

Abstract
Although bronchogenic carcinomas progress through a very well defined sequence of metaplasia, dysplasia and carcinoma in situ, very little is known about the early progression of glandular neoplasms of the lung. In particular, the early precursor lesion from which fully malignant adenocarcinomas arise has effectively eluded recognition, at least until recently. Several lines of evidence now implicate atypical adenomatous hyperplasia (AAH) as an initial morphologic stage in multistep lung tumorigenesis. Despite its small size, AAH can be appreciated at the light microscopic level and characterized at the molecular genetic level. Indeed, the genetic characterization of AAH promises to further our understanding of lung cancer development and might facilitate the design of novel strategies for early detection of lung cancer.
Keywords: adenocarcinoma, atypical adenomatous hyperplasia, atypical alveolar hyperplasia, K-ras, loss of heterozygosity, lung cancer, p53, telomerase
Introduction
Lung cancer is the leading cause of cancer death in the industrialized world [1]. A dismally low cure rate largely reflects the propensity of lung cancer to present as clinically advanced tumors: most lung cancers are discovered late during their clinical course, by which time the options for effective therapeutic intervention are limited. Of these lung cancers, adenocarcinoma is now the most common subtype [2]. This subtype is particularly notorious for eluding early detection. Even vigilant screening of sputum samples for morphologic evidence of early glandular neoplasia in individuals at risk is ineffective in curbing mortality rates [3]. Adenocarcinomas of the lung tend to arise peripherally, and malignant cells are not shed into the sputum until these tumors have developed into sizeable and frankly invasive cancers. The inability to detect early glandular neoplasia has not only frustrated efforts to treat patients effectively but has also hampered attempts to address fundamental questions about how normal alveolar cells become malignant. Understanding the early steps of lung tumorigenesis at the molecular genetic level might facilitate the development of more effective strategies for the prevention, early diagnosis, and treatment of lung cancer.
Historical concepts
Unlike central bronchogenic tumors, in which unwavering progression through the metaplasia-dysplasia-carcinoma in situ sequence has long been recognized, the morphologic progression of early glandular neoplasia has been shrouded in mystery. Earliest attempts to explain the origin of lung adenocarcinoma date back at least to the observation by Friedrich in 1939 that peripheral lung cancers often coincide with subpleural anthrocotic scars [4]. According to the 'scar cancer' concept, scar formation acts as an inciting focus for epithelial regeneration, proliferation, and ultimate neoplastic transformation. Over time, this concept has fallen into disrepute. Studies indicate that tumor-associated scarring, in most instances, seems to be a consequence of invasive tumor growth rather than a pre-existing soil in which neoplasms germinate and grow [5,6,7,8].
The inability to understand the precursor lesion of glandular neoplasia has perpetuated the desperate concept of de novo carcinoma, a vague notion that fully malignant lung adenocarcinoma materializes spontaneously in the absence of a precursor lesion. The concept of de novo carcinoma has now been rendered obsolete by the contemporary model of tumorigenesis. Simply stated, the accumulation of specific genetic alterations drives tumorigenesis forward through a sequential progression of well-defined morphologic steps [9]. The current model of tumorigenesis stipulates the presence, no matter how transient and elusive, of a glandular precursor to peripheral lung adenocarcinoma.
In 1988, Miller et al [10] observed that nearly 10% of all lungs resected for adenocarcinomas also harbored minute discrete foci of cytologically atypical bronchiolalveolar cells. In light of the cytologic atypia and the strong association with lung adenocarcinomas, they postulated that these small lesions represented the elusive precursor lesion: a glandular carcinoma in situ possessing the potential to evolve into a frankly malignant adenocarcinoma. These atypical proliferations are now commonly referred to as atypical adenomatous hyperplasia (AAH) or atypical alveolar hyperplasia.
