
To gain insight on how the C-terminal peptide GGL27 interacts with the IgG Fc domain, we first modeled the peptide's three-dimensional (3D) structure. The biophysical properties of GGL27 have been previously studied [7]. A preliminary secondary structure prediction of the TFPI-1 C-terminal residues was conducted with the web servers PSIPRED v. 3.3 [50, 51] and JPred4 [52] using the whole human TFPI-1 sequence (UniProt access code: P10646) and the GGL27 C-terminus only. The initial peptide fold was predicted by the PEPFOLD 3 web server [53–55] using 200 simulations and the sOPEP energy function (Maupetit et al., 2007; 2010) for sorting the models. The best-ranked model 1 was submitted to a 20-ns molecular dynamics (MD) simulation in an octahedral TIP3P water [56] box using the AMBER 16 molecular simulations package [57] and the AMBER ff03 force field [58]. The stepwise MD protocol for the simulation system (preliminary energy minimization, equilibration and the production simulation) followed the protocol reported by Mirza et al [59]. and the final frame structure was energy minimized similarly to the last step of the initial minimization without any positional constraints.
The obtained peptide structure was then docked without restraints with the ClusPro 2.0 protein-protein docking tool [60, 61] to the human IgG1 crystal structures that were retrieved from the Protein Data Bank (PDB; www.rcsb.org/pdb). Both the separate Fc domain (PDB ID: 4W4N; resolution 1.8 Å [62];) and the intact immunoglobulin structure of IgG1 b12 (PDB ID: 1HZH; resolution 2.7 Å [63];) were used as receptors.
Phagocytosis assay
RAW cells were reseeded every 2-3 days in DMEM medium under GLP conditions. According to the manufacturer's instructions (Vybrant ™ Phagocytosis Assay Kit (V-6694)), RAW cells (80,000 cells per well) were seeded into 96 well plate (Nunc). After adherence, 100 µl E. coli bio-particles were pre-incubated with or without 10 µM GGL27 and added to 20% human plasma. After 1 h of incubation, medium suspensions were aspirated, 100 µl of trypan blue were added to each well, and incubated for 1 min at room temperature. Fluorescence measurements were performed using Victor3 TM Multilabel Counter (PerkinElmer). Phagocytosis activity were measured according to the equation:
Statistical analysis. Values are shown as mean ± SEM. Viable count and radial diffusion assay data are presented as mean ± SD. All graphs were generated using the GraphPad Prism software 6.0.
Supplementary Material
Competing interests
The authors declare that they do not have any competing interests.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from Alfred Österlund Foundation, Knut and Alice Wallenberg Foundation, Ragnar Söderberg Foundation, the Medical Faculty, Lund University, the Swedish Foundation for Strategic Research, the Swedish Research Council, the Crafoord Foundation, Carl Trygger's Foundation, and The Royal Physiographic Society in Lund. Professor Mark S. Johnson is thanked for the excellent computing facilities at the Åbo Akademi University. Use of Biocenter Finland infrastructure at Åbo Akademi (bioinformatics) is acknowledged. The authors wish also acknowledge CSC IT Center for Science, Finland, for computational resources.
Author contributions
PP designed the research. GK, ES, SA, OS-A, CG and PP performed the experiments. AE, PP, SH, AB and HH contributed analytic tools/reagents/materials/analysis tools. PP analyzed the data and HH and PP wrote the paper.