Atypical adenomatous hyperplasia: a morphologic manifestation of early glandular neoplasia
On meticulous inspection, AAH can sometimes be seen with the naked eye as tan-to-gray firm nodules that range in size from about 1 to 7 mm (Fig. 1). By light microscopy, they are characterized by the proliferation of cuboidal to columnar epithelial cells with various degrees of cytologic atypia (Fig. 2). These cells line intact, albeit thickened, alveolar septa [11]. In contrast to reactive pneumocyte hyperplasia, they are not associated with significant inflammation and fibrosis of the surrounding lung parenchyma; unlike most lung adenocarcinomas they do not demonstrate infiltrative growth with disruption of normal lung architecture. The range of cytoarchitectural atypia falls along a spectrum of increasing severity. Low-grade lesions are characterized by a single layer of round to cuboidal cells with low cellular density, small cell size, and minimal variation in nuclear size, shape, and chromaticity. In low-grade lesions there is minimal thickening of the alveolar septum. With progressive atypia, AAH demonstrates a higher cellular density with stratification of the epithelial cells, more significant cytological atypia, and more extensive fibrosis and thickening of the alveolar septa. Because of problems with reproducibility, grading of AAH is not routinely employed [12]. Moreover, the precise point at which a high-grade AAH becomes a bronchioloalveolar carcinoma remains to be defined satisfactorily.
Figure 1.
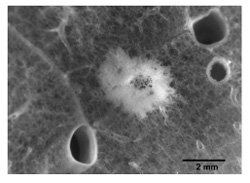
Cut surface of lung showing a 4 mm nodule of atypical adenomatous hyperplasia (magnification ×14).
Figure 2.
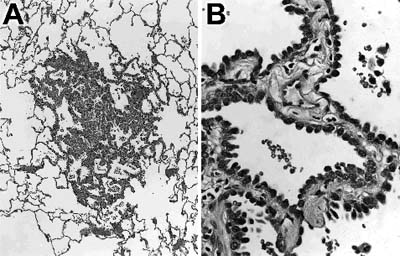
Histologic section of an atypical adenomatous hyperplasia. (A) The lesion shows preservation of normal lung architecture and is surrounded by normal lung parenchyma. (B) Higher magnification shows a single layer of cuboidal cells with cytologic atypia lines septae that are mildly thickened.
Several lines of evidence have bolstered the proposition that AAH is a precursor lesion from which lung adenocarcinomas arise. First, there is guilt by association: lungs with adenocarcinomas often also harbor one or several AAHs [10,11,13, 14,15,16]. Indeed, foci of AAH are found in as many as 25% of lungs resected for primary adenocarcinoma [15] but in only 2% of post-mortem lungs from patients without lung neoplasms [17]. Second, AAH is usually encountered in lungs of patients who smoke tobacco, a known initiator and promoter of lung carcinogenesis [11,14]. Third, AAH seems to bridge the gap between reactive pneumocyte hyperplasia and well-differentiated adenocarcinoma. AAH occupies some middle ground in terms of mean nuclear size [18,19,20], DNA content [18,21,22], cell proliferation [22,23,24,25], expression of carcinoembryonic antigen [19,26], and altered expression of important regulators of the cell cycle such as the p53 protein product [19,25,27,28] and the p27/Kip 1 protein product [29].
Molecular genetic characterization of AAH
Despite mounting evidence, the true nature of AAH could not be unequivocally established in the absence of molecular genetic characterization. On morphologic grounds alone, AAH was difficult to distinguish from a reactive proliferation. Reactive pneumocyte hyperplasia can be seen in response to various types of lung injury, and the degree of reactive cytologic atypia can even exceed the neoplastic atypia of some lung cancers. At the other extreme, AAH could not be readily distinguished from an advanced pattern of tumor spread such as creeping extension from an adjacent carcinoma or metastatic implantation from a non-contiguous carcinoma. For example, the advancing edge of non-mucinous bronchioloalveolar carcinomas is morphologically indistinguishable from AAH, and the pattern of multiple small nodules due to metastatic implantation is a characteristic feature of mucinous bronchioloalveolar carcinomas. Thus, the initial role of molecular genetic analysis in the evaluation of AAH was to establish its nature as an early glandular neoplasm in distinction to a reactive proliferation or some form of advanced tumor spread.
Although AAHs have long been recalcitrant to molecular analysis owing to their small size, the refinement of microdissection techniques coupled with improvements in PCR amplification has made it possible to study them at the molecular genetic level. Initial strategies focused on the K-ras proto-oncogene as a likely target of mutational activation, for several reasons: first, activating mutations of K-ras are detected in 24-50% of lung adenocarcinomas, but they are much less frequent in other subtypes of lung cancer [30,31]; second, K-ras mutations are easy to detect because they involve straightforward base substitutions that predictably target codon 12 [30]; and third, models of lung tumorigenesis addressing the chronological sequence of mutational events predict that K-ras activation is an early event that precedes malignant growth [32,33,34].
As expected for an early glandular neoplasm of the lung, AAHs were found to harbor activating mutations at codon 12 of the K-ras oncogene [14,35]. Furthermore, the mutational profile of these K-ras alterations clearly points to a relationship between AAHs and their fully malignant counterpart. We found that the frequencies of codon 12 K-ras mutations in AAHs and lung adenocarcinomas are comparable (39% and 41%, respectively), and the predominant type of mutation is a G→T transversion at position 1 or 2 for both AAH and lung adenocarcinomas [14]. The clonal nature of AAH has since been directly confirmed by X chromosome inactivation analysis. In their evaluation of AAHs from female patients, Niho et al [36] found that all 10 informative cases of AAH were mono-clonal. The finding that AAH is a clonal proliferation of cells that harbor activating mutations of a well-recognized oncogene clearly points to its neoplastic nature.
When AAHs and their synchronous lung carcinomas are compared for patterns of base substitutions at codon 12 of the K-ras oncogene, the paired lesions often do not share the same base changes [14]. Indeed, the pattern of base changes can be conspicuously divergent for any individual patient. The discordant nature of these base changes suggests that AAHs arising in patients with primary lung carcinomas are independent lesions. They do not reflect direct extension from a nearby carcinoma or metastatic implantation from a non-contiguous carcinoma.
With confirmation of AAH as an early stage in glandular neoplasia, the timing and sequence of specific genetic alterations now can be delineated along the entire pathway of lung tumorigenesis. In an effort to separate early from late mutational events, AAH has been evaluated for key genetic alterations that are commonly present in lung adenocarcinomas, including loss of heterozygosity (LOH) at selected chromosomal arms, alterations of the p53 tumor suppressor gene, and activation of telomerase.
LOHs on chromosomes 3p, 9p and 17p are common in adenocarcinomas of the lung [37,38] and are believed to point to the inactivation of certain tumor suppressor genes that are critical in lung tumorigenesis. In lung cancer, LOH at 3p, 9p and 17p probably targets the fragile histidine triad gene (i.e. FHIT), the p16 gene (i.e. MTS1), and the p53 gene, respectively. Deletions on chromosomal arms 3p, 9p and 17p are believed to occur during the earliest stages of lung tumorigenesis, largely on the basis of the observation that these alterations might be widespread throughout the lungs of smokers, even in the absence of overt histopathologic changes in epithelial dysplasia [39,40]. Not surprisingly, then, LOH of these chromosomal arms has been identified in early as well as advanced stages of glandular neoplasia [24,41,42]. Among the 20 AAHs that Kitaguchi et al evaluated [24], LOH was present at 3p (18%), 9p (13%) and 17p (6%).
Mutational inactivation of the p53 tumor suppressor gene is a frequent genetic event in lung cancer. About half of all non-small cell lung cancers harbor mutations of the p53 gene [43]. However, alteration of the p53 tumor suppressor gene is probably not a common event in the earliest stages of glandular neoplasia. Using immunohistochemical detection of p53 protein accumulation as an indirect measurement of p53 gene alterations, we observed p53 protein accumulation in only 11% of AAHs [27], a frequency within the range of 8-17% reported by Kitamura et al [19,25]. Moreover, immunohistochemical staining of p53 often overestimates the true frequency of actual mutations in the p53 gene [44]. Of four AAHs demonstrating high levels of p53 protein accumulation, we were able to document a mutation in the p53 gene by DNA sequence analysis in only one of these cases [27].
Telomerase activity is required for the perpetuation of cancer cells. Activation of telomerase is universally present in malignant tumors including lung adenocarcinomas. In contrast to these fully malignant neoplasms, telomerase activity was not detected in any of the six AAHs evaluated by Yashima et al [45].