References
- [1].Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci. 2002. February;27(2):67–74. doi: 10.1016/S0968-0004(01)02007-2 [DOI] [PubMed] [Google Scholar]
- [2].Frick IM, Akesson P, Herwald H, et al.. The contact system–a novel branch of innate immunity generating antibacterial peptides. Embo J. 2006. November 29;25(23):5569–78. doi: 10.1038/sj.emboj.7601422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tang YQ, Yeaman MR, Selsted ME. Antimicrobial peptides from human platelets. Infect Immun. 2002. December;70(12):6524–33. doi: 10.1128/IAI.70.12.6524-6533.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Riedel T, Suttnar J, Brynda E, et al.. Fibrinopeptides A and B release in the process of surface fibrin formation. Blood. 2011. February 3;117(5):1700–6. doi: 10.1182/blood-2010-08-300301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Papareddy P, Rydengard V, Pasupuleti M, et al.. Proteolysis of human thrombin generates novel host defense peptides. PLoS Pathog. 2010. April;6(4):e1000857. doi: 10.1371/journal.ppat.1000857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Malmstrom E, Morgelin M, Malmsten M, et al.. Protein C inhibitor–a novel antimicrobial agent. PLoS Pathog. 2009. December;5(12):e1000698. doi: 10.1371/journal.ppat.1000698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Papareddy P, Kalle M, Kasetty G, et al.. C-terminal peptides of tissue factor pathway inhibitor are novel host defense molecules. J Biol Chem. 2010. September 3;285(36):28387–98. doi: 10.1074/jbc.M110.127019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Papareddy P, Kalle M, Sorensen OE, et al.. Tissue factor pathway inhibitor 2 is found in skin and its C-terminal region encodes for antibacterial activity. PloS one. 2012;7(12):e52772. doi: 10.1371/journal.pone.0052772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Papareddy P, Kalle M, Sorensen OE, et al.. The TFPI-2 derived peptide EDC34 improves outcome of gram-negative sepsis. PLoS Pathog. 2013. December;9(12):e1003803. doi: 10.1371/journal.ppat.1003803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kalle M, Papareddy P, Kasetty G, et al.. Proteolytic activation transforms heparin cofactor II into a host defense molecule. J Immunol. 2013. June 15;190(12):6303–10. doi: 10.4049/jimmunol.1203030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Papareddy P, Kalle M, Bhongir RK, et al.. Antimicrobial effects of helix D-derived peptides of human antithrombin III. J Biol Chem. 2014. October 24;289(43):29790–800. doi: 10.1074/jbc.M114.570465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nordahl EA, Rydengård V, Nyberg P, et al.. Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci U S A. 2004. November 30;101(48):16879–84. doi: 10.1073/pnas.0406678101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Powers JP, Hancock RE. The relationship between peptide structure and antibacterial activity. Peptides. 2003. November;24(11):1681–91. doi: 10.1016/j.peptides.2003.08.023 [DOI] [PubMed] [Google Scholar]
- [14].Bulet P, Stocklin R, Menin L. Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev. 2004. April;198:169–84. doi: 10.1111/j.0105-2896.2004.0124.x [DOI] [PubMed] [Google Scholar]
- [15].Yount NY, Bayer AS, Xiong YQ, et al.. Advances in antimicrobial peptide immunobiology. Biopolymers. 2006. May 30;84(5):435–458. doi: 10.1002/bip.20543 [DOI] [PubMed] [Google Scholar]
- [16].Durr UH, Sudheendra US, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta. 2006. April 4;1758(9):1408–25. doi: 10.1016/j.bbamem.2006.03.030 [DOI] [PubMed] [Google Scholar]
- [17].Lehrer RI, Ganz T. Cathelicidins: a family of endogenous antimicrobial peptides. Curr Opin Hematol. 2002. January;9(1):18–22. doi: 10.1097/00062752-200201000-00004 [DOI] [PubMed] [Google Scholar]
- [18].Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389–95. doi: 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- [19].Lwaleed BA, Bass PS. Tissue factor pathway inhibitor: structure, biology and involvement in disease. J Pathol. 2006. February;208(3):327–39. doi: 10.1002/path.1871 [DOI] [PubMed] [Google Scholar]
- [20].Girard TJ, Warren LA, Novotny WF, et al.. Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989. April 6;338(6215):518–20. doi: 10.1038/338518a0 [DOI] [PubMed] [Google Scholar]
- [21].Mine S, Yamazaki T, Miyata T, et al.. Structural mechanism for heparin-binding of the third Kunitz domain of human tissue factor pathway inhibitor. Biochemistry. 2002. January 8;41(1):78–85. doi: bi011299g [pii] [DOI] [PubMed] [Google Scholar]