Genetic progression model for glandular neoplasia of the lung
The concept that AAH constitutes a lone step in early lung tumorigenesis might be too limited. Instead, AAH might encompass a heterogeneous population of lesions representing different points along the progression toward overt lung adenocarcinoma [25]. Under the light microscope, the cytological and architectural atypia that characterize AAH falls along a morphologic spectrum ranging from mild to severe. Accordingly, some groups have attempted to subclassify AAH based on the severity of this atypia. In addition to low-grade and high-grade categories of AAH, some schemes also recognize a borderline lesion that, on morphological grounds, seems to be in transition from high-grade AAH to early bronchioloalveolar carcinoma (Fig. 3) [19,25,46]. These distinct histologic stages might be driven by sequential genetic events. Activating mutations of the K-ras oncogene tend to be uniformly distributed across all histopathologic grades of AAH [27]. In contrast, LOHs at 3p, 9p and 17p tend to be restricted to AAHs with moderate or severe cytologic atypia [24]. The frequency of p53 protein accumulation also increases in proportion to the severity of atypia [19,25,27,28]. We noted that p53 protein accumulation is generally not observed in AAHs with low-grade morphologic features, but high-level p53 protein accumulation is present in half of those lesions with severe atypia [27].
Figure 3.
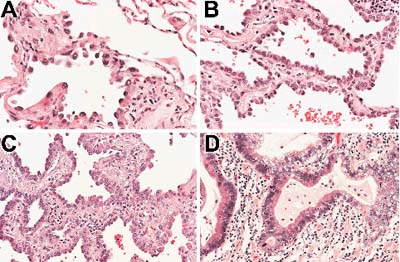
Histologic progression of atypical adenomatous hyperplasia (AAH). (A) Low-grade AAH characterized by small atypical cells lining a delicate interstitium. (B) In this high-grade AAH the cells are more crowded, and there is increased thickening of the septal walls. (C) This lesion demonstrates morphologic transition between high-grade AAH and an early bronchioloalveolar carcinoma. It is characterized by further cellular crowding, increased cytologic atypia and more prominent fibrosis of the septal walls. (D) In contrast to AAH, this well-differentiated adenocarcinoma shows severe cytologic atypia and overt infiltration of the lung parenchyma.
Consistent with the current model of tumorigenesis is the observation that the development of AAH and its progression to lung adenocarcinoma depends on the sequential accumulation of specific genetic alterations (Fig. 4). Activation of mutations of the K-ras oncogene seems to be an initiating event. Progression to increasing degrees of morphologic dysplasia might require the silencing of key tumor suppressor genes such as p16 and FHIT. Ultimately, the inactivation of p53 coupled with the activation of telomerase might participate in the critical transition from premalignant to a malignant growth. Further unraveling of these sequential genetic alterations should facilitate rationale strategies for more effective tumor detection and surveillance. Molecular analysis of exfoliated cells is now feasible in the detection of clinically silent neoplasms. K-ras mutations [47,48], p53 gene mutations [47,48], telomerase activity [49], and alterations of microsatellite markers [48,50] have all been idenitified in sputums and lavages from patients with cancers of the respiratory tract.
Figure 4.
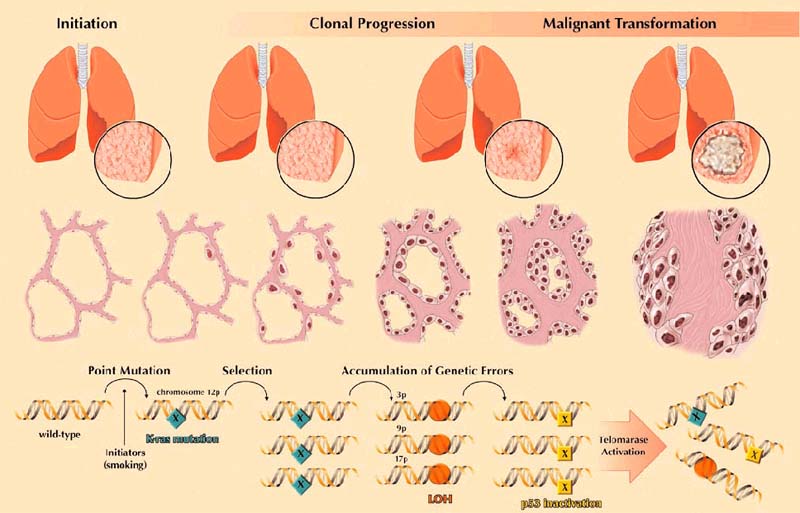
Progression model for glandular neoplasia of the lung. The clinical (row 1) and histologic (row 2) progression of glandular neoplasia is driven at the genetic level (row 3) by the accumulation of specific alterations of oncogenes and tumor suppressor genes. These genetic alterations tend to follow a consistent, but not invariable, chronological order. LOH, loss of heterozygosity.
Clinical significance of AAH
Although the neoplastic nature of AAH has been confirmed at the molecular genetic level, the frequency and pace of its clinical progression to overt malignancy are largely unknown. AAH tends to be a multifocal process such that the finding of AAH in a resection specimen strongly indicates the presence of other unresected AAHs. Potentially, these could serve as a source of cancer relapse. However, for patients who undergo the surgical removal of low-stage lung cancer, the presence of AAH does not unfavorably affect prognosis [13,16,51]. Indeed, Takigawa et al [13] identified concurrent AAH in lungs resected for low-stage adenocarcinomas as a favorable prognostic indicator. Although much remains to be learned of the natural history of AAH, its presence does not necessarily indicate sure and unremitting progression to an overtly malignant adenocarcinoma.
Continuing technological improvements now permit the pre-operative identification of AAH with high-resolution imaging techniques (that is, high-resolution spiral computed tomography). These non-invasive techniques permit the longitudinal observation of AAH and thus provide valuable insight into its natural history. The Early Lung Cancer Action Project found that out of 99 small (that is, 1–5 mm) lung nodules detected by repeat computed tomography screening in patients at high risk for lung cancer, only one nodule (1%) enlarged during follow-up and was ultimately confirmed as a lung cancer [52]. At present, the presence of AAH does not merit aggressive surgical intervention. Instead, the detection of AAH in a patient at risk for lung cancer should initiate close follow-up, mandate the cessation of smoking, and prompt consideration of chemopreventive therapy.
Concluding remarks
Most patients with lung cancer are first diagnosed when their tumors are already clinically and biologically advanced. For a long time the early non-invasive stages of glandular neoplasia eluded recognition. Recently, various lines of evidence have converged to identify AAH as a non-invasive precursor lesion from which fully malignant adenocarcinomas arise. Admittedly, AAH is a microscopic lesion that is incidentally noted in lungs resected for primary lung cancer. Given its small size, it is virtually invisible to conventional methods of screening for early asymptotic lung cancer. Nevertheless, unraveling the molecular genetic alterations that initiate the development and drive the progression of these lesions might provide new insight into the biology of lung cancer, and might facilitate the design of novel strategies for early detection of lung cancer. The natural history of AAH, namely the frequency and pace of its progression to an overtly malignant carcinoma, is a focus of continuing and future investigations. The refinement of high-resolution imaging should help to clarify this critical unresolved issue.
References
- Samet JM. The epidemiology of lung cancer. Chest. 1993;103:20S–29S. doi: 10.1378/chest.103.1_supplement.20s. [DOI] [PubMed] [Google Scholar]
- Travis WD, Travis LB, Devesa SS. Lung cancer. Cancer . 1995;75:191–202. doi: 10.1002/1097-0142(19950101)75:1+<191::aid-cncr2820751307>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Frost JK, Ball WC, Jr, Levin ML, Tockman MS, Baker RR, Carter D, Eggleston JC, Erozan YS, Gupta PK, Khouri NF. Early lung cancer detection: results of the initial (prevalence) radiologic and cytologic screening in the Johns Hopkins study. Am Rev Respir Dis. 1984;130:549–554. doi: 10.1164/arrd.1984.130.4.549. [DOI] [PubMed] [Google Scholar]
- Friedrich G. Periphere Lungenkrebse auf dem boden pleuranaher Narben. Virchows Arch (Pathol Anat) 1939;304:230–247. [Google Scholar]
- Madri JA, Carter D. Scar cancers of the lung: origin and significance. Hum Pathol. 1984;15:625–631. doi: 10.1016/s0046-8177(84)80286-5. [DOI] [PubMed] [Google Scholar]
- Shimosato Y, Suzuki A, Hashimoto T, Nishiwaki Y, Kodama T, Yoneyama T, Kameya T. Prognostic implications of fibrotic focus (scar) in small peripheral lung cancers. Am J Surg Pathol. 1980;4:365–373. doi: 10.1097/00000478-198008000-00005. [DOI] [PubMed] [Google Scholar]
- Kolin A, Koutoulakis T. Role of arterial occlusion in pulmonary scar cancers. Hum Pathol. 1988;19:1161–1167. doi: 10.1016/s0046-8177(88)80147-3. [DOI] [PubMed] [Google Scholar]
- Kung IT, Lui IO, Loke SL, Khin MA, Mok CK, Lam WK, So SY. Pulmonary scar cancer. A pathologic reappraisal. Am J Surg Pathol. 1985;9:391–400. doi: 10.1097/00000478-198506000-00001. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Miller RR, Nelems B, Evans KG, Muller NL, Ostrow DN. Glandular neoplasia of the lung: a proposed analogy to colonic tumors. . Cancer. 1988;61:1009–1014. doi: 10.1002/1097-0142(19880301)61:5<1009::aid-cncr2820610525>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Nakanishi N. Alveolar epithelial hyperplasia and adenocarcinoma of the lung. Arch Pathol Lab Med. 1990;114:363–368. [PubMed] [Google Scholar]
- Travis WD, Colby TV, Corrin B, Shimosato Y, Brambilla E. Histologic Typing of Lung and Pleural Tumours, 3rd edition; from World Health Organization International Histological classification of tumours. 1999.