- [22].Crawley JT, Lane DA. The haemostatic role of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2008. February;28(2):233–42. doi: 10.1161/ATVBAHA.107.141606 [DOI] [PubMed] [Google Scholar]
- [23].Ohkura N, Enjyoji K, Kamikubo Y, et al.. A novel degradation pathway of tissue factor pathway inhibitor: incorporation into fibrin clot and degradation by thrombin. Blood. 1997. September 1;90(5):1883–92. [PubMed] [Google Scholar]
- [24].Li A, Wun TC. Proteolysis of tissue factor pathway inhibitor (TFPI) by plasmin: effect on TFPI activity. Thromb Haemost. 1998. September;80(3):423–7. [PubMed] [Google Scholar]
- [25].Cunningham AC, Hasty KA, Enghild JJ, et al.. Structural and functional characterization of tissue factor pathway inhibitor following degradation by matrix metalloproteinase-8. Biochem J. 2002. October 15;367(Pt 2):451–8. doi: 10.1042/BJ20020696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yun TH, Cott JE, Tapping RI, et al.. Proteolytic inactivation of tissue factor pathway inhibitor by bacterial omptins. Blood. 2009. January 29;113(5):1139–48. doi: 10.1182/blood-2008-05-157180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Park CT, Creasey AA, Wright SD. Tissue factor pathway inhibitor blocks cellular effects of endotoxin by binding to endotoxin and interfering with transfer to CD14. Blood. 1997. June 15;89(12):4268–74. [PubMed] [Google Scholar]
- [28].Hembrough TA, Ruiz JF, Swerdlow BM, et al.. Identification and characterization of a very low density lipoprotein receptor-binding peptide from tissue factor pathway inhibitor that has antitumor and antiangiogenic activity. Blood. 2004. May 1;103(9):3374–80. doi: 10.1182/blood-2003-07-2234 [DOI] [PubMed] [Google Scholar]
- [29].Wesselschmidt R, Likert K, Girard T, et al.. Tissue factor pathway inhibitor: the carboxy-terminus is required for optimal inhibition of factor Xa. Blood. 1992. April 15;79(8):2004–10. [PubMed] [Google Scholar]
- [30].Ettelaie C, Adam JM, James NJ, et al.. The role of the C-terminal domain in the inhibitory functions of tissue factor pathway inhibitor. FEBS Lett. 1999. December 17;463(3):341–4. doi: 10.1016/S0014-5793(99)01663-4 [DOI] [PubMed] [Google Scholar]
- [31].Maroney SA, Ferrel JP, Pan S, et al.. Temporal expression of alternatively spliced forms of tissue factor pathway inhibitor in mice. J Thromb Haemost. 2009. July;7(7):1106–13. doi: 10.1111/j.1538-7836.2009.03454.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].DeLano WL, Ultsch MH, de Vos AM, et al.. Convergent solutions to binding at a protein-protein interface. Science. 2000. February 18;287(5456):1279–83. doi: 10.1126/science.287.5456.1279 [DOI] [PubMed] [Google Scholar]
- [33].von Pawel-Rammingen U Johansson BP, Bjorck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. The EMBO journal. 2002. April 2;21(7):1607–15. doi: 10.1093/emboj/21.7.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cope I, Mitchell P. Plasmin and plasminogen levels in pregnancy. Aust N Z J Obstet Gynaecol. 1965. August;5(3):152–5. doi: 10.1111/j.1479-828X.1965.tb00310.x [DOI] [PubMed] [Google Scholar]
- [35].Ponczek MB, Gailani D, Doolittle RF. Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost. 2008. November;6(11):1876–83. doi: 10.1111/j.1538-7836.2008.03143.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Conway EM. Reincarnation of ancient links between coagulation and complement. J Thromb Haemost. 2015. June;13(Suppl 1):S121–32. doi: 10.1111/jth.12950 [DOI] [PubMed] [Google Scholar]
- [37].Ranasinghe S, McManus DP. Structure and function of invertebrate Kunitz serine protease inhibitors. Dev Comp Immunol. 2013. March;39(3):219–27. doi: 10.1016/j.dci.2012.10.005 [DOI] [PubMed] [Google Scholar]
- [38].Maroney SA, Mast AE. New insights into the biology of tissue factor pathway inhibitor. J Thromb Haemost. 2015. June;13(Suppl 1):S200–7. doi: 10.1111/jth.12897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Keizer MP, Pouw RB, Kamp AM, et al.. TFPI inhibits lectin pathway of complement activation by direct interaction with MASP-2. Eur J Immunol. 2015. February;45(2):544–50. doi: 10.1002/eji.201445070 [DOI] [PubMed] [Google Scholar]
- [40].Francischetti IM, Valenzuela JG, Andersen JF, et al.. Ixolaris, a novel recombinant tissue factor pathway inhibitor (TFPI) from the salivary gland of the tick, Ixodes scapularis: identification of factor X and factor Xa as scaffolds for the inhibition of factor VIIa/tissue factor complex. Blood. 2002. May 15;99(10):3602–12. doi: 10.1182/blood-2001-12-0237 [DOI] [PubMed] [Google Scholar]