- Takigawa N, Segawa Y, Nakata M, Saeki H, Mandai K, Kishino D, Shimono M, Ida M, Eguchi K. Clinical investigation of atypical adenomatous hyperplasia of the lung. Lung Cancer. 1999;25:115–121. doi: 10.1016/S0169-5002(99)00055-0. [DOI] [PubMed] [Google Scholar]
- Westra WH, Baas IO, Hruban RH, Askin FB, Wilson K, Offerhaus GJA, Slebos RJC. K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res. 1996;56:2224–2228. [PubMed] [Google Scholar]
- Rao SK, Fraire AE. Alveolar cell hyperplasia in association with adenocarcinoma of lung. Mod Pathol. 1995;8:165–169. [PubMed] [Google Scholar]
- Chapman AD, Kerr KM. The association between atypical adenomatous hyperplasia and primary lung cancer. Br J Cancer . 2000;83:632–636. doi: 10.1054/bjoc.2000.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DJ, Mori M, Roggli VL, Fraire AE. Prevalence of pulmonary atypical alveolar cell hyperplasia in an autopsy population: a study of 100 cases. Mod Pathol. 1997;10:469–473. [PubMed] [Google Scholar]
- Yokozaki M, Kodama T, Yokose T, Matsumoto T, Mukai K. Differentiation of atypical adenomatous hyperplasia and adenocarcinoma of the lung by use of DNA ploidy and morphometric analysis. . Mod Pathol. 1996;9:1156–1164. [PubMed] [Google Scholar]
- Kitamura H, Kameda Y, Nakamura N, Inayama Y, Nakatani Y, Shibagaki T, Ito T, Hayashi H, Kimura H, Kanisawa M. Atypical adenomatous hyperplasia and bronchoalveolar lung carcinoma. Analysis by morphometry and the expressions of p53 and carcinoembryonic antigen. Am J Surg Pathol. 1996;20:553–562. doi: 10.1097/00000478-199605000-00002. [DOI] [PubMed] [Google Scholar]
- Kodama T, Biyajima S, Watanabe S, Shimosato Y. Morphometric study of adenocarcinomas and hyperplastic epithelial lesions in the peripheral lung. Am J Clin Pathol. 1986;85:146–151. doi: 10.1093/ajcp/85.2.146. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Noguchi M, Tsuchiya R, Kodama T, Shimosato Y. Clonal growth of atypical adenomatous hyperplasia of the lung: cytofluorometric analysis of nuclear DNA content. Mod Pathol. 1990;3:314–320. [PubMed] [Google Scholar]
- Nakanishi K, Hiroi S, Kawai T, Suzuki M, Torikata C. Argyrophilic nucleolar-organizer region counts and DNA status in bronchioloalveolar epithelial hyperplasia and adenocarcinoma of the lung. . Hum Pathol. 1998;29:235–239. doi: 10.1016/s0046-8177(98)90041-7. [DOI] [PubMed] [Google Scholar]
- Kerr KM, Carey FA, King G, Lamb D. Atypical alveolar hyperplasia: relationship with pulmonary adenocarcinoma, p53, and c-erbB-2 expression. J Pathol. 1994;174:249–256. doi: 10.1002/path.1711740404. [DOI] [PubMed] [Google Scholar]
- Kitaguchi S, Takeshima Y, Nishisaka T, Inai K. Proliferative activity, p53 expression and loss of heterozygosity on 3p, 9p and 17p in atypical adenomatous hyperplasia of the lung. Hiroshima J Med Sci . 1998;47:17–25. [PubMed] [Google Scholar]
- Kitamura H, Kameda Y, Nakamura N, Nakatani Y, Inayama Y, Iida M, Noda K, Ogawa N, Shibagaki T, Kanisawa M. Proliferative potential and p53 overexpression in precursor and early stage lesions of bronchioloalveolar lung carcinoma. Am J Pathol. 1995;146:876–887. [PMC free article] [PubMed] [Google Scholar]
- Mori M, Tezuka F, Chiba R, Funae Y, Watanabe M, Nukiwa T, Takahashi T. Atypical adenomatous hyperplasia and adenocarcinoma of the human lung: their heterology in form and analogy in immunohistochemical characteristics. Cancer. 1996;77:665–674. doi: 10.1002/(SICI)1097-0142(19960215)77:4<665::AID-CNCR12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Slebos RJC, Baas IO, Clement MJ, Offerhaus GJA, Askin FB, Hruban RH, Westra WH. p53 alterations in atypical alveolar hyperplasia of the human lung. Hum Pathol. 1998;29:801–808. doi: 10.1016/s0046-8177(98)90448-8. [DOI] [PubMed] [Google Scholar]
- Yokose T, Ito Y, Ochiai A. High prevalence of atypical adenomatous hyperplasia of the lung in autopsy specimens from elderly patients with malignant neoplasms. Lung Cancer. 2000;29:125–130. doi: 10.1016/S0169-5002(00)00101-X. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Ito T, Yazawa T, Ikeda M, Inayama Y, Nakatani Y, Kameda Y, Nakamura N, Kitamura H. Reduced expression of p27/Kip1 is associated with the development of pulmonary adenocarcinoma. J Pathol. 2000;192:26–31. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH653>3.3.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Rodenhuis S, Slebos RJ. Clinical significance of ras oncogene activation in human lung cancer. . Cancer Res. 1992;52:2665–2669. [PubMed] [Google Scholar]
- Mills NE, Fishman CL, Rom WN, Dubin N, Jacobson DR. Increased prevalence of K-ras oncogene mutations in lung adenocarcinomas. Cancer Res. 1995;55:1444–1447. [PubMed] [Google Scholar]
- Li Z-H, Zheng J, Weiss LM, Shibata D. c-K-ras and p53 mutations occur very early in adenocarcinoma of the lung. . Am J Pathol. 1994;144:303–309. [PMC free article] [PubMed] [Google Scholar]
- Westra WH, Slebos RJC, Offerhaus GJA, Goodman SN, Evers SG, Kensler TW, Askin FB, Rodenhuis S, Hruban RH. K-ras oncogene activation in lung adenocarcinomas from former smokers: evidence that K-ras mutations are an early and irreversible event in the development of adenocarcinoma of the lung. . Cancer. 1993;72:432–438. doi: 10.1002/1097-0142(19930715)72:2<432::aid-cncr2820720219>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Kumar R, Sukumar S, Barbacid M. Activation of ras oncogenes preceding the onset of neoplasia. . Science. 1990;248:1101–1104. doi: 10.1126/science.2188364. [DOI] [PubMed] [Google Scholar]
- Cooper CA, Carby FA, Bubb VJ, Lamb D, Kerr KM, Wyllie AH. The pattern of K-ras mutation in pulmonary adenocarcinoma defines a new pathway of tumour development in the human lung. J Pathol. 1997;181:401–404. doi: 10.1002/(SICI)1096-9896(199704)181:4<401::AID-PATH799>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Niho S, Yokose T, Suzuki K, Kodama T, Nishiwaki Y, Mukai K. Monoclonality of atypical adenomatous hyperplasia of the lung. . Am J Pathol. 1999;154:249–254. doi: 10.1016/S0002-9440(10)65271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen I, Bujard M, Petersen S, Wolf G, Goeze A, Schwendel A, Langreck H, Gellert K, Reichel M, Just K, du MS, Cremer T, Dietel M, Ried T. Patterns of chromosomal imbalances in adenocarcinoma and squamous cell carcinoma of the lung. Cancer Res. 1997;57:2331–2335. [PubMed] [Google Scholar]
- Sato S, Nakamura Y, Tsuchiya E. Difference of allelotype between squamous cell carcinoma and adenocarcinoma of the lung. Cancer Res . 1994;54:5652–5655. [PubMed] [Google Scholar]
- Mao L, Lee JS, Kurie JM, Fan YH, Lippman SM, Lee JJ, Ro JY, Broxson A, Yu R, Morice RC, Kemp BL, Khuri FR, Walsh GL, Hittelman WN, Hong WK. Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst. 1997;89:857–862. doi: 10.1093/jnci/89.12.857. [DOI] [PubMed] [Google Scholar]