- [41].Modica MV, Lombardo F, Franchini P, et al.. The venomous cocktail of the vampire snail Colubraria reticulata (Mollusca, Gastropoda). BMC Genomics. 2015. June 09;16:441. doi: 10.1186/s12864-015-1648-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Campbell CL, Vandyke KA, Letchworth GJ, et al.. Midgut and salivary gland transcriptomes of the arbovirus vector Culicoides sonorensis (Diptera: Ceratopogonidae). Insect Mol Biol. 2005. April;14(2):121–36. doi: 10.1111/j.1365-2583.2004.00537.x [DOI] [PubMed] [Google Scholar]
- [43].Nonaka M, Kimura A. Genomic view of the evolution of the complement system. Immunogenetics. 2006. September;58(9):701–13. doi: 10.1007/s00251-006-0142-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nonaka M, Yoshizaki F. Evolution of the complement system. Mol Immunol. 2004. February;40(12):897–902. doi: 10.1016/j.molimm.2003.10.009 [DOI] [PubMed] [Google Scholar]
- [45].Nonaka M. Evolution of the complement system. Subcell Biochem. 2014;80:31–43. doi: 10.1007/978-94-017-8881-6_3 [DOI] [PubMed] [Google Scholar]
- [46].Sun Y, Wei Z, Li N, et al.. A comparative overview of immunoglobulin genes and the generation of their diversity in tetrapods. Dev Comp Immunol. 2013 Jan-Feb;39(1-2):103–9. doi: 10.1016/j.dci.2012.02.008 [DOI] [PubMed] [Google Scholar]
- [47].Semba U, Shibuya Y, Okabe H, et al.. Whale Hageman factor (factor XII): prevented production due to pseudogene conversion. Thromb Res. 1998. April 01;90(1):31–7. doi: 10.1016/S0049-3848(97)00307-1 [DOI] [PubMed] [Google Scholar]
- [48].Kasetty G, Smeds E, Holmberg E, et al.. Vertebrate TFPI-2 C-terminal peptides exert therapeutic applications against Gram-negative infections. BMC Microbiol. 2016;16(1):129. doi: 10.1186/s12866-016-0750-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tamura K, Stecher G, Peterson D, et al.. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013. December;30(12):2725–9. doi: 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999. September 17;292(2):195–202. doi: 10.1006/jmbi.1999.3091 [DOI] [PubMed] [Google Scholar]
- [51].Buchan DW, Minneci F, Nugent TC, et al.. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013. July;41(Web Server issue):W349–57. doi: 10.1093/nar/gkt381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Drozdetskiy A, Cole C, Procter J, et al.. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015. July 01;43(W1):W389–94. doi: 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lamiable A, Thevenet P, Rey J, et al.. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016. July 08;44(W1):W449–54. doi: 10.1093/nar/gkw329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Thevenet P, Shen Y, Maupetit J, et al.. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012. July;40(Web Server issue):W288–93. doi: 10.1093/nar/gks419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shen Y, Maupetit J, Derreumaux P, et al.. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J Chem Theory Comput. 2014. October 14;10(10):4745–58. doi: 10.1021/ct500592m [DOI] [PubMed] [Google Scholar]
- [56].WLJJCaJD. Madura. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi: 10.1063/1.445869 [DOI] [Google Scholar]
- [57].AMBER 2017, University of California, San Francisco. [Internet]. 2017. [Google Scholar]
- [58].Duan Y, Wu C, Chowdhury S, et al.. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003. December;24(16):1999–2012. doi: 10.1002/jcc.10349 [DOI] [PubMed] [Google Scholar]
- [59].Mirza MU, Rafique S, Ali A, et al.. Erratum: Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci Rep. 2017. April 11;7:44633. doi: 10.1038/srep44633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kozakov D, Beglov D, Bohnuud T, et al.. How good is automated protein docking? Proteins. 2013. December;81(12):2159–66. doi: 10.1002/prot.24403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kozakov D, Hall DR, Xia B, et al.. The ClusPro web server for protein-protein docking. Nat Protoc. 2017. February;12(2):255–278. doi: 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kiyoshi M, Caaveiro JM, Kawai T, et al.. Structural basis for binding of human IgG1 to its high-affinity human receptor FcgammaRI. Nat Commun. 2015. April 30;6:6866. doi: 10.1038/ncomms7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Saphire EO, Parren PW, Pantophlet R, et al.. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001. August 10;293(5532):1155–9. doi: 10.1126/science.1061692 [DOI] [PubMed] [Google Scholar]
- [64].Gaboriaud C, Juanhuix J, Gruez A, et al.. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem. 2003. November 21;278(47):46974–82. doi: 10.1074/jbc.M307764200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.