- Wistuba II, Lam S, Behrens C, Virmani AK, Fong KM, LeRiche J, Samet JM, Srivastava S, Minna JD, Gazdar AF. Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst. 1997;89:1366–1373. doi: 10.1093/jnci/89.18.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung J, Kishimoto Y, Sugio K, Virmani A, McIntire DD, Minna JD, Gazdar AF. Allele-specific chromosome 3p deletions occur at an early stage in the pathogenesis of lung carcinoma. J Am Med Ass. 1995;273:558–563. doi: 10.1001/jama.273.7.558. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Sugio K, Hung JY, Virmani AK, McIntire DD, Minna JD, Gazdar AF. Allele-specific loss in chromosome 9p loci in preneoplastic lesions accompanying non-small-cell lung cancers. J Natl Cancer Inst. 1995;87:1224–1229. doi: 10.1093/jnci/87.16.1224. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Taylor D, Koch WM, Zahurak M, Shah K, Sidransky D, Westra WH. Immunohistochemical detection of p53 protein accumulation in head and neck cancer: correlation with p53 gene alterations. Hum Pathol . 1999;30:1221–1225. doi: 10.1016/s0046-8177(99)90041-2. [DOI] [PubMed] [Google Scholar]
- Yashima K, Litzky LA, Kaiser L, Rogers T, Lam S, Wistuba II, Milchgrub S, Srivastava S, Piatyszek MA, Shay JW, Gazdar AF. Telomerase expression in respiratory epithelium during the multistage pathogenesis of lung carcinomas. Cancer Res. 1997;57:2373–2377. [PubMed] [Google Scholar]
- Kitamura H, Kameda Y, Ito T, Hayashi H. Atypical adenomatous hyperplasia of the lung. Implications for the pathogenesis of peripheral lung adenocarcinoma. Am J Clin Pathol. 1999;111:610–622. doi: 10.1093/ajcp/111.5.610. [DOI] [PubMed] [Google Scholar]
- Mao L, Hruban RH, Boyle JO, Tockman M, Sidransky D. Detection of oncogene mutations in sputum precedes diagnosis of lung cancer. . Cancer Res. 1994;54:1634–1637. [PubMed] [Google Scholar]
- Ahrendt SA, Chow JT, Xu LH, Yang SC, Eisenberger CF, Esteller M, Herman JG, Wu L, Decker PA, Jen J, Sidransky D. Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J Natl Cancer Inst. 1999;91:332–339. doi: 10.1093/jnci/91.4.332. [DOI] [PubMed] [Google Scholar]
- Califano J, Ahrendt SA, Meininger G, Westra WH, Koch WM, Sidransky D. Detection of telomerase activity in oral rinses from head and neck squamous cell carcinoma patients. Cancer Res. 1996;56:5720–5722. [PubMed] [Google Scholar]
- Mao L, Lee DJ, Tockman MS, Erozan YS, Askin F, Sidransky D. Microsatellite alterations as clonal markers for the detection of human cancer. Proc Natl Acad Sci USA. 1994;91:9871–9875. doi: 10.1073/pnas.91.21.9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Nagai K, Yoshida J, Yokose T, Kodama T, Takahashi K, Nishimura M, Kawasaki H, Yokozaki M, Nishiwaki Y. The prognosis of resected lung carcinoma associated with atypical adenomatous hyperplasia: a comparison of the prognosis of well-differentiated adenocarcinoma associated with atypical adenomatous hyperplasia and intrapulmonary metastasis. . Cancer. 1997;79:1521–1526. doi: 10.1002/(SICI)1097-0142(19970415)79:8<1521::AID-CNCR12>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Henschke CI, McCauley DI, Yankelevitz DF, Naidich DP, McGuinness G, Miettinen OS, Libby DM, Pasmantier MW, Koizumi J, Altorki NK, Smith JP. Early Lung Cancer Action Project: overall design and findings from baseline screening. Lancet. 1999;354:99–105. doi: 10.1016/S0140-6736(99)06093-6. [DOI] [PubMed] [Google Scholar]